1. Introduction
Skeletal muscle is a highly dynamic and plastic organ, able to respond to environmental changes and characterized by complete functional recovery upon perturbations such as endurance exercise, overload or muscle injury [
1]. These exceptional adaptive features of adult skeletal muscle are reduced or even compromised in conditions such as aging and atrophy or in genetic myopathies, such as Duchenne muscular dystrophy (DMD) [
1,
2,
3]. DMD is a lethal X-linked recessive disease that affects approximately 1/3500 boys and is caused by different mutations in the dystrophin gene, leading to the loss of the functional protein, which is crucial for the proper structure and stability of myofibers [
4]. The dystrophin-deficient mouse (C57BL/10ScSn-
DMDmdx/J), referred to as
mdx mouse, represents the most frequently used animal model to study DMD, although the pathology is less severe in this animal compared to DMD patients [
5,
6]. In both cases, this fatal myopathy leads to continuous cycles of degeneration and regeneration, resulting in high heterogeneity in fiber size and distribution as well as an increase in centrally nucleated fibers (CNFs) [
7]. Another key feature of DMD is chronic inflammation, resulting in persistent inflammatory cell infiltration, mainly macrophages, upon the degeneration of myofibers, accompanied by irreversible extracellular matrix deposition (ECM), leading to fibrosis [
8,
9].
The study of skeletal muscle physiology or pathologies mainly relies on histological analyses of muscle cross-sections. This analysis is commonly carried out by measuring the cross-sectional area (CSA) or minimum Feret diameter (MFD) of myofibers and the fiber size distribution in order to evaluate muscle fiber size and heterogeneity within the muscle [
10]. Indeed, in physiological conditions, wild-type mice show a homogenous fiber size distribution and a constant CSA in the absence of perturbations. On the contrary,
mdx mice usually show decreased CSA and an increase in fibers with a smaller caliber, together with high fiber heterogeneity, which becomes more evident with the progression of the disease. Notably, MFD quantification is usually preferable to CSA [
11]. CSA and fiber size distribution measurements are usually performed by anti-laminin immunofluorescence with the goal of detecting fiber boundaries, whereas, for CNF quantification, nuclei detection is also necessary, which can be accomplished, for example, using 4′,6-diamidino-2-phenylindole (DAPI) [
1]. This type of analysis can be performed by many software packages that can allow either manual or automated quantification, although both of these procedures have crucial pros and cons. Indeed, although manual quantification accounts for the critical assessment of the investigator concerning the biological problems under examination, this approach is undoubtedly time-consuming and highly subjective among users. On the contrary, automatized software packages are designed to save time and to standardize the procedure but often do not include a step of “manual revision” by the user, thus compromising the accuracy of the quantification. Moreover, some of these software platforms are not open-source, can be difficult to implement and require specific operating systems or a good knowledge of programming languages [
10,
12,
13,
14,
15,
16,
17,
18]. Additionally, automatized software is usually not designed for the quantification of a cell population or the extracellular matrix deposition within the muscle, two fundamental features in myobiology [
1,
17]. Different readily available software packages are often optimized towards one or more parameters, reducing the ability of the user to mine different data measurements and the versatility of the software [
19].
CellProfiler represents a robust, user-friendly and open-access software platform with algorithms and features that facilitate high-throughput work in biological research [
20]. Advanced algorithms for image analysis are organized in individual modules that can be inserted in a sequential order to generate a customizable pipeline to identify or measure biological elements, named “objects”, or quantify positive areas in acquired images [
20,
21,
22].
In the current manuscript, we propose a method to perform image analysis of muscle sections by using pipelines built with CellProfiler software, which have been recently implemented and updated to CellProfiler 4 [
23]. Specifically, we present the data obtained by using a pipeline, which we named MyoProfiler, to measure MFD, CNF, PNF, cell localization and the number of macrophages in muscle sections from
mdx mice compared with wild-type ones. We also developed another pipeline, which we named SiriusProfiler, for the precise quantification of extracellular matrix deposition. Moreover, with the goal of validating the performance of our method, we compared automatic quantification, performed using CellProfiler, with manual quantification, performed using Fiji software. The results show that these pipelines allow the automatic analysis of multiple images in a quick and reliable manner by using a single software package for multiple outputs, thus representing useful tools for the quantification of key muscle parameters in both physiological and non-physiological conditions.
