

Lung Involvement in Systemic Juvenile Idiopathic Arthritis: A Narrative Review
 ,
, 

{kind=link}
Abstract
:1. Introduction
General Aspects of SJIA-LD
2. Risk Factors
3. Clinical Features
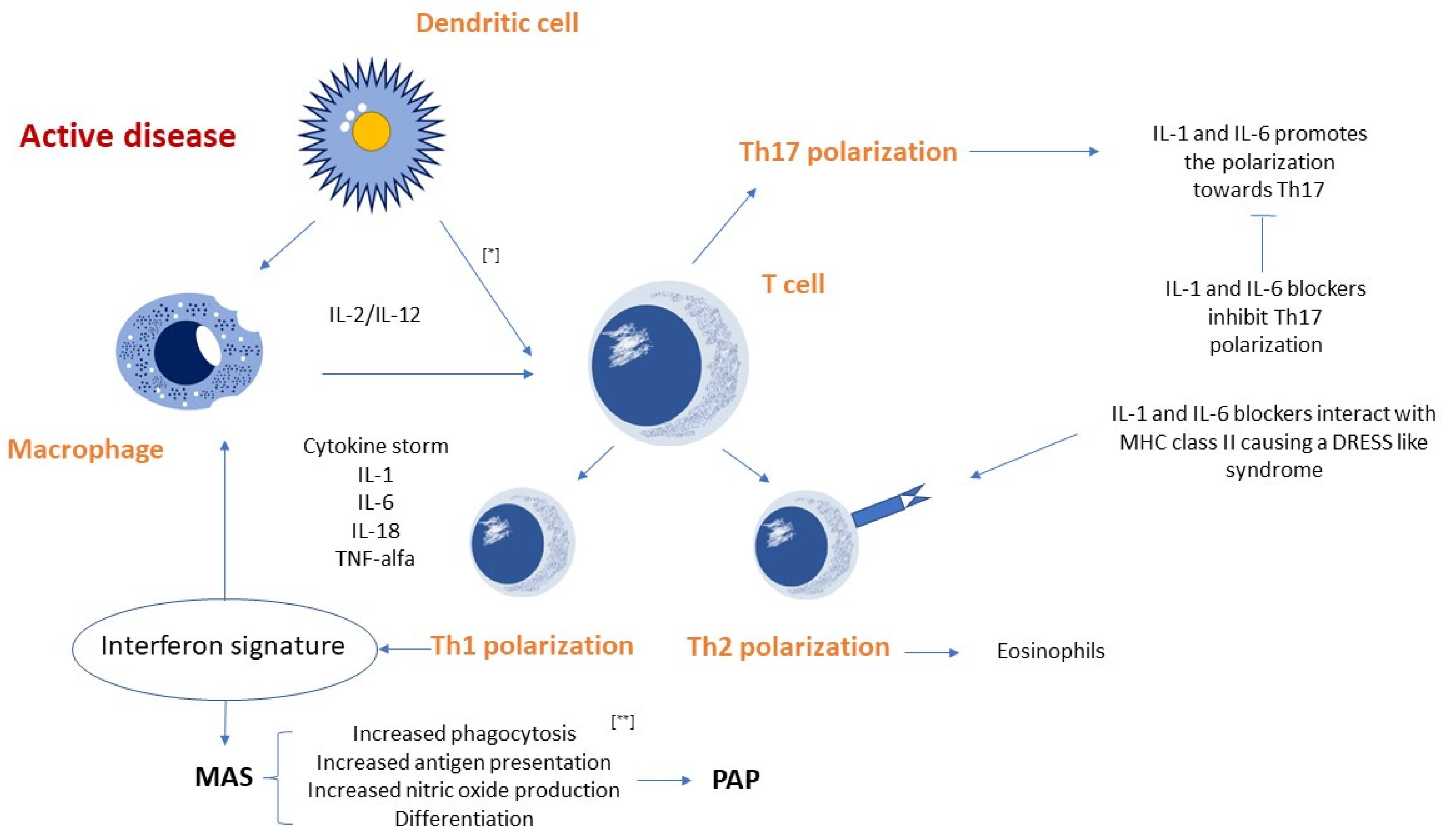
4. Pathogenetic Mechanisms
4.1. Pathogenetic Hypotheses
4.2. Potential Role of IFNγ in sJIA-LD
5. Serum Proteome Characteristics in sJIA-LD
6. Diagnostic Imaging
6.1. High Resolution Computed Tomography
6.2. Lung Ultrasound
7. Therapy
7.1. Anti-Cytokine Therapies
7.1.1. IL-1 and IL-6 Blockers
7.1.2. Etanercept
7.1.3. Abatacept
7.2. Therapeutic Strategies for Refractory sJIA Associated with Lung Disease
7.3. Therapeutic Strategies for Refractory sJIA Associated with MAS
8. Conclusions
Funding
Conflicts of Interest
Abbreviations
| JIA | Juvenile Idiopathic Arthritis; |
| sJIA | systemic Juvenile Idiopathic Arthritis; |
| ILAR | International League of Association Rheumatology; |
| MAS | Macrophage Activation Syndrome; |
| LD | Lung Disease; |
| sJIA-LD | Systemic Juvenile Idiopathic Arthritis with Lung Disease; |
| PAP | Pulmonary Alveolar Proteinosis; |
| ILD | Interstitial Lung Disease; |
| PH | Pulmonary Hypertension; |
| SAVI | STING-associated vasculopathy with onset in infancy; |
| IL-1 | Interleukin-1; |
| IL-6 | Interleukin-6; |
| IL-18 | Interleukin-18; |
| DRESS | Drug Reaction with Eosinophilia and Systemic Symptoms; |
| HLA | Human Leukocyte Antigen; |
| MHC | Major Histocompatibility Complex; |
| TCR | T-Cell Receptor; |
| DIHS | Drug-Induced Hypersensitivity Syndrome; |
| IFNγ | Interferon gamma; |
| CXCL9 | C-X-C motif chemokine ligand 9; |
| CXCL10 | C-X-C motif chemokine ligand 10; |
| BAL | Broncho-Alveolar Lavage; |
| S100A9 | S100 calcium-binding protein A9; |
| GAPDH | Glyceraldehyde 3-Phosphate Dehydrogenase; |
| LDH | Lactate Dehydrogenase; |
| ICAM 5 | Intercellular Adhesion Molecule 5; |
| MMP-7 | Matrix Metalloproteinase-7; |
| CCL11 | C-C motif chemokine Ligand 11; |
| CCL17 | C-C motif chemokine Ligand 17; |
| HRCT | High Resolution Computer Tomography; |
| CT | Computer Tomography; |
| LUS | Lung Ultrasound; |
| GC | Glucocorticoid; |
| CARRA | Childhood Arthritis and Rheumatology Research Alliance; |
| TGF-β | Transforming Growth Factor beta; |
| IL-17 | Interleukin-17; |
| FOXP3 | Forkhead box P3; |
| JAK | Janus Kinase; |
| STAT | Signal Transducer and Activator of Transcription; |
| Fc region | Fragment Crystallizable region; |
| JAK1 | Janus Kinase 1; |
| JAK2 | Janus Kinase 2. |
References
- Lee, J.J.Y.; Schneider, R. Systemic Juvenile Idiopathic Arthritis. Pediatr. Clin. N. Am. 2018, 65, 691–709. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa, S.; Okuni, M. Clinical analysis of 570 cases with juvenile rheumatoid arthritis: Results of a nationwide retrospective survey in Japan. Pediatr. Int. 1997, 39, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Seth, V.; Kabra, S.K.; Semwal, O.P.; Jain, Y. Clinico-immunological profile in juvenile rheumatoid arthritis—An Indian experience. Indian J. Pediatr. 1996, 63, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Petty, R.E.; Southwood, T.R.; Manners, P.; Baum, J.; Glass, D.N.; Goldenberg, J.; He, X.; Maldonado-Cocco, J.; Orozco-Alcala, J.; Prieur, A.-M.; et al. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: Second revision, Edmonton, 2001. J. Rheumatol. 2004, 31, 390–392. [Google Scholar] [PubMed]
