A Pilot Analysis of Circulating cfRNA Transcripts for the Detection of Lung Cancer

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
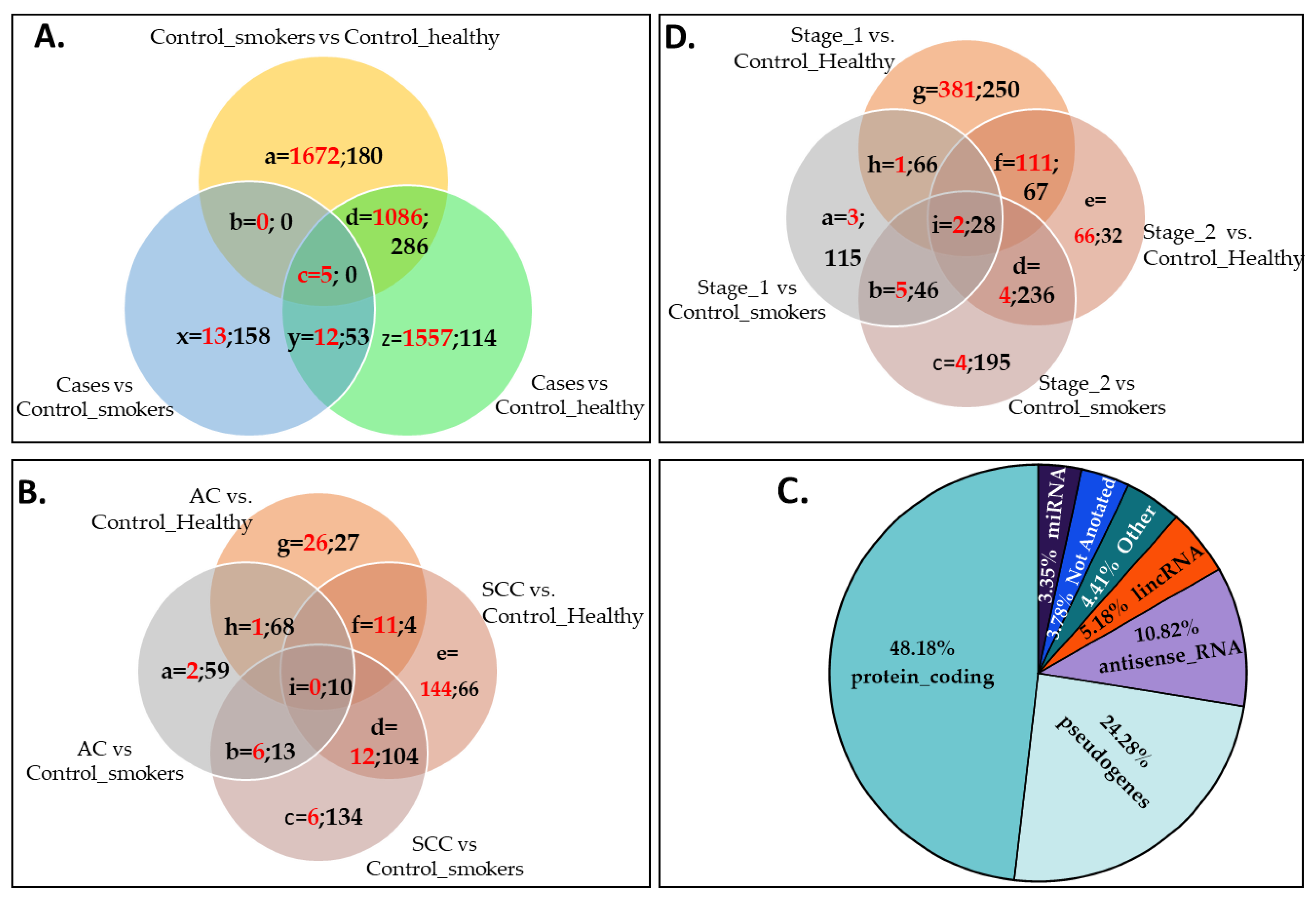
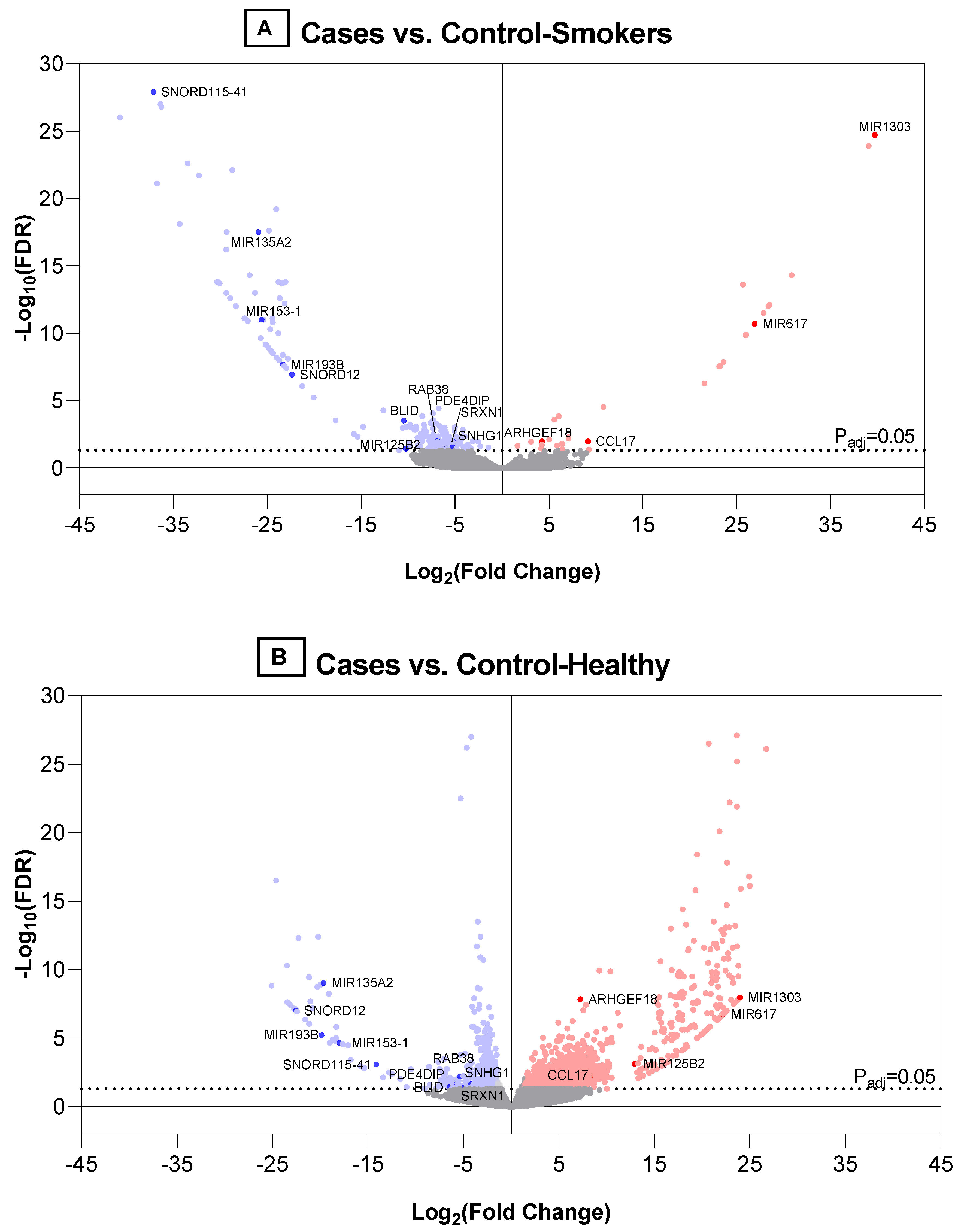
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- American Cancer Society. Facts & Figures 2022; American Cancer Society: Atlanta, Ga, USA, 2022. [Google Scholar]
- WHO Global Report on Trends in Prevalence of Tobacco Use 2000–2025, 4th ed.; World Health Organization: Geneva, Switzerland, 2021.
- Li, Y.; Xiao, X.; Li, J.; Byun, J.; Cheng, C.; Bosse, Y.; McKay, J.; Albanes, D.; Lam, S.; Tardon, A.; et al. Genome-wide interaction analysis identified low-frequency variants with sex disparity in lung cancer risk. Hum. Mol. Genet. 2022, 31, 2831–2843. [Google Scholar] [CrossRef]
- Besaratinia, A.; Caceres, A.; Tommasi, S. DNA Hydroxymethylation in Smoking-Associated Cancers. Int. J. Mol. Sci. 2022, 23, 2657. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhu, M.; Ji, M.; Fan, J.; Xie, J.; Wei, X.; Jiang, X.; Xu, J.; Chen, L.; Yin, R.; et al. Air Pollution, Genetic Factors, and the Risk of Lung Cancer: A Prospective Study in the UK Biobank. Am. J. Respir. Crit. Care Med. 2021, 204, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Bade, B.C.; Dela Cruz, C.S. Lung Cancer 2020: Epidemiology, Etiology, and Prevention. Clin. Chest. Med. 2020, 41, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Leduc, C.; Antoni, D.; Charloux, A.; Falcoz, P.E.; Quoix, E. Comorbidities in the management of patients with lung cancer. Eur. Respir. J. 2017, 49, 1601721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campling, B.G.; Collins, B.N.; Algazy, K.M.; Schnoll, R.A.; Lam, M. Spontaneous smoking cessation before lung cancer diagnosis. J. Thorac. Oncol. 2011, 6, 517–524. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA. Cancer. J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Force, U.S.P.S.T. Final Update Summary: Lung Cancer: Screening. Available online: https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/lung-cancer-screening (accessed on 4 January 2022).