2. Materials and Methods
2.1. Mice and Ethical Approval
Wild-type (C57BL/10J, The Jackson Laboratory, Bar Harbor, ME, USA) and dystrophic mdx mice (C57BL/10ScSn-DMDmdx/J, The Jackson Laboratory, Bar Harbor, ME, USA) were purchased from Charles River. Five-month-old wild-type and mdx mice were used for experiments. All experimental protocols and procedures were conducted following the National Ethical Guidelines (Italian Ministry of Health; D.L. 26, 4 March 2014), approved by the local ethics committee (protocol number 375/2019/PR). Animals were housed at controlled temperature (22 ± 1 °C) and humidity (60 ± 5%) and maintained under a 12 h/12 h light/dark cycle with ad libitum access to food and water.
Mice were euthanized and then dissected in order to carefully excise tibialis anterior (TA) muscles from the hind limbs. Collected TA muscles were mounted in Optimal Cutting Temperature (OCT, Tissue Tek®, Sakura Finetek, Alphen aan den Rijn, The Netherlands, Europe) compound and then frozen in liquid nitrogen-cooled isopentane (2-methylbutane; Sigma-Aldrich, Merck KGaA, Burlington, MA, USA). Embedded muscles were then cross-sectioned at a thickness of 8 µm using a Leica cryostat (Leica CM1850UV, Wetzlar, Germany) set at −25 °C, and sections were stored in a −80 °C freezer.
2.2. Immunofluorescence of Muscle Sections and Image Acquisition
The immunofluorescence of muscle sections was performed following a previously described procedure [
3,
24]. Primary antibodies used for this study were rabbit polyclonal antibody raised against laminin, α1 (Sigma-Aldrich, Merck KGaA, Burlington, MA, USA; Cat#: L9393, RRID:AB_477163, 1:500) and rat monoclonal antibody raised against F4/80 (Bio-Rad Laboratories, Hercules, CA, USA; Cat#: MCA497G, RRID:AB_872005, 1:300). Secondary antibodies for immunofluorescence were Alexa Fluor
® 488 goat anti-rabbit IgG (H+L; Thermo Fisher Scientific, Waltham, MA, USA; Cat#: A11034, RRID:AB_2576217, 1:500) and Alexa Fluor
® 594 goat anti-rat IgG (H+L; Molecular Probes, Eugene, OR, USA; Cat#: A11007, RRID:AB_141374, 1:500). Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Thermo Fisher Scientific, Waltham, MA, USA; Cat#: D1306, RRID:AB_2629482).
Representative images of TA muscle immunofluorescences were acquired using an Olympus confocal microscope (Olympus FV1200, Olympus, Tokyo, Japan) with 40× magnification and visualized with FV10-ASW software (version 4.2; Olympus, Tokyo, Japan). Images for histological analysis were acquired using an Olympus BX53 microscope mounting an XM10 cam (Olympus, Tokyo, Japan) and using “cellSens Standard” software (version 1.17; Olympus, Tokyo, Japan). We acquired adjacent images at 10× magnification of the whole muscle section from both WT and mdx mice. A few fields with evident histological defects were removed before the analysis in order to avoid artifacts. Images were saved and exported as 16-bit images (grayscale images).
2.3. Staining for Extracellular Matrix Deposition and Image Acquisition
Sirius red staining is commonly used to detect extracellular matrix deposition and fibrosis within tissue sections. Briefly, muscle cryosections were thawed and then fixed with Bouin’s solution (Sigma-Aldrich, Merk KGaA, Burlington, MA, USA; Cat#: HT10132) for 1 h, washed and then stained with Picrosirius red dye (Direct Red 80; Sigma-Aldrich, Merk KGaA, Burlington, MA, USA; Cat#: CI 35780) for 1 h, followed by sequential dehydration in 90%, 100% ethanol and xylene and then mounted with EUKITT (Sigma-Aldrich, Merk KGaA, Burlington, MA, USA; Cat#: 03989). Images were acquired using an Olympus BX-41 microscope (Olympus, Tokyo, Japan) with 10× magnification and visualized using “cellSens Entry” software (version 3.1.1, Olympus, Tokyo, Japan). Specifically, we acquired adjacent images at 10× magnification of entire muscle sections from both genotypes.
2.4. CellProfiler-Based Pipelines for Muscle Analysis
CellProfiler, developed by the Carpenter Lab at the Broad Institute of Harvard and MIT, is open-access software and available for Windows and macOS [
20,
21]. CellProfiler code was written using Python [
22], and an updated, faster version of CellProfiler was recently released (CellProfiler 4) [
23]. Java (
www.java.com, accessed on 20 October 2018) installation and update are required prior to CellProfiler installation. Inexperienced users are encouraged to read the CellProfiler manual before using it. For the analysis of data described in this paper, we used the latest version (4.2.1) of CellProfiler downloaded from the official CellProfiler website (
www.cellprofiler.org, accessed on 11 October 2021) and installed it on a laptop computer (Intel
® Core™ i7 4500 U CPU @1.80 GHz 2.40 GHz, 8.00 GB RAM, and 64 bit Windows 10 Home operating system). CellProfiler can process a wide range of image formats using the BioFormats library (complete list of formats permissible here
https://docs.openmicroscopy.org/bio-formats/5.9.2/supported-formats.html, accessed on 20 February 2018).