- Lang, B.A.; Schneider, R.; Reilly, B.J.; Silverman, E.D.; Laxer, R.M. Radiologic features of systemic onset juvenile rheumatoid arthritis. J. Rheumatol. 1995, 22, 168–173. [Google Scholar]
- Lomater, C.; Gerloni, V.; Gattinara, M.; Mazzotti, J.; Cimaz, R.; Fantini, F. Systemic onset juvenile idiopathic arthritis: A retrospective study of 80 consecutive patients followed for 10 years. J. Rheumatol. 2000, 27, 491–496. [Google Scholar]
- Schulert, G.S.; Grom, A.A. Pathogenesis of Macrophage Activation Syndrome and Potential for Cytokine- Directed Therapies. Annu. Rev. Med. 2015, 66, 145–159. [Google Scholar] [CrossRef] [Green Version]
- Minoia, F.; Davì, S.; Horne, A.; Demirkaya, E.; Bovis, F.; Li, C.; Lehmberg, K.; Weitzman, S.; Insalaco, A.; Wouters, C.; et al. Clinical Features, Treatment, and Outcome of Macrophage Activation Syndrome Complicating Systemic Juvenile Idiopathic Arthritis: A Multinational, Multicenter Study of 362 Patients: Macrophage Activation Syndrome in Systemic JIA. Arthritis Rheumatol. 2014, 66, 3160–3169. [Google Scholar] [CrossRef]
- Schulert, G.S.; Yasin, S.; Carey, B.; Chalk, C.; Do, T.; Schapiro, A.H.; Husami, A.; Watts, A.; Brunner, H.I.; Huggins, J.; et al. Systemic Juvenile Idiopathic Arthritis–Associated Lung Disease: Characterization and Risk Factors. Arthritis Rheumatol. 2019, 71, 1943–1954. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.T.; Wang, C.T.; Gershwin, M.E.; Chiang, B.L. The pathogenesis of oligoarticular/polyarticular vs. systemic juvenile idiopathic arthritis. Autoimmun. Rev. 2011, 10, 482–489. [Google Scholar] [CrossRef]
- Volpi, S.; Picco, P.; Caorsi, R.; Candotti, F.; Gattorno, M. Type I interferonopathies in pediatric rheumatology. Pediatr. Rheumatol. 2016, 14, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, Y.; Weiss, J.E.; Haroldson, K.L.; Lee, T.; Punaro, M.; Oliveira, S.; Rabinovich, E.; Riebschleger, M.; Anton, J.; Blier, P.R.; et al. Pulmonary Hypertension and Other Potentially Fatal Pulmonary Complications in Systemic Juvenile Idiopathic Arthritis: Pulmonary Complications in Systemic JIA. Arthritis Care Res. 2013, 65, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Ter Haar, N.M.; Dijkhuizen, E.H.P.; Swart, J.F.; Van Royen-Kerkhof, A.; El Idrissi, A.; Leek, A.P.; De Jager, W.; De Groot, M.C.H.; Haitjema, S.; Holzinger, D.; et al. Treatment to Target Using Recombinant Interleukin-1 Receptor Antagonist as First-Line Monotherapy in New-Onset Systemic Juvenile Idiopathic Arthritis: Results From a Five-Year Follow-Up Study. Arthritis Rheumatol. 2019, 71, 1163–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saper, V.E.; Ombrello, M.J.; Tremoulet, A.H.; Montero-Martin, G.; Prahalad, S.; Canna, S.; Shimizu, C.; Deutsch, G.; Tan, S.Y.; Remmers, E.F.; et al. Severe delayed hypersensitivity reactions to IL-1 and IL-6 inhibitors link to common HLA-DRB1*15 alleles. Ann. Rheum. Dis. 2022, 81, 406–415. [Google Scholar] [CrossRef] [PubMed]