- Force, U.S.P.S.T.; Krist, A.H.; Davidson, K.W.; Mangione, C.M.; Barry, M.J.; Cabana, M.; Caughey, A.B.; Davis, E.M.; Donahue, K.E.; Doubeni, C.A.; et al. Screening for Lung Cancer: US Preventive Services Task Force Recommendation Statement. JAMA 2021, 325, 962–970. [Google Scholar] [CrossRef]
- Jonas, D.E.; Reuland, D.S.; Reddy, S.M.; Nagle, M.; Clark, S.D.; Weber, R.P.; Enyioha, C.; Malo, T.L.; Brenner, A.T.; Armstrong, C.; et al. Screening for Lung Cancer With Low-Dose Computed Tomography: Updated Evidence Report and Systematic Review for the US Preventive Services Task Force. JAMA 2021, 325, 971–987. [Google Scholar] [CrossRef]
- Tanner, N.T.; Aggarwal, J.; Gould, M.K.; Kearney, P.; Diette, G.; Vachani, A.; Fang, K.C.; Silvestri, G.A. Management of Pulmonary Nodules by Community Pulmonologists: A Multicenter Observational Study. Chest 2015, 148, 1405–1414. [Google Scholar] [CrossRef]
- Pinzani, P.; D’Argenio, V.; Del Re, M.; Pellegrini, C.; Cucchiara, F.; Salvianti, F.; Galbiati, S. Updates on liquid biopsy: Current trends and future perspectives for clinical application in solid tumors. Clin. Chem. Lab. Med. 2021, 59, 1181–1200. [Google Scholar] [CrossRef] [PubMed]
- Li, R.Y.; Liang, Z.Y. Circulating tumor DNA in lung cancer: Real-time monitoring of disease evolution and treatment response. Chin. Med. J. Engl. 2020, 133, 2476–2485. [Google Scholar] [CrossRef] [PubMed]
- Gale, D.; Heider, K.; Ruiz-Valdepenas, A.; Hackinger, S.; Perry, M.; Marsico, G.; Rundell, V.; Wulff, J.; Sharma, G.; Knock, H.; et al. Residual ctDNA after treatment predicts early relapse in patients with early-stage non-small cell lung cancer. Ann. Oncol. 2022, 33, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Larson, M.H.; Pan, W.; Kim, H.J.; Mauntz, R.E.; Stuart, S.M.; Pimentel, M.; Zhou, Y.; Knudsgaard, P.; Demas, V.; Aravanis, A.M.; et al. A comprehensive characterization of the cell-free transcriptome reveals tissue- and subtype-specific biomarkers for cancer detection. Nat. Commun. 2021, 12, 2357. [Google Scholar] [CrossRef]
- Sorber, L.; Zwaenepoel, K.; Jacobs, J.; De Winne, K.; Goethals, S.; Reclusa, P.; Van Casteren, K.; Augustus, E.; Lardon, F.; Roeyen, G.; et al. Circulating Cell-Free DNA and RNA Analysis as Liquid Biopsy: Optimal Centrifugation Protocol. Cancers 2019, 11, 458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, S.; Janke, F.; Dietz, S.; Sultmann, H. Circulating MicroRNAs as Potential Biomarkers for Lung Cancer. Recent Results Cancer Res. 2020, 215, 299–318. [Google Scholar] [CrossRef]
- De Fraipont, F.; Gazzeri, S.; Cho, W.C.; Eymin, B. Circular RNAs and RNA Splice Variants as Biomarkers for Prognosis and Therapeutic Response in the Liquid Biopsies of Lung Cancer Patients. Front. Genet. 2019, 10, 390. [Google Scholar] [CrossRef] [Green Version]
- Peng, W.; Wang, J.; Shan, B.; Peng, Z.; Dong, Y.; Shi, W.; He, D.; Cheng, Y.; Zhao, W.; Zhang, C.; et al. Diagnostic and Prognostic Potential of Circulating Long Non-Coding RNAs in Non Small Cell Lung Cancer. Cell Physiol. Biochem. 2018, 49, 816–827. [Google Scholar] [CrossRef]
- Gao, L.; Ma, J.; Mannoor, K.; Guarnera, M.A.; Shetty, A.; Zhan, M.; Xing, L.; Stass, S.A.; Jiang, F. Genome-wide small nucleolar RNA expression analysis of lung cancer by next-generation deep sequencing. Int. J. Cancer 2015, 136, E623–E629. [Google Scholar] [CrossRef]
- Available online: https://www.illumina.com/products/by-type/sequencing-kits/library-prep-kits/rna-prep-enrichment.html (accessed on 4 January 2022).