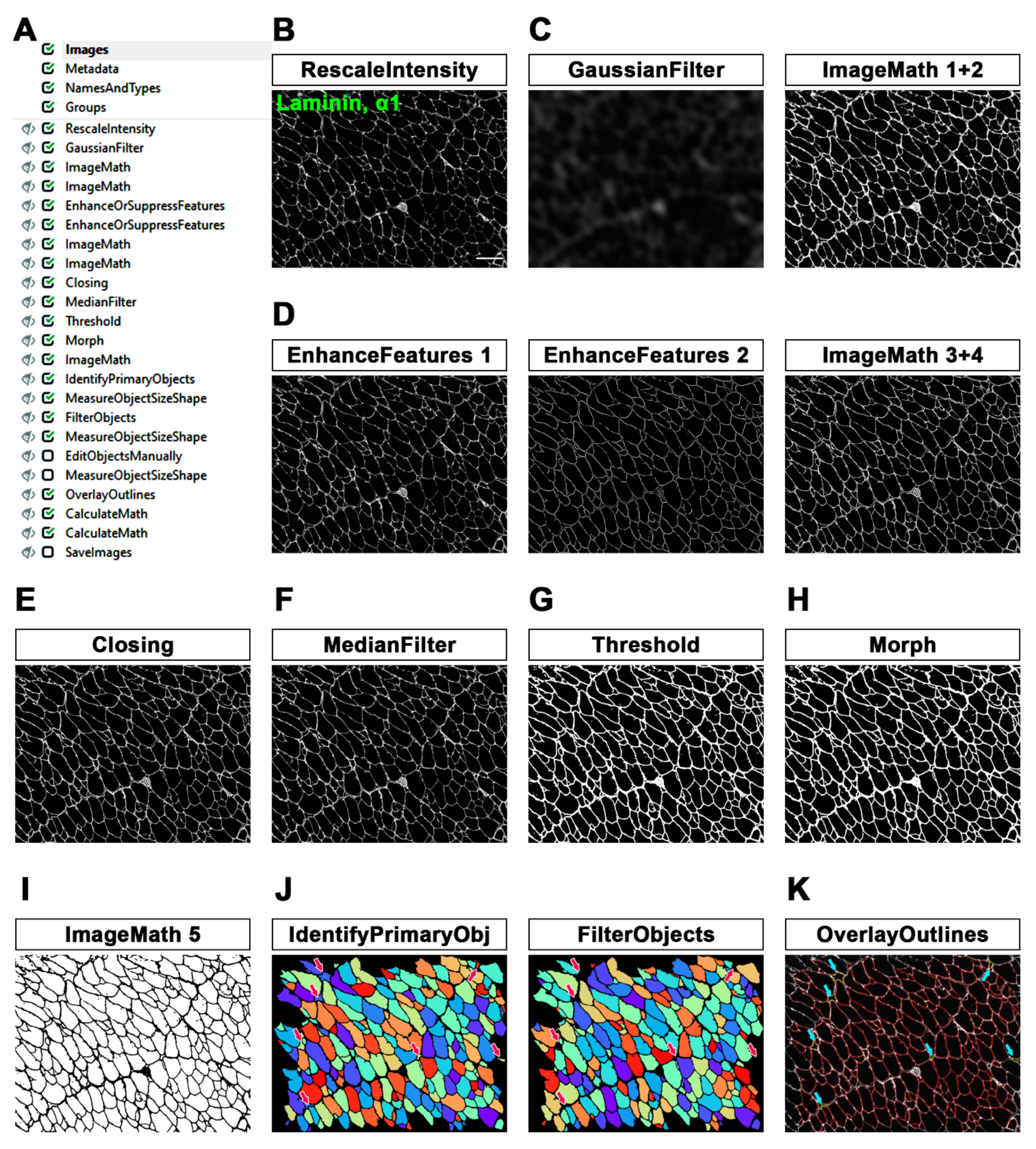
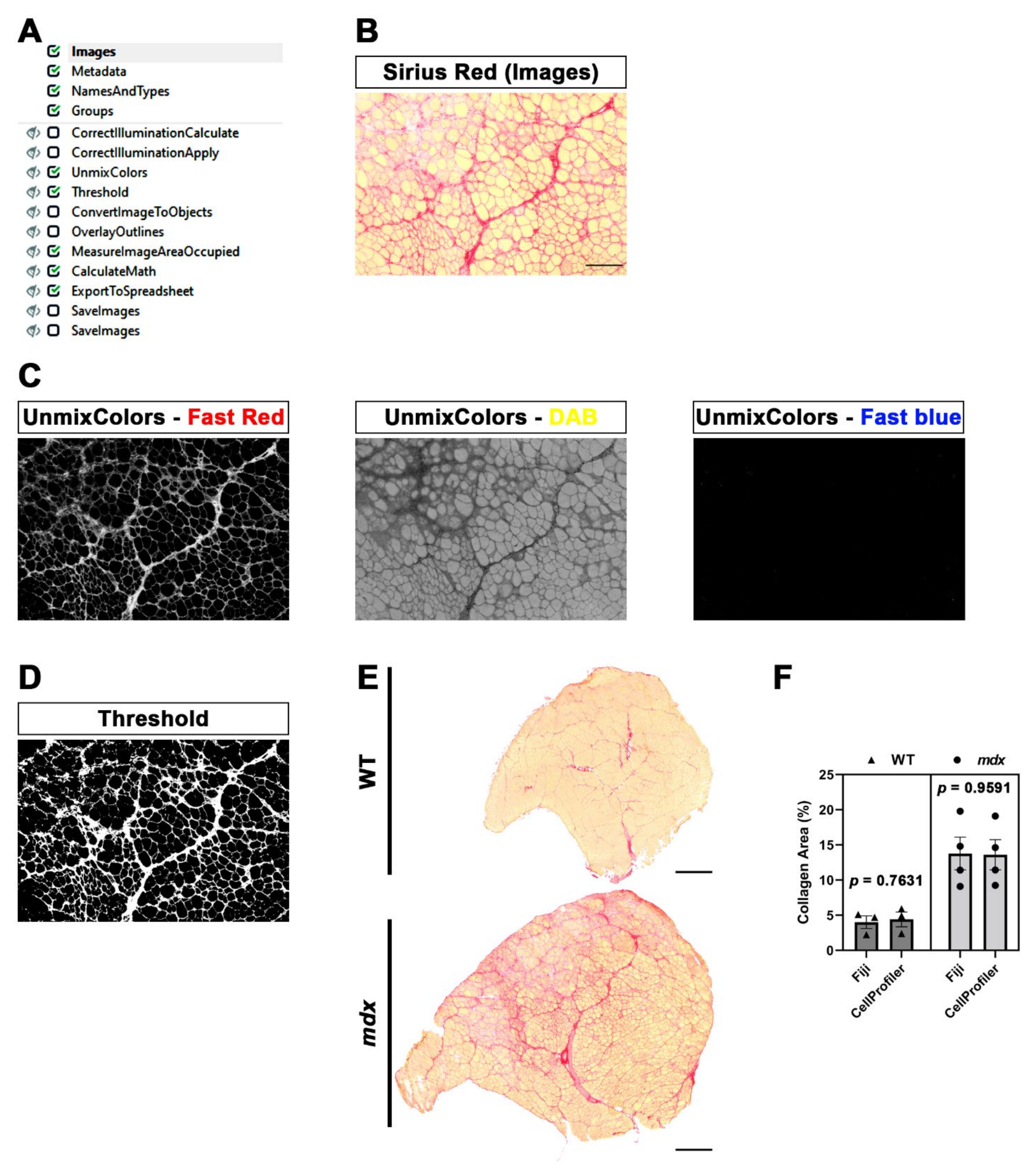
To use a pipeline, the user has to run CellProfiler (version 4.2.1), go to “file”, select “open project” and run the project corresponding to the pipeline of interest. Alternatively, .cpproj or .cppipe files can be run. Then, a list of images can be dropped into the Images module. Image processing and data extraction can be performed through Metadata, NamesAndTypes and Groups modules: for each module, a caption with detailed information is available. Together with these four standard modules, custom modules are displayed as soon as the pipeline/project is opened. We also added captions for each module of both pipelines that we designed. The shared workflow proceeds with data processing, including the pipeline of interest (composed of defined modules), and then with the test mode (Start Test Mode) in order to check the result of each module, followed by image analysis (Analyze Images). Test mode is particularly convenient when the user is designing a new pipeline or implementing an old one in order to check how the pipeline itself works with different image sets. Before running these commands, modules can also be selected/deselected (checkmark) or hidden, depending on the outputs that have to be displayed. The analysis ends with the generation of output data and a spreadsheet. It is important to define input and output folders before image processing. Pipelines developed in our lab are available in the
Supplementary Materials section (Supplementary Files S1–S3). Additionally, in the
Supplementary Materials section, we provide a troubleshooting guide (Troubleshooting_guide,
Supplementary File S4).
