Abstract
In systemic sclerosis (SSc) mortality is mainly linked to lung involvement which is characterized by interstitial lung disease (ILD) and pulmonary hypertension (PH). In SSc, PH may be due to different etiologies, including ILD, chronic thromboembolic disease, pulmonary veno-occlusive disease, and pulmonary arterial hypertension (PAH). The main tool to screen PAH is transthoracic echocardiography (TTE), which has a sensitivity of 90%, even if definitive diagnosis should be confirmed by right heart catheterization (RHC). The radiological evaluation (i.e., HRTC) plays an important role in defining the possible causes and in monitoring the evolution of lung damage. For PAH, identifying individuals who have borderline elevation of pulmonary arterial pressure needs to be appropriately managed and followed. In the past few years, the strategy for the management of PAH has significantly evolved and new trials are underway to test other therapies. This review provides an overview of the tools to evaluate PAH in SSc patients and on treatment options for these patients.
1. Introduction
Systemic sclerosis (SSc) is a connective tissue disease characterized by immune and endothelial dysfunction, inflammation and fibrosis [1,2,3,4,5,6,7,8]. The main cause of death in SSc patients is the effect that collagen overproduction has on the internal organs, such as the pulmonary system [9,10,11,12]. Lung involvement in SSc patients includes fibrosis or pulmonary hypertension (PH) [1,13,14,15,16,17,18,19,20]. In 2018 it was proposed to lower the cut-off for the diagnosis of PH to a mean pulmonary artery pressure (mPAP > 20 mmHg) [12,13,14,21,22,23,24,25,26,27]. PH can be classified as pre- and post-capillary, and more precisely pre-capillary PH is characterized by a pulmonary artery wedge pressure (PAWP) ≤ 15 mmHg and a pulmonary vascular resistance (PVR) ≥ 3 WU, while post-capillary PH is defined by a PAWP > 15 mmHg with normal PVR [12,13,14]. The increase in afterload due to a chronic elevation of pressure in the pulmonary circulation can progressively affect the right ventricle (RV), leading to RV dysfunction [10,11,12,13,14,15,16]. Five categories have been proposed, on the basis of clinical characteristics and pathophysiological mechanisms: Group 1 (PAH); Group 2 (PH due to left heart disease); Group 3 (PH due to lung disease or hypoxia); Group 4 (chronic thromboembolic pulmonary hypertension (CTEPH)); and Group 5 (PH due to unclear or multi-factorial mechanisms). Patients in Group 1, Group 3 and Group 4 present a pre-capillary form of PH, while those in Group 2 may present both an isolated post-capillary PH or a combined PH with a pre-capillary component, given by an elevated diastolic pressure gradient or an increase in pulmonary vascular resistance [10,11,12,13,14,15,16]. In SSc, PH can be correlated by: vasculopathy of the small pulmonary arteries (Group 1 PAH); interstitial lung disease (Group 3); and left ventricular systolic or diastolic dysfunction (Group 2) [10,11,12,13,14,15,16]. In SSc, pulmonary hypertension phenotyping should be achieved to provide a correct treatment regimen [10,11,12,13,14,15,16,17,18,19,20]. It is likely that PAH (classified by the WHO as group I) is present in a consistent number of SSc patients, but its mixed forms (either WHO group III with interstitial lung disease) also exist, while some patients may also have post-capillary PH (WHO group II) [10,11,12,13,14,15,16,21,22,23]. Pulmonary hypertension associated with ILD generally derives from an involvement of more than >20% of the lung parenchyma [10,11,12,13,14,15,16,21,22,23]. Some patients may also develop Pulmonary Veno-Occlusive Disease (WHO group 1) or Chronic Thrombo-embolic PH (WHO group 4) (Table 1) [10,11,12,13,14,15,16,21,22,23]. However, PH frequency varies across different reports depending upon the duration of follow-up [10,11,12,13,14,15,16,23,24,25,26,27,28,29,30,31,32,33,34].

Table 1.
Different phenotypes of PH in SSc patients.
The SSc limited cutaneous subset has been associated with PAH [10,11,12,13,14,15,16,23,24,25,26,27,28,29,30,31,32,33,34], and develops in approximately 10% of individuals with SSc [14,21,22,23]. These patients are more likely to have severe intrinsic right ventricular dysfunction, high BNP (B-type natriuretic factor), low DLCO and poor survival [10,11,12,13,14,15,16,23,24,25,26,27,28,29,30,31,32,33,34]. Lungs from patients with SSc-PAH present a characteristic vascular pathology where perivascular lymphocytes and intimal fibrosis are the hallmarks [10,11,12,13,14,15,16,23,24,25,26,27,28,29,30,31,32,33,34,35]. The complex clinical presentation and the distinct vascular pathology of SSc-PAH suggest a unique biology among group I PH conditions [10,11,12,13,14,15,16,23,24,25,26,27,28,29,30,31,32,33,34]. There have been increasing efforts to understand the relationship between autoimmunity, inflammation and the evolution of PAH [10,11,12,13,14,15,16,23,24,25,26,27,28,29,30,31,32,33,34]. The role of autoimmunity has also suggested the use of immunosuppressors as potential therapeutic candidates after standard-of-care vasoactive and vasodilating therapy [16,34].
This review provides an overview on the screening and diagnostic tools and on the treatment options for PAH in SSc patients.
2. Screening Procedures in SSc
Recommendations and systematic reviews have proposed different methods of screening and follow-up of PAH. In particular, transthoracic echocardiography, natriuretic peptide NT-proBNP, and spirometry are suggested for screening/follow up while right heart catheterization is reserved for diagnosis (Table 2).
Table 2.
Investigation tools to properly categorize different types of systemic sclerosis–pulmonary hypertension (SSc–PH).
2.1. Transthoracic Echocardiography
Transthoracic echocardiography (TTE) is the gold standard for the screening of PAH in SSc patients. In ESC/ERS Guidelines and in literature, a TTE is recommended every year in SSc patients, with or without symptoms [13,14]. The usual parameters used to determine the probability of developing PH are: the tricuspid valve regurgitation velocity (TRV), the dilation of the right section of the heart, the presence of pericardial effusion and the dilation of the inferior vena cava. The 2D-speckle tracking echocardiography estimates the strain on the right atrium (RA) and the right ventricle (RV) of the heart [13,14]. Recently a study demonstrated that a deformity of systolic longitudinal peak of 14.48% at the apical segment of the lateral wall of RV has a 100% specificity for PAH development in SSc [13,14]. However, echocardiography has a low predictive value (e.g., the ultra-sound method is operator-dependent).
2.2. Right Heart Catheterization
Right heart catheterization (RHC) is the best technique for PAH diagnosis [10,11,12,13,14,15]. The RHC provides useful information on the degree of hemodynamic damage, determines response to treatments and establishes prognosis of PAH [19,33,34,35,36,37,38,39,40,41,42,43]. The RCH evaluation is recommended when patients have an intermediate or high risk of developing PH, based on ETT evaluation, defined as a TRV peak >2.8 ms−1, or a TRV <2.8 ms−1 (or not measurable) with other variables suggestive of PH [38].