- Shetty, A.C.; Adkins, R.S.; Chatterjee, A.; McCracken, C.L.; Hodges, T.; Creasy, H.H.; Giglio, M.; Mahurkar, A.; White, O. CAVERN: Computational and visualization environment for RNA-seq analyses. In Proceedings of the 69th Annual Meeting, Houston, TX, USA, 15–19 October 2019; American Society of Human Genetics: Houston, TX, USA, 2019. [Google Scholar]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Society. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Yamashita, S.; Chujo, M.; Miyawaki, M.; Tokuishi, K.; Anami, K.; Yamamoto, S.; Kawahara, K. Combination of p53AIP1 and survivin expression is a powerful prognostic marker in non-small cell lung cancer. J. Exp. Clin. Cancer Res. 2009, 28, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, T.; Zhang, X.; Dong, Y.; Liu, J.; Zhang, W.; Wu, F.; Bo, H.; Shao, H.; Zhang, R.; Shen, H. Chemokine CCL17 Affects Local Immune Infiltration Characteristics and Early Prognosis Value of Lung Adenocarcinoma. Front. Cell Dev. Biol. 2022, 10, 816927. [Google Scholar] [CrossRef]
- Yang, J.; Jia, Y.; Wang, B.; Yang, S.; Du, K.; Luo, Y.; Li, Y.; Zhu, B. Circular RNA CHST15 Sponges miR-155-5p and miR-194-5p to Promote the Immune Escape of Lung Cancer Cells Mediated by PD-L1. Front. Oncol. 2021, 11, 595609. [Google Scholar] [CrossRef]
- Song, C.; Gao, Y.; Tian, Y.; Han, X.; Chen, Y.; Tian, D.L. Expression of p114RhoGEF predicts lymph node metastasis and poor survival of squamous-cell lung carcinoma patients. Tumour. Biol. 2013, 34, 1925–1933. [Google Scholar] [CrossRef]
- Hsieh, J.J.; Hou, M.M.; Chang, J.W.; Shen, Y.C.; Cheng, H.Y.; Hsu, T. RAB38 is a potential prognostic factor for tumor recurrence in non-small cell lung cancer. Oncol. Lett. 2019, 18, 2598–2604. [Google Scholar] [CrossRef]
- Chang, J.W.; Wei, N.C.; Su, H.J.; Huang, J.L.; Chen, T.C.; Wu, Y.C.; Yu, C.T.; Hou, M.M.; Hsieh, C.H.; Hsieh, J.J.; et al. Comparison of genomic signatures of non-small cell lung cancer recurrence between two microarray platforms. Anticancer Res. 2012, 32, 1259–1265. [Google Scholar]
- Weng, T.Y.; Wang, C.Y.; Hung, Y.H.; Chen, W.C.; Chen, Y.L.; Lai, M.D. Differential Expression Pattern of THBS1 and THBS2 in Lung Cancer: Clinical Outcome and a Systematic-Analysis of Microarray Databases. PLoS ONE 2016, 11, e0161007. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Wang, L.; Ma, Z.; Ma, Y.; Zhao, J.; Peng, B.O.; Qiao, Z. Sequencing study on familial lung squamous cancer. Oncol. Lett. 2015, 10, 2634–2638. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Yu, Z.; Huo, S.; Chen, Z.; Ou, Z.; Mai, J.; Ding, S.; Zhang, J. Overexpression of ELF3 facilitates cell growth and metastasis through PI3K/Akt and ERK signaling pathways in non-small cell lung cancer. Int. J. Biochem. Cell Biol. 2018, 94, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, X.; Li, W.; Wu, L.; Chang, L.; Chen, H. miRNA-124 modulates lung carcinoma cell migration and invasion. Int J Clin. Pharmacol. Ther. 2016, 54, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Jiang, H.; Xiao, Z.; Baker, A.; Young, M.R.; Veenstra, T.D.; Colburn, N.H. Sulfiredoxin-Peroxiredoxin IV axis promotes human lung cancer progression through modulation of specific phosphokinase signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 7004–7009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Jiang, G.; Xu, E.; Zhou, J.; Liu, L.; Yang, Q. Identification of SRXN1 and KRT6A as Key Genes in Smoking-Related Non-Small-Cell Lung Cancer Through Bioinformatics and Functional Analyses. Front. Oncol. 