2.5. Validation of CellProfiler-Based Pipelines by Fiji
Quantifications obtained with pipelines designed with CellProfiler were validated using Fiji software [
25,
26]. The quantification of the minimum Feret diameter (MFD) of muscle fibers began using anti-laminin, α1-labeled images. DAPI-labeled nuclei and anti-F480-stained macrophages were manually counted using the Cell Counter plugin. CNFs were quantified by combining anti-laminin, α1-labeled images and DAPI-labeled images. All numerical data were exported to Excel files and used for final quantifications. Sirius red quantifications with Fiji were performed using the Color Deconvolution plugin, and the red image was thresholded using the Otsu threshold method [
27]. All analyzed images and samples used for quantifications in CellProfiler were used for the validation with Fiji.
2.6. Statistical Analysis
Data are presented as means plus/minus standard error of the mean (SEM). Output data were compared by 2-tailed unpaired Student’s t-test. Results with p value < 0.05 were considered statistically significant. All “p values” are indicated on the graphs in the figures. All data analyses were performed using GraphPad Prism 9.3 (GraphPad Software, San Diego, CA, USA).
4. Discussion
Dystrophic muscles show high fiber heterogeneity, an elevated number of centrally nucleated fibers (CNFs) and heterogeneous cell populations and dynamics, especially in close proximity to injury sites. Moreover, due to massive extracellular matrix (ECM) deposition, significant interstitial spaces are detectable, especially at late stages of the disease [
30]. For this reason, the full automation of fiber quantification can be tricky and inaccurate, and hence, a further step of manual adjustment and revision is required [
15].
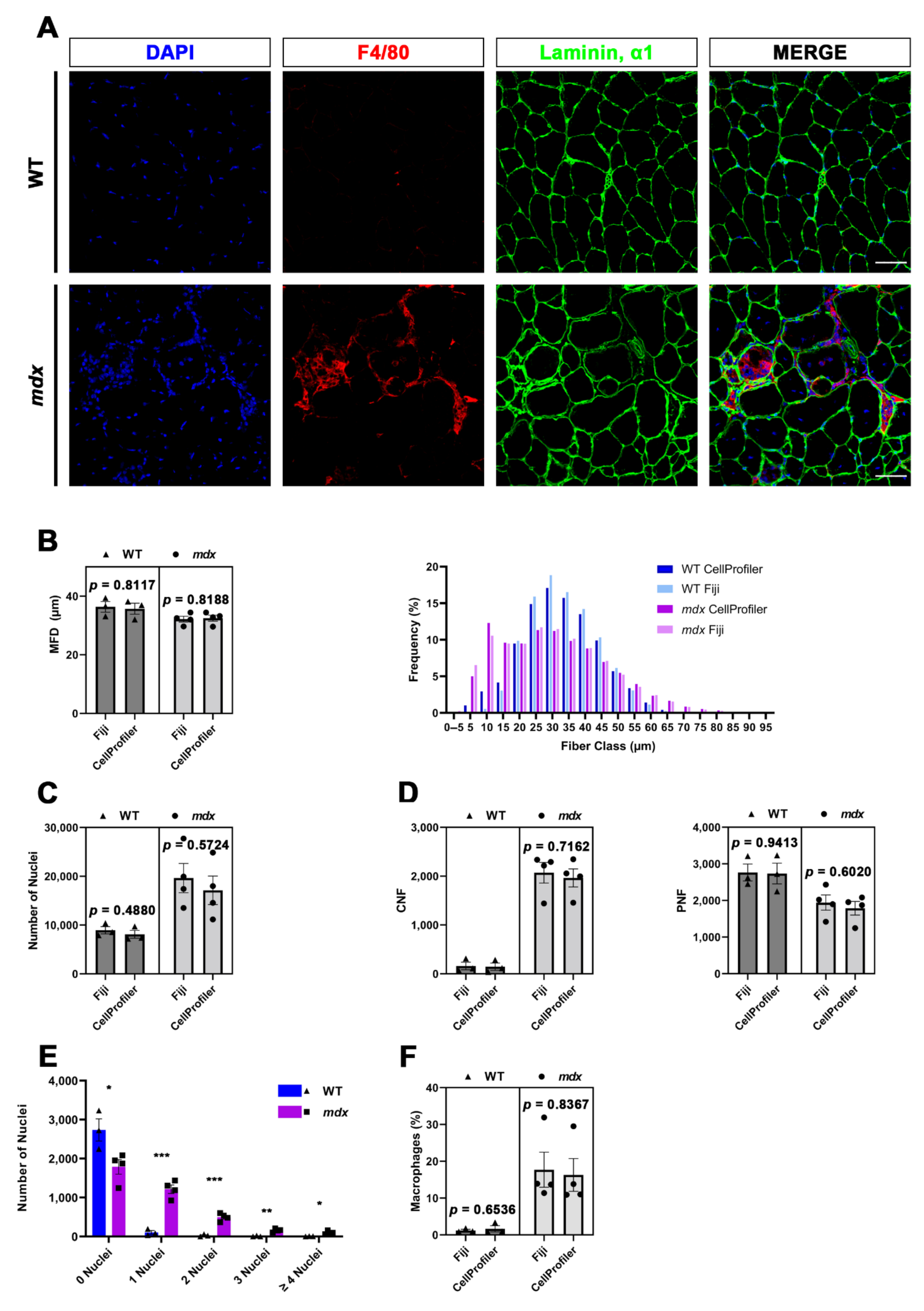
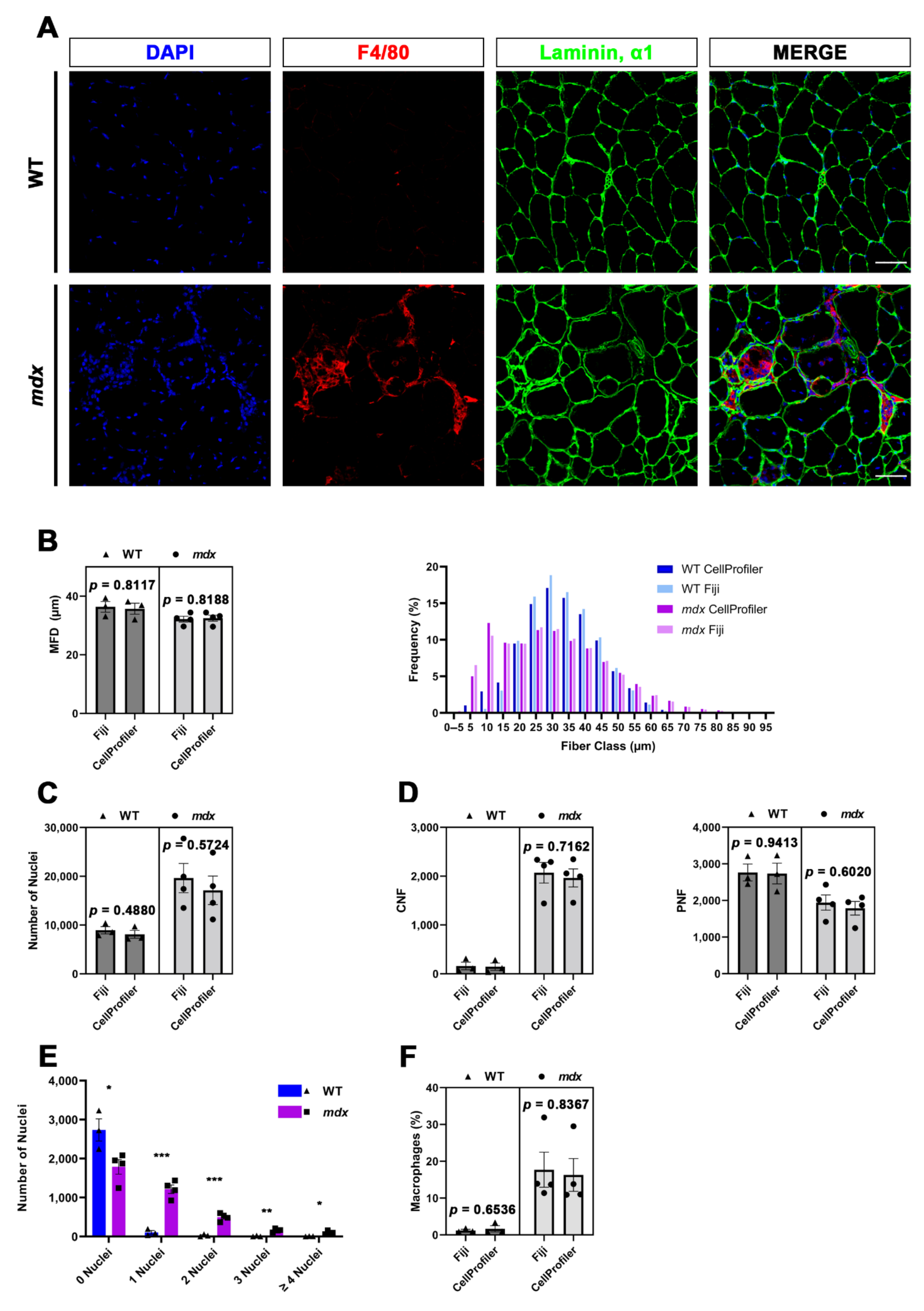
In the present study, by taking advantage of CellProfiler software, we developed two pipelines for the fully automated histological analysis of specific muscle hallmarks, starting from tibialis anterior (TA) sections of mdx mice, a widely used animal model for Duchenne muscular dystrophy (DMD), and their wild-type (WT) littermates. The quantifications performed with CellProfiler were then compared to those performed with Fiji in order to validate the robustness and