The assessment of pulmonary arterial wedge pressure could be over- or under-estimated (Figure 1). In fact, there are no internationally accepted clinical guidelines presenting the best practice for performing RHC. However, it has also been proposed that the standardization of RHC should be completed, to optimize the use of this technique in routine clinical practice [10,11,12,13,14,15].
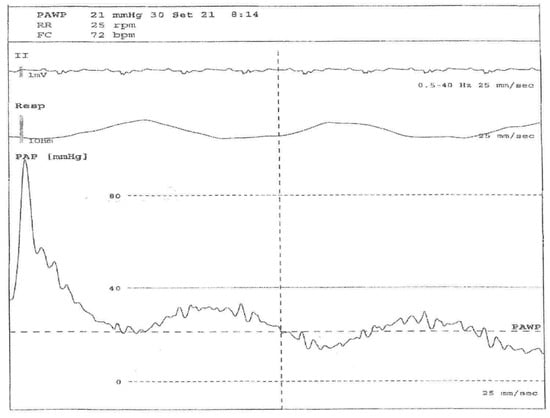
Figure 1.
Right heart catheterization (RHC) evaluation in a 56-year-old systemic sclerosis patient with pulmonary arterial hypertension (PAH). Legend: From below: pulmonary arterial pressure, respiratory and EKG waveforms during arterial catheterization. The first part of the pressure trace reflects the pressure in a pulmonary artery (large swings, dicrotic notch), then the balloon is inflated and the tip of the Swan–Ganz catheter floats until it wedges in a small artery (small swings synchronous with respiratory rate), allowing a pulmonary arterial wedge pressure (PAWP) to be obtained, which is an indirect measure of left ventricle pressure.
2.3. Spirometry
In SSc-PAH patients, there is a frequent reduction in the diffusion of lung carbon monoxide (DLCO) with normal forced vital capacity values (FVC) [26,27]. The reduction of DLCO during the follow-up of SSc patients could be considered as a sign of possible PAH presence [26,27]. A DLCO value < 50% has a high specificity and positive predictive value, but a normal DLCO value does not exclude the presence of PAH [26,27]. A more accurate assessment can be obtained by evaluating the FVC%/DLCO% ratio, as suggested in the DETECT algorithm [23,26,27].
2.4. Radiological Tools
Radiological evaluation plays an important role in defining the possible causes and in monitoring the evolution of the disease.
2.4.1. Chest X-ray
The chest X-ray can show classical radiological findings of PH only in the late stage of the disease. Typical radiological findings are represented by central pulmonary arterial dilation, increase of the diameter of the right interlobar artery (>16 mm) and reduction of the retrosternal space related to right ventricular enlargement [36]. Moreover, chest X-ray can be useful in underlying pulmonary parenchymal disease.
A CT scan is more accurate than chest X-ray in recognizing the radiological features of pulmonary hypertension and a systematic approach is recommended in evaluating CT scans, to promptly recognize parenchymal, vascular and cardiac signs of pulmonary hypertension [37].
2.4.2. High-Resolution Computed Tomography (HRTC)
Parenchymal signs. There are different forms of pulmonary involvement in SSc. HRCT plays a central role in the diagnosis of ILD (interstitial lung disease), in recognizing the pattern of lung involvement and in providing information regarding the extension of the interstitial involvement [3] (Figure 2).
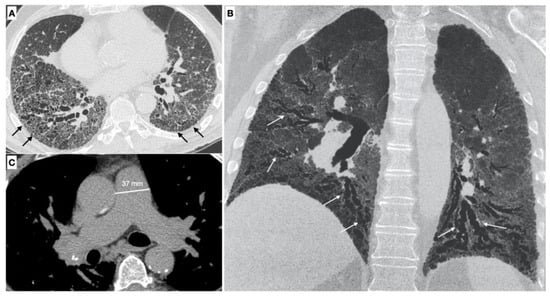
Figure 2.
(A,B) Parenchymal signs: axial high resolution CT scan shows a fibrotic NSIP pattern due to the presence of diffuse irregular septal thickening (black arrows), bronchiectasis and bronchiolectasis on a background of diffuse ground glass opacities. Both lower lobes are symmetrically involved by these alterations. Bronchiectasis and bronchiolectasis (white arrows) are better visualized on the MinIP coronal plane reconstruction; (C) Vascular signs: a main pulmonary artery with a diameter greater than 36 mm measured in a scanning plane of its bifurcation is a sign of pulmonary hypertension.
It is possible to distinguish two different phenotypes of patients with SSc based on the association between ILD and pulmonary hypertension: the first one occurs when PH is associated with ILD, the second one when PH is not related with the extension of the ILD [38]. The association of ILD and PH increase the mortality risk to five-fold in SSc-PAH [39].
Vascular signs. The pulmonary vascular involvement in SSc can cause PAH. Typical radiological findings are represented by a main pulmonary artery with a diameter equal of greater than 29 mm measured in a scanning plane of its bifurcation, this sign has a positive predictive value of 97%, sensitivity of 87% and specificity of 89% for the presence of PH [37,40,41].
Meanwhile a diameter of the main pulmonary artery that is greater than that of the ascending aorta (PA/AA > 1) is another vascular sign of pulmonary hypertension, with a appositive predictive value of 96% and a specificity of 92% [42].
2.4.3. Classification in Different WHO Group of Pulmonary Hypertension
HRCT and CT angiography are particularly useful to subdivide patients into different groups, according to underlying causes of pulmonary hypertension (HP).
Interstitial lung disease (WHO Group III of pulmonary hypertension). ILD is present in up to 90% of patients with SSc [43]. The most common ILD in the SSc patient is the NSIP pattern, characterized by a different degree of inflammation and fibrosis (cellular and fibrotic NSIP). The cellular NSIP pattern is characterized by the presence of ground glass opacities with a bilateral and symmetrical distribution; when fibrotic changes occur (fibrotic NSIP pattern), irregular septal thickening, bronchiectasis and bronchiolectasis can be recognized on HRCT as superimposed features on a background of pre-existing ground glass opacities [43]. The usual interstitial pneumonia (UIP) pattern is less common in SSc and this pattern is characterized by the presence of irregular septal thickening in the peripheral and subpleural regions of the lungs, honeycombing, traction bronchiectasis and pulmonary volume loss. This pattern has a worst prognosis and a faster progression [3] (Figure 3).
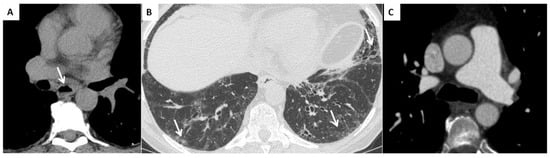
Figure 3.