2021, 11, 810301. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, X.; Zhang, M.; Bai, X.; Li, H.; Kan, L.; Niu, H.; He, P. ARID1A is downregulated in non-small cell lung cancer and regulates cell proliferation and apoptosis. Tumour. Biol. 2014, 35, 5701–5707. [Google Scholar] [CrossRef]
- Wang, N.; Zhang, T. Downregulation of MicroRNA-135 Promotes Sensitivity of Non-Small Cell Lung Cancer to Gefitinib by Targeting TRIM16. Oncol. Res. 2018, 26, 1005–1014. [Google Scholar] [CrossRef]
- Hu, F.; Li, C.; Zheng, X.; Zhang, H.; Shen, Y.; Zhou, L.; Yang, X.; Han, B.; Zhang, X. Lung adenocarcinoma resistance to therapy with EGFRtyrosine kinase inhibitors is related to increased expression of cancer stem cell markers SOX2, OCT4 and NANOG. Oncol. Rep. 2020, 43, 727–735. [Google Scholar] [CrossRef]
- Choi, K.H.; Shin, C.H.; Lee, W.J.; Ji, H.; Kim, H.H. Dual-strand tumor suppressor miR-193b-3p and -5p inhibit malignant phenotypes of lung cancer by suppressing their common targets. Biosci. Rep. 2019, 39, BSR20190634. [Google Scholar] [CrossRef] [Green Version]
- She, K.; Yan, H.; Huang, J.; Zhou, H.; He, J. miR-193b availability is antagonized by LncRNA-SNHG7 for FAIM2-induced tumour progression in non-small cell lung cancer. Cell Prolif. 2018, 51, e12406. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.J.; Zhang, E.N.; Zhong, Z.K.; Jiang, M.Z.; Yang, X.F.; Zhou, D.M.; Wang, X.W. MicroRNA-153 expression and prognosis in non-small cell lung cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 8671–8675. [Google Scholar]
- Yuan, Y.; Du, W.; Wang, Y.; Xu, C.; Wang, J.; Zhang, Y.; Wang, H.; Ju, J.; Zhao, L.; Wang, Z.; et al. Suppression of AKT expression by miR-153 produced anti-tumor activity in lung cancer. Int. J. Cancer. 2015, 136, 1333–1340. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, H.G.; Fan, M.J.; Lv, Z.Q.; Shen, X.M.; He, X.X. Expressions of connexin 32 and 26 and their correlation to prognosis of non-small cell lung cancer. Ai Zheng 2009, 28, 173–176. [Google Scholar] [PubMed]
- Shan, N.; Shen, L.; Wang, J.; He, D.; Duan, C. MiR-153 inhibits migration and invasion of human non-small-cell lung cancer by targeting ADAM19. Biochem. Biophys. Res. Commun. 2015, 456, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Lim, N.J.; Jang, S.G.; Lee, G.K. miR-592 and miR-552 can distinguish between primary lung adenocarcinoma and colorectal cancer metastases in the lung. Anticancer Res. 2014, 34, 2297–2302. [Google Scholar] [PubMed]
- Huang, S.P.; Jiang, Y.F.; Yang, L.J.; Yang, J.; Liang, M.T.; Zhou, H.F.; Luo, J.; Yang, D.P.; Mo, W.J.; Chen, G.; et al. Downregulation of miR-125b-5p and Its Prospective Molecular Mechanism in Lung Squamous Cell Carcinoma. Cancer Biother. Radiopharm. 2022, 37, 125–140. [Google Scholar] [CrossRef]