efficiency of our method. It is therefore clear that this work did not aim to address differences between the two genotypes, which have been widely investigated in a multitude of previous works. We are interested in the validation of our CellProfiler-based pipelines. The first pipeline, named MyoProfiler, was designed for enhancing and redefining myofiber boundaries, starting from images of muscle cross-sections stained with laminin, α1 antibody; then, once the signal was satisfactorily identified, muscle sections were segmented, and their cross-sectional area (CSA) and minimum Feret diameter (MFD) were measured. The second part of MyoProfiler is aimed at identifying and segmenting DAPI-stained nuclei and at classifying segmented myofibers into CNFs and peripherally nucleated fibers (PNFs). The last part of the pipeline included the detection of macrophages, starting from the detection of F4/80, a pan-macrophage-specific surface marker. Finally, the SiriusProfiler pipeline was developed with the goal of identifying and quantifying ECM from Sirius red-stained muscle sections. To this end, we used the same samples used for the analysis by MyoProfiler. Concerning the time needed for analysis, Myoprofiler takes ~10 min to analyze 10 images, while an analysis performed with Fiji takes several hours for the user. Meanwhile, SiriusProfiler takes ~80 s to analyze 10 images, while manual quantification with Fiji takes ~13 min.
In the MyoProfiler pipeline, one of the most critical steps to deal with is the identification of muscle fibers and their appropriate segmentation. Muscle fiber integrity and laminin, α1 signals rely on the quality of muscle sections. Moreover, the laminin signal identifies not only myofiber boundaries but also nerve bundles, capillaries, veins, arteries and interstitial space (among fiber boundaries). It is therefore necessary to enhance the laminin signal, fix interrupted fibers where possible and reduce the background. All of the steps preceding the Threshold module resolve those problems. Moreover, we used the three-class Otsu method [
27], in which we assigned the middle-class to the background in order to exclusively select the positive signal. Object segmentation (IdentifyPrimaryObjects module) was performed using the minimum-cross entropy threshold method [