(A) Esophageal involvement in a 56-year-old male with a diagnosis of SSc. Axial image with a mediastinal window setting shows a dilatation of the esophagus (diameter >1.2 cm) with an air-fluid level (white arrows); (B) Parenchymal signs: axial high resolution CT scan shows a fibrotic NSIP pattern due to the presence of diffuse irregular septal thickening, bronchiectasis and bronchiolectasis on a background of diffuse ground glass opacities (white arrows); (C) Vascular signs: a main pulmonary artery with a diameter greater than 36 mm measured in a scanning plane of its bifurcation is a sign of pulmonary hypertension.
PH can be detected in patients with ILD, in particular in those patients with an extension of fibrotic changes greater than 20% of the lung volume [3], and this is an indicator of morbidity and mortality [3].
Combined pulmonary fibrosis and emphysema syndrome (WHO Group III of pulmonary hypertension). This is a relatively new entity characterized by the presence of emphysema, both paraseptal and centrilobular, in upper lobes in association with lung fibrosis in lung bases. It occurs most often in smokers. This syndrome is associated with a higher risk of developing pulmonary hypertension and is described in patients with connective tissue diseases, including SSc patients [44]. Moreover, it can be identified in SSc patients with no history of smoking exposure [45].
Pulmonary veno-occlusive disease (WHO Group I of pulmonary hypertension). This rare condition is characterized by a diffuse obstruction of the small pulmonary veins. Typical HRCT findings are represented by the presence of centrilobular ground glass opacities, septal thickening, and lymph nodes enlargement. POVD-like aspects are common in patients with SSc-PH [46].
SSc patients with pulmonary hypertension and radiological signs of POVD have a worst prognosis compared with those patients without them therefore it is important to distinguish this phenotype not only at the baseline CT examination but also during follow-up [47].
Chronic thromboembolic pulmonary hypertension (WHO Group IV of pulmonary hypertension). SSc patients are at higher risk of developing chronic thromboembolic disease, and this form of PH that can be surgically treated. Typical vascular signs of CTEPH can be detected on CT angiography, including vascular stenosis, retraction with total or subtotal obstruction, recanalization or residual bands within the vascular lumen. Dilatation of systemic supply is another feature, which is the abnormal enlargement of bronchial, intercostal, phrenic and internal mammary arteries [48].
Cardiac features of PH are represented by a right ventricle hypertrophy, defined as a wall thickening of more than 4 mm or leftward bowing of the interventricular septum, and right ventricular dilatation, defined as a right ventricle to left ventricle diameter ratio of more than 1:1 at the midventricular level on axial image. [49].
Moreover, linear bands, due to previous pulmonary infarction, mosaic pattern of perfusion, bronchial wall thickening and cylindrical airway dilatation (segmental and subsegmental bronchi) are typical parenchymal features of chronic pulmonary hypertension detectable on CT scan [37,50].
2.4.4. Cardiac Magnetic Resonance Imaging
Cardiac involvement is very common in SSc patients. The fibrotic myocardiopathy can cause a dysfunction of the left heart and then an increase of the risk of arrhythmic and ischemic complications, associated with the development of post-capillary pulmonary hypertension (group 2 of the diagnostic classification) [10,11,12,13,51]. The myocardial fibrosis is observed in 50–80% of post-mortem cases [10,11,12,13,52].
The structural alterations are better visualized on cardiac magnetic resonance imaging (MRI) [51]. MRI can also demonstrate pericardial effusion [52]. Several studies have demonstrated that cardiac MR is a very sensitive procedure for the evaluation of myocardial fibrosis in SSc patients and can help to distinguish group 1 (PAH) and group 2 pulmonary hypertension [10,11,12,13]. At the time of writing, the role of cardiac MRI in the evaluation of PAH is not well established.
2.5. Biomarkers
The natriuretic peptide (NT-proBNP) is the most frequent used biomarker in SSc, but it is not specific for right ventricular dysfunctions [19,33,34,35,36,37,38,39,40,41,42,43]. This biomarker may be useful in combination with other screening techniques and in screening algorithms [19,33,34,35,36,37,38,39,40,41,42,43]. The increase of serum levels of uric acid has been included in the DETECT algorithm [19,33,34,35,36,37,38,39,40,41,42,43]. Further studies are still needed to demonstrate the utility of blood markers in PAH diagnosis.
3. Screening Algorithms
The use of algorithms within SSc-PAH screening is associated with a better outcome. Here, we describe three (Table 2) of the most widely used algorithms for PAH screening in SSc [37,53,54,55,56,57,58,59,60].
ESC/ERS Guidelines recommend early echocardiographic screening in asymptomatic SSc patients, followed by the measurement of biomarkers and DLCO [14].
DETECT algorithm is based on two steps: first, the evaluation of a group of functional and blood parameters related to high risk; in case of positivity with a high-risk score, the patient undergoes the second step which evaluates echocardiography parameters. If the second step score is still elevated, the patient will then receive a RHC [23].
ASIG is based on two steps, evaluating DLCO, FVC/DLCO and NT-proBNP values. If either or both parameters (step A) are present patients must then undergo echocardiography associated with other confirmatory tests like HRTC and 6 MWT (step B). If there is no parenchyma lung involvement, a RHC should be performed on the patient [19,37,53,54,55,56,57,58,59].
Impact of Screening for SSc-PAH
There are encouraging data to support the fact that survival from SSc-PAH has significantly improved over time, including through the detection of milder disease. Furthermore, new and combination therapies (discussed later) have dramatically changed the treatment paradigm of SSc-PAH. In the study by Humbert et al., which included two cohorts of SSc-PAH from the same management era, patients enrolled in a systematic PAH evaluation program had less advanced pulmonary vascular disease compared to those who were symptomatic and diagnosed by SSc-PAH by RHC [61]. Furthermore, survival rates were considerably higher in patients who were identified by the PAH detection evaluation at 1-year (100% vs. 75%), 3-years (81% vs. 31%), 5-years (73% vs. 25%) and 8-years (64% vs. 17%) [61]. Similarly, in a recent meta-analysis of randomized, controlled trials (n = 11) and observational registries (n = 19), the survival rate of CTD-PAH patients treated after (compared to before) 2010 was higher at 3 years (73% vs. 65%) [62]. The authors also highlighted that, although survival in CTD-PAH has improved over the last 10 years, the risk of death was still higher than in those with PAH overall [62]. However, in a recent meta-analysis study examining trends in vascular disease in SSc over time, there was a significant improvement in PAH 1-year mortality, but not in PAH 3-year or 5-year mortality [63]. The authors speculated that it will likely require more time to assemble enough treated patients to determine improvement in outcomes. Furthermore, changes in treatment (e.g., upfront combination therapy) are relatively recent and therefore could not translate into the results of the meta-analyses. The authors also observed that there was trend for improvement in three surrogate measures (mPAP, PVR and RAP) of SSc-PAH severity, which supports that regular screening for PAH is an effective strategy to identify and likely treat patients with milder disease [63].