- Wang, J.; Chen, H.; Liao, Y.; Chen, N.; Liu, T.; Zhang, H.; Zhang, H. Expression and clinical evidence of miR-494 and PTEN in non-small cell lung cancer. Tumour. Biol. 2015, 36, 6965–6972. [Google Scholar] [CrossRef]
- Wang, M.; Zhu, X.; Sha, Z.; Li, N.; Li, D.; Chen, L. High expression of kinesin light chain-2, a novel target of miR-125b, is associated with poor clinical outcome of elderly non-small-cell lung cancer patients. Br. J. Cancer 2015, 112, 874–882. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Li, D.; Zheng, H.; He, Z.; Qian, F.; Wu, X.; Yin, Z.; Bao, P.T.; Jin, M. A novel lncRNA-miRNA-mRNA competing endogenous RNA regulatory network in lung adenocarcinoma and kidney renal papillary cell carcinoma. Thorac. Cancer 2021, 12, 2526–2536. [Google Scholar] [CrossRef]
- Tan, J.; Wang, W.; Song, B.; Song, Y.; Meng, Z. Integrative Analysis of Three Novel Competing Endogenous RNA Biomarkers with a Prognostic Value in Lung Adenocarcinoma. Biomed. Res. Int. 2020, 2020, 2837906. [Google Scholar] [CrossRef]
- Shi, S.L.; Zhang, Z.H. Long non-coding RNA SNHG1 contributes to cisplatin resistance in non-small cell lung cancer by regulating miR-140-5p/Wnt/beta-catenin pathway. Neoplasma 2019, 66, 756–765. [Google Scholar] [CrossRef]
- Mullins, K.; Seneviratne, C.; Shetty, A.; Jiang, F.; Christenson, R.; Stass, S. Proof of Concept: Detection of cell free RNA from EDTA plasma in patients with lung cancer and non-cancer patients. medRxiv 2022. [Google Scholar] [CrossRef]
- Rasmussen, M.; Reddy, M.; Nolan, R.; Camunas-Soler, J.; Khodursky, A.; Scheller, N.M.; Cantonwine, D.E.; Engelbrechtsen, L.; Mi, J.D.; Dutta, A.; et al. RNA profiles reveal signatures of future health and disease in pregnancy. Nature 2022, 601, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Terry, S.J.; Elbediwy, A.; Zihni, C.; Harris, A.R.; Bailly, M.; Charras, G.T.; Balda, M.S.; Matter, K. Stimulation of cortical myosin phosphorylation by p114RhoGEF drives cell migration and tumor cell invasion. PLoS ONE 2012, 7, e50188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.; Shewan, A.M.; Ewald, A.J.; Werb, Z.; Mostov, K.E. p114RhoGEF governs cell motility and lumen formation during tubulogenesis through a ROCK-myosin-II pathway. J. Cell Sci. 2015, 128, 4317–4327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leiro-Fernandez, V.; Mouronte-Roibas, C.; Garcia-Rodriguez, E.; Botana-Rial, M.; Ramos-Hernandez, C.; Torres-Duran, M.; Ruano-Ravina, A.; Fernandez-Villar, A.; On behalf of the Lung Cancer Group at the Álvaro Cunqueiro Hospital in Vigo. Predicting delays in lung cancer diagnosis and staging. Thorac. Cancer 2019, 10, 296–303. [Google Scholar] [CrossRef] [Green Version]
- Hellyer, J.A.; Patel, M.I. Sex disparities in lung cancer incidence: Validation of a long-observed trend. Transl. Lung Cancer Res. 2019, 8, 543–545. [Google Scholar] [CrossRef]
- Molina, A.J.; Garcia-Martinez, L.; Zapata-Alvarado, J.; Alonso-Orcajo, N.; Fernandez-Villa, T.; Martin, V. Trends in Lung Cancer Incidence in a Healthcare Area. Arch Bronconeumol. 2015, 51, e53–e55. [Google Scholar] [CrossRef]