29]. Segmented fibers can be filtered out (FilterObjects module) if they do not achieve specified filter values (area, form factor and solidity). Finally, the automation of the procedure can be supported by a step of manual revision (EditObjects), necessary for an accurate evaluation of the quantification. Interestingly, the lack of this step was one of the limitations of the previously published MuscleAnalyzer pipeline [
32].
Contrary to myofibers, the identification and segmentation of nuclei are easily affordable, as is nuclei segmentation from 2D cell culture images. This is possible since nuclei show a relatively uniform morphology, dimension and contrast due to the high contrast of the DAPI signal relative to the background. Once we reduced the salt-and-pepper background noise (MedianFilter) and increased the speckle features (EnhanceOrSuppressFeatures), we identified nuclei using an adaptive minimum-cross entropy threshold [
29]. The quantification of nuclei worked appropriately, and the values obtained were comparable to those obtained with Fiji, although with Fiji, we detected slightly more nuclei. This is probably because in CellProfiler analysis, we excluded objects touching the borders. The identified nuclei were then shrunk to one point (ExpandOrShrinkObjects) and masked with 5-pixel-shrunken fibers (MaskObjects) in order to detect CNFs and PNFs with RelateObjects and ClassifyObjects modules. The strategy of identification and quantification of CNFs and PNFs, as with myofiber and nuclei, worked efficiently.
We also used the identified nuclei for the detection and quantification of resident (in WT mice) and infiltrating (in mdx mice) macrophages. Once the background noise had been reduced (MedianFilter) and the F4/80 signal had been enhanced (EnhanceOrSuppressFeatures), we once again used an adaptive minimum cross-entropy threshold for macrophage segmentation. Since F4/80 is a membrane surface marker, we chose to expand nuclei dimensions by 2 pixels and then expanded segmented nuclei with macrophages in order to better detect them. To finely select true macrophages, we decided to discard all objects that were partially masked (10% of masking region). Despite the challenge in detecting macrophages, especially in mdx cross-sections, we found overall comparable values in CellProfiler vs. Fiji analysis.