4. Treatment of SSc-PAH
4.1. Overview of SSc-PAH Treatment
Patients should be managed by expert specialist PAH centers. Many valuable authoritative reviews have been written on the management of PAH and guidelines developed by the ESC/ERS for the diagnosis and treatment of PAH [14,64]. Giordano et al. [65], review the relevance and impact of the 2015 ESC/ERS guidelines to patients with SSc-PAH. In this review we will focus on the major drug classes, including combination therapy, relevant to SSc-vasculopathy in SSc. Of interest, a unified vascular endophenotype has been proposed in SSc where vascular-acting therapies could be deployed with a potential disease-modifying effect before the onset of irreversible tissue fibrosis and organ dysfunction [66,67,68].
4.2. Principes of the Treatment Strategy of PAH
The ESC/ERS guidelines divide the treatment strategy of PAH into three steps [14]. First is general measures and supportive therapies (oxygen, anticoagulation, diuretics and digoxin) [14]. Patients should be referred to expert specialist centers and undergo acute vasoreactivity testing for an indication of the need for treatment with calcium channel blockers. The second step consists of calcium channel blockers in vasoreactive patients and PAH-approved therapies in non-vasoreactive patients, either as mono- or combination-therapy according to the prognostic risk [14]. The third step is dictated by the response to the initial treatment strategy. If there is an inadequate response, then combination therapy or lung transplantation should be considered [14].
4.3. General Measures
Patient education is essential and should be delivered by a dedicated multi-disciplinary team. Patients require high quality and appropriate education throughout the course of their disease [69]. General measures include (but are not limited to) supervised exercise training and rehabilitation, avoiding pregnancy, immunizations (influenza and pneumococcal) and psychosocial support [14]. In elective surgery, an epidural is preferred to general anesthesia whenever possible [14].
4.4. Drugs Therapies for SSc-PAH
The target mechanisms, drug classes and examples of the major therapies used for the treatment of SSc-PAH are presented in Table 3.
Table 3.
Target mechanisms, drug classes and examples of therapies used for the treatment of SSc-PAH.
4.4.1. Nitric Oxide Pathway
Nitric oxide is an endogenous vasodilator and also has anti-proliferative effects. Inhibition of the cyclic guanosine monophosphate (cGMP) degrading enzyme phosphodiesterase type-5 (PDE5) results in vasodilation through increasing the local bioavailability of nitric oxide. PDE5 inhibitors approved for the treatment of SSc-PAH are sildenafil (SUPER-1 and SUPER-2 trials) [70,71] and tadalafil (PHIRST-1 and PHIRST-2 trials) [72,73]. Riociguat is an approved guanylate cyclase stimulator which augments the NO-cGMP pathway [74]. The combination of PDE5 inhibitors and riociguat is strictly contraindicated due to the potential for significant hypotension.
4.4.2. Prostacyclin Pathway Agonists
Prostacyclin is a powerful vasodilator and also inhibits platelet aggregation and cell proliferation. Prostacyclin analogues can be delivered by different routes of administration: intravenously, subcutaneous, orally and via inhalation. The drugs include Iloprost, epoprostenol, treprostinil which are all approved for the treatment of SSc-PAH. Currently, intravenous iloprost is only available in Europe (and not the United States). Selexipag is an oral selective IP prostacyclin receptor-agonist. In a phase 3, event-driven randomized, double-blind, placebo-controlled (‘GRIPHON’) trial, which included patients with SSc, the risk of death or complications related to PAH was significantly lower with selexipag [75].
4.4.3. Endothelin Receptor Antagonists
Endothelin-1 is a potent vasoconstrictor, including within the pulmonary vasculature and smooth muscle mitogen. Bosentan (BREATH-1 trial), macitentan (SERAPHIN trial) and ambrisentan (ARIES 1 and 2 trials) are endothelin receptor-antagonists which are approved for the treatment of SSc-PAH [76,77,78,79,80].
4.4.4. Investigational Therapies
Considering the poor prognosis and complex pathobiology of SSc-PAH, other novel therapeutic targets are being actively investigated including immunosuppressive/modulatory therapies (e.g., mycophenolate mofetil) [81]. For example, in a multicenter, double-blind, randomized, placebo-controlled, proof-of-concept trial (n = 57), B-cell depletion with rituximab (1000 mg administered 2 weeks apart) was found to be safe and effective, and, although the primary end-point (mean of change in 6 MWD) favored treatment, this did not reach statistical significance [82].
5. Conclusions
In conclusion, many conditions can cause pulmonary hypertension in patients with SSc, and sometimes different causes can co-exist in the same patient, therefore all the tools to evaluate the damage play an important role in defining the possible causes and in monitoring the evolution of the disease. The correct and early elevation of pulmonary arterial pressure is essential to decide the appropriate management and follow-up. In fact, recently, there was an increase in strategies for the management of PAH and other potential therapies. Furthermore, new drugs are being studied in several trials to provide robust evidence of their role in the improvement of long-term SSc-PH outcomes.