- Nigrovic, P.A. Storm Warning: Lung Disease in Systemic Juvenile Idiopathic Arthritis. Arthritis Rheumatol. 2019, 71, 1773–1775. [Google Scholar] [CrossRef]
- Binstadt, B.A.; Nigrovic, P.A. The Conundrum of Lu.ng Disease and Drug Hypersensitivity-like Reactions in Systemic Juvenile Idiopathic Arthritis. Arthritis Rheumatol. 2022, 74, 1122–1131. [Google Scholar] [CrossRef]
- Weiss, E.S.; Girard-Guyonvarc’h, C.; Holzinger, D.; De Jesus, A.A.; Tariq, Z.; Picarsic, J.; Schiffrin, E.J.; Foell, D.; Grom, A.A.; Ammann, S.; et al. Interleukin-18 diagnostically distinguishes and pathogenically promotes human and murine macrophage activation syndrome. Blood 2018, 131, 1442–1455. [Google Scholar] [CrossRef]
- Maeno, N.; Takei, S.; Nomura, Y.; Imanaka, H.; Hokonohara, M.; Miyata, K. Highly elevated serum levels of interleukin-18 in systemic juvenile idiopathic arthritis but not in other juvenile idiopathic arthritis subtypes or in Kawasaki disease: Comment on the article by Kawashima et al. Arthritis Rheum. 2002, 46, 2539–2541. [Google Scholar] [CrossRef]
- Saper, V.E.; Chen, G.; Deutsch, G.H.; Guillerman, R.P.; Birgmeier, J.; Jagadeesh, K.; Canna, S.; Schulert, G.; Deterding, R.; Xu, J.; et al. Emergent high fatality lung disease in systemic juvenile arthritis. Ann. Rheum. Dis. 2019, 78, 1722–1731. [Google Scholar] [CrossRef]
- Zaripova, L.N.; Midgley, A.; Christmas, S.E.; Beresford, M.W.; Baildam, E.M.; Oldershaw, R.A. Juvenile idiopathic arthritis: From aetiopathogenesis to therapeutic approaches. Pediatr. Rheumatol. 2021, 19, 135. [Google Scholar] [CrossRef]
- Chen, G.; Deutsch, G.H.; Schulert, G.S.; Zheng, H.; Jang, S.; Trapnell, B.; Lee, P.Y.; Macaubas, C.; Ho, K.; Schneider, C.; et al. Identification of Distinct Inflammatory Programs and Biomarkers in Systemic Juvenile Idiopathic Arthritis and Related Lung Disease by Serum Proteome Analysis. Arthritis Rheumatol. 2022, 74, 1271–1283. [Google Scholar] [CrossRef] [PubMed]
- Erkens, R.; Esteban, Y.; Towe, C.; Schulert, G.; Vastert, S. Pathogenesis and Treatment of Refractory Disease Courses in Systemic Juvenile Idiopathic Arthritis. Rheum. Dis. Clin. N. Am. 2021, 47, 585–606. [Google Scholar] [CrossRef] [PubMed]
- Scholl, P.R.; Diez, A.; Karr, R.; Sekaly, R.P.; Trowsdale, J.; Geha, R.S. Effect of isotypes and allelic polymorphism on the binding of staphylococcal exotoxins to MHC class II molecules. J. Immunol. 1990, 144, 226–230. [Google Scholar] [PubMed]