The SiriusProfiler pipeline, in contrast to MyoProfiler, works on brightfield images. This pipeline consists of a few crucial steps: the splitting of the input image into red, yellow and blue components and then the application of a Threshold module to the image corresponding to the red component. In this case, we used the three-class Otsu method [
27], in which we assigned the middle-class intensity to the foreground, since Sirius red staining always shows a bright red signal and a less intense one. For Fiji analysis, we basically used the same approach, and we found no differences between the two methods, demonstrating the effectiveness of this method.
Occasionally, experimental errors can occur during the histological preparation of muscle samples, such as the wrong orientation of the sectioning angle (i.e., oblique sectioning), thus resulting in muscle fibers with a non-polygonal/non-circular aspect. This fact results in the incorrect measurement of muscle area by the CSA of myofibers, as has been previously demonstrated [
33]. This inconvenient issue can be overcome using the minimal Feret diameter (MFD) as a parameter for the analysis of muscle fibers. Indeed, MFD is defined as the distance between the two parallel planes restricting the object perpendicular to that direction, so it is independent of the sectioning angle of the sample [
33]. Using MyoProfiler, we quantified both CSA and MFD, but we present only the MFD quantification (
Figure 3B). Moreover, isolated muscles can undergo poor inclusion or inappropriate storage before the sectioning. Finally, histological artifacts can occur during the sectioning of muscles, or they can have an uneven or irregular signal pattern due to errors occurring during the staining. Even one of these events can make the identification of objects and cellular components difficult, thus also affecting the quantification. This identification is allowed by CellProfiler thanks to the IdentifyPrimaryObjects module, which relies on a thresholding method that needs to be finely tuned in order to realize correct image segmentation. Fortunately, CellProfiler provides a test mode that makes it possible to test the pipeline on selected image sets and correct or change specific parameters once the output image has been generated. Of course, it is always better to test the pipeline, with a selected parameter, on a large set of images and on images of perfect or poorer quality, thus making the pipeline more robust. Finally, it is recommended to use appropriate conversion factors depending on camera properties and magnification, as well as parameters set in IdentifyPrimaryObjects, if necessary.
Undoubtedly, an automatic or semi-automatic approach should only be applied using good or average–good staining and images to avoid, for instance, the quantification of interstitial spaces or of other non-fiber structures. MyoProfiler proficiently quantifies muscle fibers with minimal error and ensures the possibility of an automated method to decrease the time required for quantification and, at the same time, offers a step of manual editing in order to maximize the efficiency and reliability of this approach, if needed.
In recent years, several other semi-automatic software packages or tools have been described [
10,
15,
17,
33]. Some of the issues with these approaches include the necessity of programming skills, the need for images of very high quality and the lack of implementation and of batch analysis. Moreover, Lau and colleagues proposed a method to detect and quantify muscle fibers and CNFs by using a previous version of CellProfiler. As also stated by the authors, their pipeline is not designed to identify specific cell populations and does not have a manual editing step [
32]. Finally, Sanz and colleagues proposed a useful pipeline for the detection of fibers and the capillary-to-muscle fiber interface on muscle biopsies. Nevertheless, the pipeline has been designed using only muscles from healthy patients and images acquired with 20× magnification, thus reducing the reproducibility if input images are, for instance, 10× magnification-acquired images from dystrophic muscles [
34]. Furthermore, the benefit of using CellProfiler for automated image analysis relies on its flexibility and the possibility of custom modifications that can also be applied by non-expert users. It can distinguish subtle changes and measure multiple properties at once. Moreover, it can perform batch analysis (thousands of images), and the latest version (CellProfiler 4) has been demonstrated to be faster and less tedious than previous ones [
23]. Finally, the use of CellProfiler hints at the possibility to build user-friendly tools that are able to adapt and perform their tasks without needing to use long and more complex tools based on machine learning or deep learning.
The applicability of our pipelines relies on the possibility of also using them on images acquired from histological sections of human biopsies in order to obtain robust and valuable quantifications of histological parameters in both healthy and diseased patients (e.g., DMD patients). Indeed, as stated in
Appendix A.4, our pipelines can be used also for quantifications on human muscle sections. Therefore, it would be possible, for instance, to histologically visualize the effect of corticosteroids, a widely used therapy in muscular dystrophies [
35]. This should aid research and preclinical studies concerning muscle diseases.
To conclude, future directions starting from this work could include the development of novel CellProfiler-based pipelines aimed at quantifying other histological features of muscle histology as well as the detection and counting of other cell populations infiltrating or residing in skeletal muscle upon immunofluorescence for specific cell markers.



{kind=link}
{kind=link}
{kind=link}
{kind=link}