Author Contributions
Conceptualization, B.R., M.H., F.S., E.B., M.C., M.M.-C. and M.C.; writing—original draft preparation, B.R., M.H., E.B., P.C., P.G. and R.P.; writing—review and editing, C.T., R.B.; supervision, M.C. and M.M.-C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Smith, V.; Scirè, C.A.; Talarico, R.; Airo, P.; Alexander, T.; Allanore, Y.; Bruni, C.; Codullo, V.; Dalm, V.; De Vries-Bouwstra, J.; et al. Systemic sclerosis: State of the art on clinical practice guidelines. RMD Open 2019, 4, e000782. [Google Scholar] [CrossRef] [Green Version]
- Ruaro, B.; Confalonieri, M.; Matucci-Cerinic, M.; Salton, F.; Confalonieri, P.; Santagiuliana, M.; Citton, G.; Baratella, E.; Bruni, C. The Treatment of Lung Involvement in Systemic Sclerosis. Pharmaceuticals 2021, 14, 154. [Google Scholar] [CrossRef] [PubMed]
- Denton, C.P.; Wells, A.U.; Coghlan, J.G. Major lung complications of systemic sclerosis. Nat. Rev. Rheumatol. 2018, 14, 511–527. [Google Scholar] [CrossRef] [PubMed]
- Trombetta, A.C.; Smith, V.; Pizzorni, C.; Meroni, M.; Paolino, S.; Cariti, C.; Ruaro, B.; Sulli, A.; Cutolo, M. Quantitative Alterations of Capillary Diameter Have a Predictive Value for Development of the Capillaroscopic Systemic Sclerosis Pattern. J. Rheumatol. 2016, 43, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Codina, A.F.; Walker, K.M.; Pope, J.E. The Scleroderma Algorithm Group Treatment Algorithms for Systemic Sclerosis According to Experts. Arthritis Rheumatol. 2018, 70, 1820–1828. [Google Scholar] [CrossRef] [Green Version]
- Attanasio, U.; Cuomo, A.; Pirozzi, F.; Loffredo, S.; Abete, P.; Petretta, M.; Marone, G.; Bonaduce, D.; De Paulis, A.; Rossi, F.W.; et al. Pulmonary Hypertension Phenotypes in Systemic Sclerosis: The Right Diagnosis for the Right Treatment. Int. J. Mol. Sci. 2020, 21, 4430. [Google Scholar] [CrossRef] [PubMed]
- Ruaro, B.; Santiago, T.; Hughes, M.; Lepri, G.; Poillucci, G.; Baratella, E.; Salton, F.; Confalonieri, M. The Updated Role of Ultrasound in Assessing Dermatological Manifestations in Systemic Sclerosis. Open Access Rheumatol. Res. Rev. 2021, 13, 79–91. [Google Scholar] [CrossRef]
- Ruaro, B.; Casabella, A.; Paolino, S.; Pizzorni, C.; Alessandri, E.; Seriolo, C.; Botticella, G.; Molfetta, L.; Odetti, P.; Smith, V.; et al. Correlation between bone quality and microvascular damage in systemic sclerosis patients. Rheumatology 2018, 57, 1548–1554. [Google Scholar] [CrossRef] [Green Version]
- Tyndall, A.J.; Bannert, B.; Vonk, M.; Airò, P.; Cozzi, F.; Carreira, P.E.; Bancel, D.F.; Allanore, Y.; Müller-Ladner, U.; Distler, O.; et al. Causes and risk factors for death in systemic sclerosis: A study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann. Rheum. Dis. 2010, 69, 1809–1815. [Google Scholar] [CrossRef] [Green Version]
- Chung, L.; Liu, J.; Parsons, L.; Hassoun, P.M.; McGoon, M.; Badesch, D.B.; Miller, D.P.; Nicolls, M.R.; Zamanian, R.T. Characterization of Connective Tissue Disease-Associated Pulmonary Arterial Hypertension From REVEAL. Chest 2010, 138, 1383–1394. [Google Scholar] [CrossRef] [Green Version]
- Poudel, D.R.; Jayakumar, D.; Danve, A.; Sehra, S.T.; Derk, C.T. Determinants of mortality in systemic sclerosis: A focused review. Rheumatol. Int. 2017, 38, 1847–1858. [Google Scholar] [CrossRef] [PubMed]
- Elhai, M.; Meune, C.; Boubaya, M.; Avouac, J.; Hachulla, E.; Balbir-Gurman, A.; Riemekasten, G.; Airò, P.; Joven, B.; Vettori, S.; et al. Mapping and predicting mortality from systemic sclerosis. Ann. Rheum. Dis. 2017, 76, 1897–1905. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Montani, D.; Celermajer, D.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef] [PubMed]
- Galiè, N.; Humbert, M.; Vachiery, J.-L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Heart J. 2016, 37, 67–119. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Rivas, M.; Homs, N.A.; Cuartero, D.; Corbella, X. The prevalence and incidence rate of pulmonary arterial hypertension in systemic sclerosis: Systematic review and meta-analysis. Autoimmun. Rev. 2021, 20, 102713. [Google Scholar] [CrossRef] [PubMed]
- Ruaro, B.; Confalonieri, M.; Salton, F.; Wade, B.; Baratella, E.; Geri, P.; Confalonieri, P.; Kodric, M.; Biolo, M.; Bruni, C. The Relationship between Pulmonary Damage and Peripheral Vascular Manifestations in Systemic Sclerosis Patients. Pharmaceuticals 2021, 14, 403. [Google Scholar] [CrossRef]
- Orlandi, M.; Landini, N.; Sambataro, G.; Nardi, C.; Tofani, L.; Bruni, C.; Randone, S.B.-; Blagojevic, J.; Melchiorre, D.; Hughes, M.; et al. The role of chest CT in deciphering interstitial lung involvement: Systemic sclerosis versus COVID-19. Rheumatology 2021. [Google Scholar] [CrossRef]
- Soldano, S.; Trombetta, A.C.; Contini, P.; Tomatis, V.; Ruaro, B.; Brizzolara, R.; Montagna, P.; Sulli, A.; Paolino, S.; Pizzorni, C.; et al. Increase in circulating cells coexpressing M1 and M2 macrophage surface markers in patients with systemic sclerosis. Ann. Rheum. Dis. 2018, 77, 1842–1845. [Google Scholar] [CrossRef]
- Ruaro, B.; Soldano, S.; Smith, V.; Paolino, S.; Contini, P.; Montagna, P.; Pizzorni, C.; Casabella, A.; Tardito, S.; Sulli, A.; et al. Correlation between circulating fibrocytes and dermal thickness in limited cutaneous systemic sclerosis patients: A pilot study. Rheumatol. Int. 2019, 39, 1369–1376. [Google Scholar] [CrossRef]
- Baratella, E.; Ruaro, B.; Giudici, F.; Wade, B.; Santagiuliana, M.; Salton, F.; Confalonieri, P.; Simbolo, M.; Scarpa, A.; Tollot, S.; et al. Evaluation of Correlations between Genetic Variants and High-Resolution Computed Tomography Patterns in Idiopathic Pulmonary Fibrosis. Diagnostics 2021, 11, 762. [Google Scholar] [CrossRef]
- Hemnes, A.R.; Humbert, M. Pathobiology of pulmonary arterial hypertension: Understanding the roads less travelled. Eur. Respir. Rev. 2017, 26, 170093. [Google Scholar] [CrossRef] [PubMed]