- Henderson, L.A.; Hoyt, K.J.; Lee, P.Y.; Rao, D.A.; Jonsson, A.H.; Nguyen, J.P.; Rutherford, K.; Julé, A.M.; Charbonnier, L.-M.; Case, S.; et al. Th17 reprogramming of T cells in systemic juvenile idiopathic arthritis. JCI Insight 2020, 5, e132508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bracaglia, C.; de Graaf, K.; Pires Marafon, D.; Guilhot, F.; Ferlin, W.; Prencipe, G.; Caiello, I.; Davì, S.; Schulert, G.; Ravelli, A.; et al. Elevated circulating levels of interferon-γ and interferon-γ-induced chemokines characterise patients with macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. Ann. Rheum. Dis. 2017, 76, 166–172. [Google Scholar] [CrossRef]
- Schulert, G.S. The IL-18/IFNγ axis in systemic JIA and MAS—New answers, more questions. Rheumatology 2021, 60, 3045–3047. [Google Scholar] [CrossRef]
- Sullivan, K.D.; Lewis, H.C.; Hill, A.A.; Pandey, A.; Jackson, L.P.; Cabral, J.M.; Smith, K.P.; Liggett, L.A.; Gomez, E.B.; Galbraith, M.D.; et al. Trisomy 21 consistently activates the interferon response. eLife 2016, 5, e16220. [Google Scholar] [CrossRef]
- Araya, P.; Waugh, K.A.; Sullivan, K.D.; Núñez, N.G.; Roselli, E.; Smith, K.P.; Granrath, R.E.; Rachubinski, A.L.; Estrada, B.E.; Butcher, E.T.; et al. Trisomy 21 dysregulates T cell lineages toward an autoimmunity-prone state associated with interferon hyperactivity. Proc. Natl. Acad. Sci. USA 2019, 116, 24231–24241. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, K.D.; Evans, D.; Pandey, A.; Hraha, T.H.; Smith, K.P.; Markham, N.; Rachubinski, A.L.; Wolter-Warmerdam, K.; Hickey, F.; Espinosa, J.M.; et al. Trisomy 21 causes changes in the circulating proteome indicative of chronic autoinflammation. Sci. Rep. 2017, 7, 14818. [Google Scholar] [CrossRef] [Green Version]
- Park, E.H.; Lee, E.Y.; Shin, K.; Kim, H.A. Tocilizumab-induced anaphylaxis in patients with adult-onset Still’s disease and systemic juvenile idiopathic arthritis: A case-based review. Rheumatol. Int. 2020, 40, 791–798. [Google Scholar] [CrossRef]
- Avau, A.; Matthys, P. Therapeutic Potential of Interferon-γ and Its Antagonists in Autoinflammation: Lessons from Murine Models of Systemic Juvenile Idiopathic Arthritis and Macrophage Activation Syndrome. Pharmaceuticals 2015, 8, 793–815. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, A.; Grom, A.A.; Behrens, E.M.; Cron, R.Q. Macrophage activation syndrome as part of systemic juvenile idiopathic arthritis: Diagnosis, genetics, pathophysiology and treatment. Genes Immun. 2012, 13, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Prencipe, G.; Bracaglia, C.; De Benedetti, F. Interleukin-18 in pediatric rheumatic diseases. Curr. Opin. Rheumatol. 2019, 31, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Iriguchi, S.; Kikuchi, N.; Kaneko, S.; Noguchi, E.; Morishima, Y.; Matsuyama, M.; Yoh, K.; Takahashi, S.; Nakauchi, H.; Ishii, Y. T-cell–restricted T-bet overexpression induces aberrant hematopoiesis of myeloid cells and impairs function of macrophages in the lung. Blood 2015, 125, 370–382. [Google Scholar] [CrossRef] [PubMed]