- Pesch, B.; Kendzia, B.; Gustavsson, P.; Jockel, K.H.; Johnen, G.; Pohlabeln, H.; Olsson, A.; Ahrens, W.; Gross, I.M.; Bruske, I.; et al. Cigarette smoking and lung cancer--relative risk estimates for the major histological types from a pooled analysis of case-control studies. Int. J. Cancer 2012, 131, 1210–1219. [Google Scholar] [CrossRef]

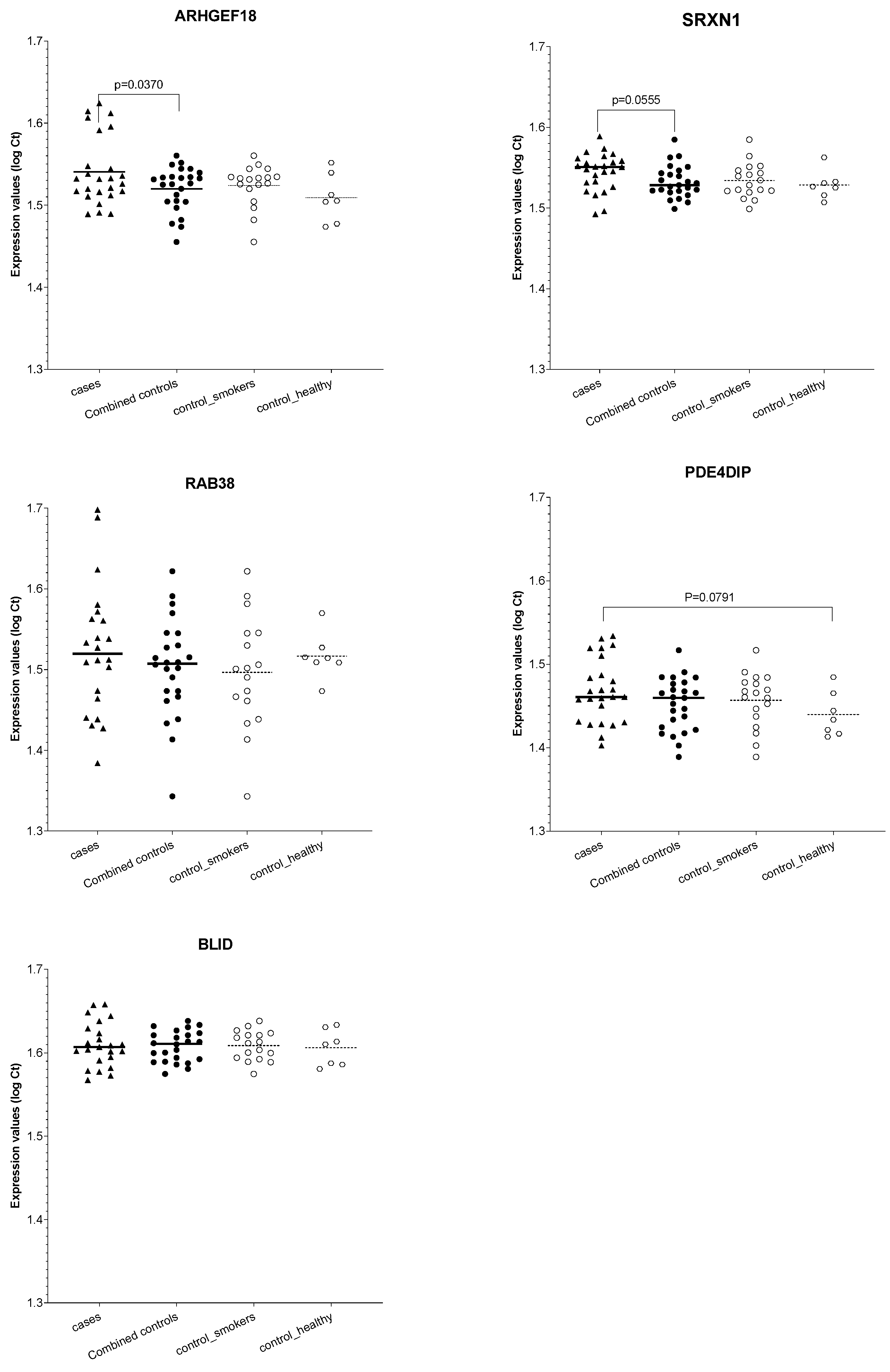

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cases | Control_ Smokers | Control_ Healthy | p-Value Cases vs. | p-Value Smokers vs. Healthy | ||
|---|---|---|---|---|---|---|
| Control_ Smokers | Control_ Healthy | |||||
| Discovery Cohort (N = 36): | ||||||
| Sample Size | 12 | 12 | 12 | |||
| Age (mean, (SD)) | 67.17 (8.99) | 68.44 (10.01) | 40.17 (4.99) | 0.728 | <0.0001 | <0.0001 |
| Gender (Male, N (%)) | 11 (91.67) | 9 (75) | 7 (58.33) | 0.3144 | 0.0480 | 0.3144 |
| Race (Caucasian, N (%)) | 5 (4.67) | 5 (4.67) | 5 (4.67) | ns | ns | ns |
| Stage | ||||||
| Stage I (N) | 7 (AC = 5) | |||||
| Stage II (N) | 4 (AC = 1) | |||||
| Stage III-IV (N) | 1 (AC = 0) | |||||
| Histological Type | ||||||
| AC (N) | 6 | |||||
| SCC (N) | 6 | |||||
| Average Plasma Volumes Used (mL) | 1.6 | 1.6 | 1.54 | ns | ns | ns |
| Validation Cohort (N = 50): | ||||||
| Sample Size | 25 | 18 | 7 | |||
| Age (mean, (SD)) | 64.60 (8.97) | 61.28 (10.23) | 58.14(17.38) | ns | ns | ns |
| Gender (Male, N (%)) | 19 (76.00) | 13 (72.22) | 5 (71.43) | ns | ns | ns |
| Race (Caucasian, N (%)) | 52 (50.99) | 11 (61.11) | 3 (60.00) | ns | ns | ns |
| Stage | ||||||
| Stage I (N) | 4 (AC = 2) | |||||
| Stage II (N) | 2 (AC = 0) | |||||
| Stage III-IV (N) | 9 (AC = 8) | |||||
| Missing Data | 10 (AC = 7) | |||||
| Histological Type | ||||||
| AC (N) | 13 | |||||
| SCC (N) | 12 | |||||
| Average Plasma Volumes Used (mL) | 0.5 | 0.5 | 0.5 | |||
| Gene Name | Gene ID | Gene Type | Compared with Control_Healthy Group | Compared with Control_Smokers Group | Ref * | %Detected; % CV 1 | %Detected; % CV 2 | %Detected; % CV 3 | Differentially Expressed in Stage I? | Differentially Expressed in Stage II? | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| log2FoldChange | p-Value | p-Adj | log2FoldChange | p-Value | p-Adj | |||||||||
| ENSG00000102970 | CCL17 | protein | 8.7116 | 1.5 × 10−4 | 5.2 × 10−3 | 9.1579 | 5.0 × 10−5 | 1.1 × 10−2 | [28,29,30] | 27.23; | 25.00; | 41.67; | - | - |
| coding | 42.76 | 30.72 | 56.66 | |||||||||||
| ENSG00000104880 | ARHGEF18 | Protein | 7.2400 | 4.6 × 10−11 | 1.4 × 10−8 | 4.2579 | 4.7 × 10−5 | 1.1 × 10−2 | [31] | 81.82; | 91.67; | 91.67; | Vs._ control_healthy | |
| coding | 27.96 | 20.43 | 45.07 | |||||||||||
| ENSG00000123892 | RAB38 | protein | −5.6801 | 1.0 × 10−3 | 1.9 × 10−2 | −6.8853 | 4.6 × 10−5 | 1.0 × 10−2 | [32,33] | 54.55; | 66.67; | 41.67; | Vs. both controls | |
| coding | 45.28 | 48.13 | 26.48 | |||||||||||
| ENSG00000178104 | PDE4DIP | protein | −5.3701 | 1.9 × 10−4 | 6.3 × 10−3 | −5.2635 | 1.9 × 10−4 | 3.0 × 10−2 | [34,35] | 63.64; | 83.33; | 50.00; | Vs. control_healthy | |
| coding | 43.90 | 36.32 | 15.50 | |||||||||||