- Hachulla, E.; de Groote, P.; Gressin, V.; Sibilia, J.; Diot, E.; Carpentier, P.; Mouthon, L.; Hatron, P.-Y.; Jego, P.; Allanore, Y.; et al. The three-year incidence of pulmonary arterial hypertension associated with systemic sclerosis in a multicenter nationwide longitudinal study in France. Arthritis Care Res. 2009, 60, 1831–1839. [Google Scholar] [CrossRef] [PubMed]
- Coghlan, J.G.; Denton, C.P.; Grünig, E.; Bonderman, D.; Distler, O.; Khanna, D.; Müller-Ladner, U.; Pope, J.E.; Vonk, M.C.; Doelberg, M.; et al. Evidence-based detection of pulmonary arterial hypertension in systemic sclerosis: The DETECT study. Ann. Rheum. Dis. 2014, 73, 1340–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakkar, V.; Nikpour, M.; Stevens, W.M.; Proudman, S.M. Prospects for improving outcomes in systemic sclerosis-related pulmonary hypertension. Intern. Med. J. 2015, 45, 248–254. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, B.E.; Valerio, C.J.; Keir, G.J.; Handler, C.; Wells, A.U.; Denton, C.P.; Coghlan, J.G. Improving the detection of pulmonary hypertension in systemic sclerosis using pulmonary function tests. Arthritis Care Res. 2011, 63, 3531–3539. [Google Scholar] [CrossRef]
- Mihai, C.; Antic, M.; Dobrota, R.; Bonderman, D.; Chadha-Boreham, H.; Coghlan, J.G.; Denton, C.P.; Doelberg, M.; Grünig, E.; Khanna, D.; et al. Factors associated with disease progression in early-diagnosed pulmonary arterial hypertension associated with systemic sclerosis: Longitudinal data from the DETECT cohort. Ann. Rheum. Dis. 2017, 77, 128–132. [Google Scholar] [CrossRef]
- Visovatti, S.H.; Distler, O.; Coghlan, J.G.; Denton, C.P.; Grünig, E.; Bonderman, D.; Müller-Ladner, U.; Pope, J.E.; Vonk, M.C.; Seibold, J.R.; et al. Borderline pulmonary arterial pressure in systemic sclerosis patients: A post-hoc analysis of the DETECT study. Arthritis Res. Ther. 2014, 16, 493. [Google Scholar] [CrossRef] [Green Version]
- Bae, S.; Saggar, R.; Bolster, M.B.; Chung, L.; Csuka, M.E.; Derk, C.; Domsic, R.; Fischer, A.; Frech, T.; Goldberg, A.; et al. Baseline characteristics and follow-up in patients with normal haemodynamics versus borderline mean pulmonary arterial pressure in systemic sclerosis: Results from the PHAROS registry. Ann. Rheum. Dis. 2012, 71, 1335–1342. [Google Scholar] [CrossRef]
- Lammi, M.R.; Saketkoo, L.A.; Gordon, J.K.; Lauto, P.; Fagan, K.; Steen, V.D. PHAROS Investigators Clinical characteristics and survival of systemic sclerosis patients with pulmonary hypertension and elevated wedge pressure: Observations from the PHAROS cohort. Respirology 2017, 22, 1386–1392. [Google Scholar] [CrossRef] [Green Version]
- Opitz, C.F.; Hoeper, M.; Gibbs, J.S.R.; Kaemmerer, H.; Pepke-Zaba, J.; Coghlan, J.G.; Scelsi, L.; D’Alto, M.; Olsson, K.M.; Ulrich, S.; et al. Pre-Capillary, Combined, and Post-Capillary Pulmonary Hypertension. J. Am. Coll. Cardiol. 2016, 68, 368–378. [Google Scholar] [CrossRef] [Green Version]
- Chung, L.; Farber, H.W.; Benza, R.; Miller, D.; Parsons, L.; Hassoun, P.M.; McGoon, M.; Nicolls, M.R.; Zamanian, R.T. Unique Predictors of Mortality in Patients with Pulmonary Arterial Hypertension Associated with Systemic Sclerosis in the REVEAL Registry. Chest 2014, 146, 1494–1504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, O.A.; Goh, N.; Corte, T.; Rouse, H.; Hennessy, O.; Thakkar, V.; Byron, J.; Sahhar, J.; Roddy, J.; Gabbay, E.; et al. Extent of disease on high-resolution computed tomography lung is a predictor of decline and mortality in systemic sclerosis-related interstitial lung disease. Rheumatology 2012, 52, 155–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, A.; Swigris, J.J.; Bolster, M.B.; Chung, L.; Csuka, M.E.; Domsic, R.; Frech, T.; Hinchcliff, M.; Hsu, V.; Hummers, L.K.; et al. Pulmonary hypertension and interstitial lung disease within PHAROS: Impact of extent of fibrosis and pulmonary physiology on cardiac haemodynamic parameters. Clin. Exp. Rheumatol. 2014, 32, S109–S114. [Google Scholar]
- Thakkar, V.; Stevens, W.; Prior, D.; Youssef, P.; Liew, D.; Gabbay, E.; Roddy, J.; Walker, J.; Zochling, J.; Sahhar, J.; et al. The inclusion of N-terminal pro-brain natriuretic peptide in a sensitive screening strategy for systemic sclerosis-related pulmonary arterial hypertension: A cohort study. Arthritis Res. Ther. 2013, 15, R193. [Google Scholar] [CrossRef] [Green Version]
- Ghataorhe, P.; Rhodes, C.; Harbaum, L.; Attard, M.; Wharton, J.; Wilkins, M.R. Pulmonary arterial hypertension—Progress in understanding the disease and prioritizing strategies for drug development. J. Intern. Med. 2017, 282, 129–141. [Google Scholar] [CrossRef]
- Fleischner, F.G. Pulmonary embolism. Clin. Radiol. 1962, 13, 169–182. [Google Scholar] [CrossRef]
- Peña, E.; Dennie, C.; Veinot, J.; Muñiz, S.H. Pulmonary Hypertension: How the Radiologist Can Help. Radiographics 2012, 32, 9–32. [Google Scholar] [CrossRef]
- Zompatori, M.; Leone, M.B.; Giannotta, M.; Galiè, N.; Palazzini, M.; Reggiani, M.L.B.; Bono, L.; Pollini, G.S. Pulmonary hypertension and systemic sclerosis: The role of high-resolution computed tomography. Radiol. Med. 2013, 118, 1360–1372. [Google Scholar] [CrossRef]
- Mathai, S.C.; Hummers, L.K.; Champion, H.C.; Wigley, F.M.; Zaiman, A.; Hassoun, P.M.; Girgis, R.E. Survival in pulmonary hypertension associated with the scleroderma spectrum of diseases: Impact of interstitial lung disease. Arthritis Care Res. 2009, 60, 569–577. [Google Scholar] [CrossRef]
- Ruaro, B.; Baratella, E.; Confalonieri, P.; Wade, B.; Marrocchio, C.; Geri, P.; Busca, A.; Pozzan, R.; Andrisano, A.G.; Cova, M.A.; et al. High-Resolution Computed Tomography: Lights and Shadows in Improving Care for SSc-ILD Patients. Diagnostics 2021, 11, 1960. [Google Scholar] [CrossRef]