- Macaubas, C.; Wong, E.; Zhang, Y.; Nguyen, K.D.; Lee, J.; Milojevic, D.; Shenoi, S.; Stevens, A.M.; Ilowite, N.; Saper, V.; et al. Altered signaling in systemic juvenile idiopathic arthritis monocytes. Clin. Immunol. 2016, 163, 66–74. [Google Scholar] [CrossRef] [Green Version]
- de Jager, W.; Vastert, S.; Noordman, B.; Holzinger, D.; Kuis, W.; Prakken, B.; Wulffraat, N. Akinra restores the defective IL-18 NK cell axis in steroid naïve systemic onset JIA patients. Pediatr. Rheumatol. 2011, 9, O33. [Google Scholar] [CrossRef] [Green Version]
- Conant, K.; Wang, Y.; Szklarczyk, A.; Dudak, A.; Mattson, M.P.; Lim, S.T. Matrix metalloproteinase-dependent shedding of intercellular adhesion molecule-5 occurs with long-term potentiation. Neuroscience 2010, 166, 508–521. [Google Scholar] [CrossRef] [Green Version]
- O’Dwyer, D.N.; Norman, K.C.; Xia, M.; Huang, Y.; Gurczynski, S.J.; Ashley, S.L.; White, E.S.; Flaherty, K.R.; Martinez, F.J.; Murray, S.; et al. The peripheral blood proteome signature of idiopathic pulmonary fibrosis is distinct from normal and is associated with novel immunological processes. Sci. Rep. 2017, 7, 46560. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, B.; Branagan, P.; Moloney, F.; Haroon, M.; O’Connell, O.J.; O’Connor, T.M.; O’Regan, K.; Harney, S.; Henry, M.T. Biomarkers to identify ILD and predict lung function decline in scleroderma lung disease or idiopathic pulmonary fibrosis. Sarcoidosis Vasc. Diffus. Lung Dis. Off. J. Wasog. 2015, 32, 228–236. [Google Scholar]
- On behalf of the IPF-PRO Registry Investigators; Todd, J.L.; Neely, M.L.; Overton, R.; Durham, K.; Gulati, M.; Huang, H.; Roman, J.; Newby, L.K.; Flaherty, K.R.; et al. Peripheral blood proteomic profiling of idiopathic pulmonary fibrosis biomarkers in the multicentre IPF-PRO Registry. Respir. Res. 2019, 20, 227. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Jeong, Y.; de Frías, S.P.; Easthausen, I.; Hoffman, K.; Oromendia, C.; Taheri, S.; Esposito, A.J.; Arias, L.Q.; Ayaub, E.A.; et al. Serum proteomic profiling of rheumatoid arthritis–interstitial lung disease with a comparison to idiopathic pulmonary fibrosis. Thorax 2022, 77, 1041–1044. [Google Scholar] [CrossRef] [PubMed]
- Deterding, R.R.; Wagner, B.D.; Harris, J.K.; DeBoer, E.M. Pulmonary Aptamer Signatures in Children’s Interstitial and Diffuse Lung Disease. Am. J. Respir. Crit. Care Med. 2019, 200, 1496–1504. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.A.; Hay, T.; Newell, J.D.; Divgi, V.D.; Fan, L.L. Pediatric diffuse lung disease: Diagnosis and classification using high-resolution CT. Am. J. Roentgenol. 1999, 173, 713–718. [Google Scholar] [CrossRef] [PubMed]
- Brody, A.S. New perspectives in imaging interstitial lung disease in children. Pediatr. Radiol. 2008, 38, 205–207. [Google Scholar] [CrossRef] [PubMed]
- Klusmann, M.; Owens, C. HRCT in paediatric diffuse interstitial lung disease—A review for 2009. Pediatr. Radiol. 2009, 39, 471–481. [Google Scholar] [CrossRef]