- Foley, R.W.; Kaneria, N.; Ross, R.V.M.; Suntharalingam, J.; Hudson, B.J.; Rodrigues, J.C.; Robinson, G. Computed tomography appearances of the lung parenchyma in pulmonary hypertension. Br. J. Radiol. 2021, 94, 20200830. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.S.; Wells, A.U.; Padley, S.P.G. A CT Sign of Chronic Pulmonary Arterial Hypertension: The Ratio of Main Pulmonary Artery to Aortic Diameter. J. Thorac. Imaging 1999, 14, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Naranjo, M.; Hassoun, P. Systemic Sclerosis-Associated Pulmonary Hypertension: Spectrum and Impact. Diagnostics 2021, 11, 911. [Google Scholar] [CrossRef] [PubMed]
- Bastos, A.D.L.; Corrêa, R.D.A.; Ferreira, G.A. Tomography patterns of lung disease in systemic sclerosis. Radiol. Bras. 2016, 49, 316–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cottin, V.; Cordier, J.-F. Combined pulmonary fibrosis and emphysema in connective tissue disease. Curr. Opin. Pulm. Med. 2012, 18, 418–427. [Google Scholar] [CrossRef]
- Antoniou, K.M.; Margaritopoulos, G.A.; Goh, N.S.; Karagiannis, K.; Desai, S.; Nicholson, A.G.; Siafakas, N.M.; Coghlan, J.G.; Denton, C.P.; Hansell, D.M.; et al. Combined Pulmonary Fibrosis and Emphysema in Scleroderma-Related Lung Disease Has a Major Confounding Effect on Lung Physiology and Screening for Pulmonary Hypertension. Arthritis Rheumatol. 2016, 68, 1004–1012. [Google Scholar] [CrossRef]
- Connolly, M.J.; Abdullah, S.; Ridout, D.A.; Schreiber, B.E.; Haddock, J.A.; Coghlan, J.G. Prognostic significance of computed tomography criteria for pulmonary veno-occlusive disease in systemic sclerosis-pulmonary arterial hypertension. Rheumatology 2017, 56, 2197–2203. [Google Scholar] [CrossRef] [Green Version]
- Günther, S.; Jais, X.; Maitre, S.; Bérezné, A.; Dorfmüller, P.; Seferian, A.; Savale, L.; Mercier, O.; Fadel, E.; Sitbon, O.; et al. Computed tomography findings of pulmonary venoocclusive disease in scleroderma patients presenting with precapillary pulmonary hypertension. Arthritis Care Res. 2012, 64, 2995–3005. [Google Scholar] [CrossRef]
- Ruaro, B.; Baratella, E.; Confalonieri, P.; Confalonieri, M.; Vassallo, F.G.; Wade, B.; Geri, P.; Pozzan, R.; Caforio, G.; Marrocchio, C.; et al. High-Resolution Computed Tomography and Lung Ultrasound in Patients with Systemic Sclerosis: Which One to Choose? Diagnostics 2021, 11, 2293. [Google Scholar] [CrossRef]
- Jaramillo, F.A.; Gutierrez, F.R.; Telli, F.G.D.; Aravena, S.Y.; Javidan-Nejad, C.; Bhalla, S. Approach to Pulmonary Hypertension: From CT to Clinical Diagnosis. Radiographics 2018, 38, 357–373. [Google Scholar] [CrossRef] [Green Version]
- Castañer, E.; Gallardo, X.; Ballesteros, E.; Andreu, M.; Pallardó, Y.; Mata, J.M.; Riera, L. CT Diagnosis of Chronic Pulmonary Thromboembolism. Radiographics 2009, 29, 31–50. [Google Scholar] [CrossRef] [PubMed]
- Launay, D.; Sobanski, V.; Hachulla, E.; Humbert, M. Pulmonary hypertension in systemic sclerosis: Different phenotypes. Eur. Respir. Rev. 2017, 26, 170056. [Google Scholar] [CrossRef] [Green Version]
- Rayner, C.; Grubnic, S. Pulmonary manifestations of systemic autoimmune disease. Best Pr. Res. Clin. Rheumatol. 2004, 18, 381–410. [Google Scholar] [CrossRef] [PubMed]
- Bruni, C.; De Luca, G.; Lazzaroni, M.-G.; Zanatta, E.; Lepri, G.; Airò, P.; Dagna, L.; Doria, A.; Matucci-Cerinic, M. Screening for pulmonary arterial hypertension in systemic sclerosis: A systematic literature review. Eur. J. Intern. Med. 2020, 78, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Giuggioli, D.; Bruni, C.; Cacciapaglia, F.; Dardi, F.; De Cata, A.; Del Papa, N.; Iannone, F.; Lunardi, C.; Maglione, W.; Molinaro, F.; et al. Pulmonary arterial hypertension: Guidelines and unmet clinical needs. Reumatismo 2021, 72, 228–246. [Google Scholar] [CrossRef]
- Hao, Y.; Thakkar, V.; Stevens, W.; Morrisroe, K.; Prior, D.; Rabusa, C.; Youssef, P.; Gabbay, E.; Roddy, J.; Walker, J.; et al. A comparison of the predictive accuracy of three screening models for pulmonary arterial hypertension in systemic sclerosis. Arthritis Res. Ther. 2015, 17, 7. [Google Scholar] [CrossRef] [Green Version]
- Mukerjee, D.; George, D.S.; Knight, C.; Davar, J.; Wells, A.U.; Du Bois, R.M.; Black, C.M.; Coghlan, J.G. Echocardiography and pulmonary function as screening tests for pulmonary arterial hypertension in systemic sclerosis. Rheumatology 2004, 43, 461–466. [Google Scholar] [CrossRef] [Green Version]
- Kowal-Bielecka, O.; Fransen, J.; Avouac, J.; Becker, M.; Kulak, A.; Allanore, Y.; Distler, O.; Clements, P.; Cutolo, M.; Czirjak, L.; et al. Update of EULAR recommendations for the treatment of systemic sclerosis. Ann. Rheum. Dis. 2017, 76, 1327–1339. [Google Scholar] [CrossRef] [Green Version]
- Hsu, V.M.; Chung, L.; Hummers, L.K.; Wigley, F.; Simms, R.; Bolster, M.; Silver, R.; Fischer, A.; Hinchcliff, M.; Varga, J.; et al. Development of pulmonary hypertension in a high-risk population with systemic sclerosis in the Pulmonary Hypertension Assessment and Recognition of Outcomes in Scleroderma (PHAROS) cohort study. Semin. Arthritis Rheum. 2014, 44, 55–62. [Google Scholar] [CrossRef]
- Denton, C.P. Systemic sclerosis: From pathogenesis to targeted therapy. Clin. Exp. Rheumatol. 2015, 33, S3–S7. [Google Scholar]
- Humbert, M.; Yaici, A.; de Groote, P.; Montani, D.; Sitbon, O.; Launay, D.; Gressin, V.; Guillevin, L.; Clerson, P.; Simonneau, G.; et al. Screening for pulmonary arterial hypertension in patients with systemic sclerosis: Clinical characteristics at diagnosis and long-term survival. Arthritis Care Res. 2011, 63, 3522–3530. [Google Scholar] [CrossRef] [PubMed]