- Society, A.T.; Society, E.R. American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. Am. J. Respir. Crit. Care Med. 2002, 165, 277–304. [Google Scholar] [CrossRef] [Green Version]
- Sohn, D.I.; Laborde, H.A.; Bellotti, M.; Seijo, L. Juvenile rheumatoid arthritis and bronchiolitis obliterans organized pneumonia. Clin. Rheumatol. 2006, 26, 247–250. [Google Scholar] [CrossRef]
- García-Peña, P.; Boixadera, H.; Barber, I.; Toran, N.; Lucaya, J.; Enríquez, G. Thoracic Findings of Systemic Diseases at High-Resolution CT in Children. RadioGraphics 2011, 31, 465–482. [Google Scholar] [CrossRef]
- Schultz, R.; Mattila, J.; Gappa, M.; Verronen, P. Development of progressive pulmonary interstitial and intra-alveolar cholesterol granulomas (PICG) associated with therapy-resistant chronic systemic juvenile arthritis (CJA). Pediatr. Pulmonol. 2001, 32, 397–402. [Google Scholar] [CrossRef]
- Athreya, B.H.; Doughty, R.A.; Bookspan, M.; Schumacher, H.R.; Sewell, E.M.; Chatten, J. Pulmonary manifestations of juvenile rheumatoid arthritis. A report of eight cases and review. Clin. Chest Med. 1980, 1, 361–374. [Google Scholar] [CrossRef]
- Vega-Fernandez, P.; Ting, T.V.; Mar, D.A.; Schapiro, A.H.; Deluna, M.D.; Saper, V.E.; Grom, A.A.; Schulert, G.S.; Fairchild, R.M. Lung Ultrasound in Children with Systemic Juvenile Idiopathic Arthritis Associated Interstitial Lung Disease. Arthritis Care Res. 2022. [Google Scholar] [CrossRef]
- Attanasi, M.; Lucantoni, M.; Rapino, D.; Petrosino, M.I.; Marsili, M.; Gasparroni, G.; Di Filippo, P.; Di Pillo, S.; Chiarelli, F.; Breda, L. Lung function in children with juvenile idiopathic arthritis: A cross-sectional analysis. Pediatr. Pulmonol. 2019, 54, 1242–1249. [Google Scholar] [CrossRef] [PubMed]
- Russo, D.; Di Filippo, P.; Attanasi, M.; Lizzi, M.; Di Pillo, S.; Chiarelli, F. Biologic Therapy and Severe Asthma in Children. Biomedicines 2021, 9, 760. [Google Scholar] [CrossRef] [PubMed]
- Nigrovic, P.A. Review: Is There a Window of Opportunity for Treatment of Systemic Juvenile Idiopathic Arthritis?: Window of Opportunity in Systemic JIA. Arthritis Rheumatol. 2014, 66, 1405–1413. [Google Scholar] [CrossRef] [PubMed]
- DeWitt, E.M.; Kimura, Y.; Beukelman, T.; Nigrovic, P.A.; Onel, K.; Prahalad, S.; Schneider, R.; Stoll, M.L.; Angeles-Han, S.; Milojevic, D.; et al. Consensus treatment plans for new-onset systemic juvenile idiopathic arthritis. Arthritis Care Res. 2012, 64, 1001–1010. [Google Scholar] [CrossRef] [Green Version]
- Zielinski, C.E.; Mele, F.; Aschenbrenner, D.; Jarrossay, D.; Ronchi, F.; Gattorno, M.; Monticelli, S.; Lanzavecchia, A.; Sallusto, F. Pathogen-induced human TH17 cells produce IFN-γ or IL-10 and are regulated by IL-1β. Nature 2012, 484, 514–518. [Google Scholar] [CrossRef]