- Khanna, D.; Zhao, C.; Saggar, R.; Mathai, S.C.; Chung, L.S.; Coghlan, J.G.; Shah, M.; Hartney, J.; McLaughlin, V. Long-Term Outcomes in Patients With Connective Tissue Disease–Associated Pulmonary Arterial Hypertension in the Modern Treatment Era: Meta-Analyses of Randomized, Controlled Trials and Observational Registries. Arthritis Rheumatol. 2021, 73, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.; Zanatta, E.; Sandler, R.D.; Avouac, J.; Allanore, Y. Improvement with time of vascular outcomes in systemic sclerosis: A systematic review and meta-analysis study. Rheumatology 2021. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; McLaughlin, V.V.; Al Dalaan, A.M.; Satoh, T.; Galiè, N. Treatment of pulmonary hypertension. Lancet Respir. Med. 2016, 4, 323–336. [Google Scholar] [CrossRef]
- Giordano, N.; Corallo, C.; Chirico, C.; Brazzi, A.; Marinetti, A.; Fioravanti, A.; Valenti, R.; Nuti, R.; Pecetti, G. Pulmonary arterial hypertension in systemic sclerosis: Diagnosis and treatment according to the European Society of Cardiology and European Respiratory Society 2015 guidelines. J. Scleroderma Relat. Disord. 2019, 4, 35–42. [Google Scholar] [CrossRef]
- Bruni, C.; Frech, T.; Manetti, M.; Rossi, F.W.; Furst, D.E.; De Paulis, A.; Rivellese, F.; Guiducci, S.; Matucci-Cerinic, M.; Bellando-Randone, S. Vascular Leaking, a Pivotal and Early Pathogenetic Event in Systemic Sclerosis: Should the Door Be Closed? Front. Immunol. 2018, 9, 2045. [Google Scholar] [CrossRef]
- Hughes, M.; Khanna, D.; Pauling, J. Drug initiation and escalation strategies of vasodilator therapies for Raynaud’s phenomenon: Can we treat to target? Rheumatology 2020, 59, 464–466. [Google Scholar] [CrossRef] [Green Version]
- Hughes, M.; Allanore, Y.; Chung, L.; Pauling, J.D.; Denton, C.P.; Matucci-Cerinic, M. Raynaud phenomenon and digital ulcers in systemic sclerosis. Nat. Rev. Rheumatol. 2020, 16, 208–221. [Google Scholar] [CrossRef]
- Devgire, V.; Martin, A.F.; McKenzie, L.; Sandler, R.D.; Hughes, M. A systematic review of internet-based information for individuals with Raynaud’s phenomenon and patients with systemic sclerosis. Clin. Rheumatol. 2020, 39, 2363–2367. [Google Scholar] [CrossRef]
- Galiè, N.; Ghofrani, A.; Torbicki, A.; Barst, R.J.; Rubin, L.J.; Badesch, D.; Fleming, T.; Parpia, T.; Burgess, G.; Branzi, A.; et al. Sildenafil Citrate Therapy for Pulmonary Arterial Hypertension. N. Engl. J. Med. 2005, 353, 2148–2157. [Google Scholar] [CrossRef] [Green Version]
- Badesch, D.B.; Hill, N.S.; Burgess, G.; Rubin, L.J.; Barst, R.J.; Galiè, N.; Simonneau, G. Sildenafil for pulmonary arterial hypertension associated with connective tissue disease. J. Rheumatol. 2007, 34, 2417–2422. [Google Scholar]
- Galiè, N.; Brundage, B.H.; Ghofrani, A.; Oudiz, R.J.; Simonneau, G.; Safdar, Z.; Shapiro, S.; White, R.J.; Chan, M.; Beardsworth, A.; et al. Tadalafil Therapy for Pulmonary Arterial Hypertension. Circulation 2009, 119, 2894–2903. [Google Scholar] [CrossRef] [Green Version]
- Galiè, N.; Denton, C.P.; Dardi, F.; Manes, A.; Mazzanti, G.; Li, B.; Varanese, L.; Esler, A.; Harmon, C.; Palazzini, M. Tadalafil in idiopathic or heritable pulmonary arterial hypertension (PAH) compared to PAH associated with connective tissue disease. Int. J. Cardiol. 2017, 235, 67–72. [Google Scholar] [CrossRef]
- Ghofrani, H.-A.; Galie, N.; Grimminger, F.; Grünig, E.; Humbert, M.; Jing, Z.-C.; Keogh, A.M.; Langleben, D.; Kilama, M.O.; Fritsch, A.; et al. Riociguat for the Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2013, 369, 330–340. [Google Scholar] [CrossRef] [Green Version]
- Sitbon, O.; Channick, R.; Chin, K.; Frey, A.; Gaine, S.; Galiè, N.; Ghofrani, A.; Hoeper, M.; Lang, I.M.; Preiss, R.; et al. Selexipag for the Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2015, 373, 2522–2533. [Google Scholar] [CrossRef] [Green Version]
- Rubin, L.J.; Badesch, D.B.; Barst, R.J.; Galie, N.; Black, C.M.; Keogh, A.; Pulido, T.; Frost, A.; Roux, S.; Leconte, I.; et al. Bosentan Therapy for Pulmonary Arterial Hypertension. N. Engl. J. Med. 2002, 346, 896–903. [Google Scholar] [CrossRef]
- Galiè, N.; Olschewski, H.; Oudiz, R.J.; Torres, F.; Frost, A.; Ghofrani, A.; Badesch, D.B.; McGoon, M.D.; McLaughlin, V.V.; Roecker, E.B.; et al. Ambrisentan for the Treatment of Pulmonary Arterial Hypertension. Circulation 2008, 117, 3010–3019. [Google Scholar] [CrossRef]
- Fischer, A.; Denton, C.P.; Matucci-Cerinic, M.; Gillies, H.; Blair, C.; Tislow, J.; Nathan, S.D. Ambrisentan response in connective tissue disease-associated pulmonary arterial hypertension (CTD-PAH)—A subgroup analysis of the ARIES-E clinical trial. Respir. Med. 2016, 117, 254–263. [Google Scholar] [CrossRef]
- Pulido, T.; Adzerikho, I.; Channick, R.N.; Delcroix, M.; Galiè, N.; Ghofrani, A.; Jansa, P.; Jing, Z.-C.; Le Brun, F.-O.; Mehta, S.; et al. Macitentan and Morbidity and Mortality in Pulmonary Arterial Hypertension. N. Engl. J. Med. 2013, 369, 809–818. [Google Scholar] [CrossRef] [Green Version]
- Rhee, R.L.; Gabler, N.B.; Sangani, S.; Praestgaard, A.; Merkel, P.A.; Kawut, S.M. Comparison of Treatment Response in Idiopathic and Connective Tissue Disease–associated Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2015, 192, 1111–1117. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Li, M.; Zhang, Y.; Shi, X.; Li, L.; Jin, M. The effects and mechanisms of mycophenolate mofetil on pulmonary arterial hypertension in rats. Rheumatol. Int. 2010, 30, 341–348. [Google Scholar] [CrossRef]
- Zamanian, R.T.; Badesch, D.; Chung, L.; Domsic, R.T.; Medsger, T.; Pinckney, A.; Keyes-Elstein, L.; D’Aveta, C.; Spychala, M.; White, R.J.; et al. Safety and Efficacy of B-Cell Depletion with Rituximab for the Treatment of Systemic Sclerosis Associated Pulmonary Arterial Hypertension: A Multi-center, Double-blind, Randomized, Placebo-controlled Trial. Am. J. Respir. Crit. Care Med. 2021, 204, 209–221. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).