- Li, L.; Kim, J.; Boussiotis, V.A. IL-1β–Mediated Signals Preferentially Drive Conversion of Regulatory T Cells but Not Conventional T Cells into IL-17–Producing Cells. J. Immunol. 2010, 185, 4148–4153. [Google Scholar] [CrossRef] [Green Version]
- Nigrovic, P.A.; Mannion, M.; Prince, F.H.M.; Zeft, A.; Rabinovich, C.E.; Van Rossum, M.A.J.; Cortis, E.; Pardeo, M.; Miettunen, P.M.; Janow, G.; et al. Anakinra as first-line disease-modifying therapy in systemic juvenile idiopathic arthritis: Report of forty-six patients from an international multicenter series. Arthritis Rheum. 2011, 63, 545–555. [Google Scholar] [CrossRef]
- Horneff, G.; Schulz, A.C.; Klotsche, J.; Hospach, A.; Minden, K.; Foeldvari, I.; Trauzeddel, R.; Ganser, G.; Weller-Heinemann, F.; Haas, J.P. Experience with etanercept, tocilizumab and interleukin-1 inhibitors in systemic onset juvenile idiopathic arthritis patients from the BIKER registry. Arthritis Res. Ther. 2017, 19, 256. [Google Scholar] [CrossRef] [Green Version]
- Russo, R.A.G.; Katsicas, M.M. Clinical Remission in Patients with Systemic Juvenile Idiopathic Arthritis Treated with Anti-Tumor Necrosis Factor Agents. J. Rheumatol. 2009, 36, 1078–1082. [Google Scholar] [CrossRef]
- Verweyen, E.L.; Schulert, G.S. Interfering with interferons: Targeting the JAK-STAT pathway in complications of systemic juvenile idiopathic arthritis (SJIA). Rheumatology 2022, 61, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Guan, P.; Sprague, L.; Verbist, K.; Tedrick, P.; An, Q.A.; Cheng, C.; Kurachi, M.; Levine, R.; Wherry, E.J.; et al. Janus kinase inhibition lessens inflammation and ameliorates disease in murine models of hemophagocytic lymphohistiocytosis. Blood 2016, 127, 1666–1675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albeituni, S.; Verbist, K.C.; Tedrick, P.E.; Tillman, H.; Picarsic, J.; Bassett, R.; Nichols, K.E. Mechanisms of action of ruxolitinib in murine models of hemophagocytic lymphohistiocytosis. Blood 2019, 134, 147–159. [Google Scholar] [CrossRef]
- Maschalidi, S.; Sepulveda, F.E.; Garrigue, A.; Fischer, A.; de Saint Basile, G. Therapeutic effect of JAK1/2 blockade on the manifestations of hemophagocytic lymphohistiocytosis in mice. Blood 2016, 128, 60–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verweyen, E.; Holzinger, D.; Weinhage, T.; Hinze, C.; Wittkowski, H.; Pickkers, P.; Albeituni, S.; Verbist, K.; Nichols, K.E.; Schulert, G.; et al. Synergistic Signaling of TLR and IFNα/β Facilitates Escape of IL-18 Expression from Endotoxin Tolerance. Am. J. Respir. Crit. Care Med. 2020, 201, 526–539. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Lee, P.Y.; Yao, X.; Zheng, S.; Li, T. Tofacitinib Treatment of Refractory Systemic Juvenile Idiopathic Arthritis. Pediatrics 2019, 143, e20182845. [Google Scholar] [CrossRef]
- Bader-Meunier, B.; Hadchouel, A.; Berteloot, L.; Polivka, L.; Béziat, V.; Casanova, J.-L.; Lévy, R. Effectiveness and safety of ruxolitinib for the treatment of refractory systemic idiopathic juvenile arthritis like associated with interstitial lung disease: A case report. Ann. Rheum. Dis. 2022, 81, e20. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Hosokawa, T.; Kawashima, H. Successful treatment of plasma exchange for refractory systemic juvenile idiopathic arthritis complicated with macrophage activation syndrome and severe lung disease. Ann. Rheum. Dis. 2022, 81, e61. [Google Scholar] [CrossRef] [Green Version]
- Paim, L.B.; Landim, M.L.L.; Firmino, S.L.; Monteiro, C.M.B.E.; Cordeiro, L.R.; de Araujo, F.G.; de Carvalho, J.F. Canakinumab for Pulmonary Artery Hypertension and Macrophage Activation Syndrome Associated with Uncontrolled Systemic Juvenile Idiopathic Arthritis. Indian J. Pediatr. 2021, 88, 608–609. [Google Scholar] [CrossRef]
- Rood, J.E.; Rezk, A.; Pogoriler, J.; Finn, L.S.; Burnham, J.M.; Josephson, M.B.; Bar-Or, A.; Behrens, E.M.; Canna, S.W. Improvement of Refractory Systemic Juvenile Idiopathic Arthritis-Associated Lung Disease with Single-Agent Blockade of IL-1β and IL-18. J. Clin. Immunol. 2022. [Google Scholar] [CrossRef]
- Lily’s Fight with Macrophage Activation Syndrome: A New Drug Comes to the Rescue. Available online: https://systemicjia.org/lilys-fight-with-macrophage-activation-syndrome-mas-new-drug/ (accessed on 27 October 2022).
- Hayes, D.; Wilson, K.C.; Krivchenia, K.; Hawkins, S.M.M.; Balfour-Lynn, I.M.; Gozal, D.; Panitch, H.B.; Splaingard, M.L.; Rhein, L.M.; Kurland, G.; et al. Home Oxygen Therapy for Children. An Official American Thoracic Society Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2019, 199, e5–e23. [Google Scholar] [CrossRef] [PubMed]
- Aytaç, S.; Batu, E.D.; Ünal, Ş.; Bilginer, Y.; Çetin, M.; Tuncer, M.; Gümrük, F.; Özen, S. Macrophage activation syndrome in children with systemic juvenile idiopathic arthritis and systemic lupus erythematosus. Rheumatol. Int. 2016, 36, 1421–1429. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrongari, D.; Di Filippo, P.; Misticoni, F.; Basile, G.; Di Pillo, S.; Chiarelli, F.; Attanasi, M. Lung Involvement in Systemic Juvenile Idiopathic Arthritis: A Narrative Review. Diagnostics 2022, 12, 3095. https://doi.org/10.3390/diagnostics12123095
Petrongari D, Di Filippo P, Misticoni F, Basile G, Di Pillo S, Chiarelli F, Attanasi M. Lung Involvement in Systemic Juvenile Idiopathic Arthritis: A Narrative Review. Diagnostics. 2022; 12(12):3095. https://doi.org/10.3390/diagnostics12123095
Chicago/Turabian StylePetrongari, Duilio, Paola Di Filippo, Francesco Misticoni, Giulia Basile, Sabrina Di Pillo, Francesco Chiarelli, and Marina Attanasi. 2022. "Lung Involvement in Systemic Juvenile Idiopathic Arthritis: A Narrative Review" Diagnostics 12, no. 12: 3095. https://doi.org/10.3390/diagnostics12123095
APA StylePetrongari, D., Di Filippo, P., Misticoni, F., Basile, G., Di Pillo, S., Chiarelli, F., & Attanasi, M. (2022). Lung Involvement in Systemic Juvenile Idiopathic Arthritis: A Narrative Review. Diagnostics, 12(12), 3095. https://doi.org/10.3390/diagnostics12123095