| ENSG00000259571 | BLID | protein | −6.4355 | 3.0 × 10−3 | 3.7 × 10−2 | −10.4897 | 7.4 × 10−7 | 3.1 × 10−4 | [36] | 18.18; | 58.33; | 33.33; | Vs. control_smokers | |
| coding | 33.91 | 53.16 | 4.89 | |||||||||||
| ENSG00000271303 | SRXN1 | protein | −4.8705 | 3.3 × 10−3 | 3.9 × 10−2 | −5.9253 | 2.5 × 10−4 | 3.7 × 10−2 | [37,38,39] | 54.55; | 66.67; | 41.67; | - | - |
| coding | 54.83 | 47.06 | 27.08 | |||||||||||
| ENSG00000207586 | MIR135A2 | miRNA | −19.6805 | 2.1 × 10−12 | 8.9 × 10−10 | −25.9452 | 1.5 × 10−21 | 3.3 × 10−18 | [40,41] | 18.18; | 41.67; | 8.33; | Vs. both controls | Vs. both controls |
| 51.99 | 44.96 | 40.13 | ||||||||||||
| ENSG00000207639 | MIR193B | miRNA | −19.8578 | 4.3 × 10−8 | 6.2 × 10−6 | −23.3468 | 4.0 × 10−11 | 2.1 × 10−8 | [42,43,44] | 18.18; | 33.33; | 8.33; | Vs. both controls | Vs. both controls |
| 30.89 | 52.06 | 16.35 | ||||||||||||
| ENSG00000207647 | MIR153-1 | miRNA | −17.9600 | 1.9 × 10−7 | 2.3 × 10−5 | −25.6172 | 1.4 × 10−14 | 1.1 × 10−11 | [45,46,47,48] | 18.18; | 33.33; | 16.67; | - | - |
| 26.33 | 57.10 | 53.34 | ||||||||||||
| ENSG00000207763 | MIR617 | miRNA | 22.1588 | 8.9 × 10−10 | 1.8 × 10−7 | 26.8981 | 2.8 × 10−14 | 2.0 × 10−11 | [49] | 9.09; 33.09 | 0;0 | 33.33; 41.96 | Vs. both controls | Vs. both controls |
| ENSG00000207863 | MIR125B2 | miRNA | 12.9574 | 1.0 × 10−5 | 7.3 × 10−4 | −10.2390 | 2.7 × 10−4 | 3.8 × 10−2 | [50,51,52] | 9.09; 31.19 | 41.67; 51.38 | 16.67; 32.68 | - | - |
| ENSG00000221552 | MIR1303 | miRNA | 23.9859 | 3.3 × 10−11 | 1.1 × 10−8 | 39.7024 | 2.8 × 10−29 | 1.8 × 10−25 | [31,35] | 9.09; 37.13 | 0;0 | 33.33; 59.01 | - | - |
| ENSG00000200478 | SNORD115-41 | snoRNA | −14.1259 | 1.2 × 10−5 | 8.3 × 10−4 | −37.1348 | 3.8 × 10−33 | 1.2 × 10−28 | [22] | 9.09; 14.02 | 33.33; 43.45 | 0;0 | ** | ** |
| ENSG00000212304 | SNORD12 | snoRNA | −22.5404 | 4.4 × 10−10 | 1.0 × 10−7 | −22.3897 | 2.4 × 10−10 | 1.2 × 10−7 | [22] | 18.18; 68.86 | 25.00; 40.72 | 0;0 | Vs. both controls | Vs. both controls |
| ENSG00000255717 | SNHG1 | processed transcript | −4.2180 | 1.4 × 10−3 | 2.3 × 10−2 | −5.2295 | 5.0 × 10−5 | 1.1 × 10−2 | [53,54,55] | 63.64; 43.01 | 83.33; 44.07 | 83.33; 46.29 | Vs. control_smokers | Vs. both controls |
| ID | Description of Pathway | Gene Ratio | p-Value | p-Adjust |
|---|---|---|---|---|
| Cases vs. Control_Healthy: | ||||
| GO:0001501 | skeletal system development | 71/1685 | 8.5591 × 10−5 | 0.016903 |
| GO:0005125 | cytokine activity | 42/1656 | 6.6270 × 10−5 | 0.022704 |
| GO:0005198 | structural molecule activity | 105/1656 | 2.0920 × 10−5 | 0.009907 |
| GO:0007186 | G protein-coupled receptor signaling | 179/1685 | 5.2099 × 10−6 | 0.002827 |
| GO:0007200 | phospholipase C-activating G protein-coupled receptor signaling | 22/1685 | 4.5649 × 10−5 | 0.011269 |
| GO:0007399 | nervous system development | 258/1685 | 0.0003 | 0.037022 |
| GO:0008154 | actin polymerization or depolymerization | 34/1685 | 0.0004 | 0.046481 |
| GO:0009888 | tissue development | 248/1685 | 1.0524 × 10−6 | 0.001336 |
| GO:0009953 | dorsal/ventral pattern formation | 19/1685 | 0.0003 | 0.044695 |
| GO:0010454 | negative regulation of cell fate commitment | 7/1685 | 2.6616 × 10−5 | 0.007885 |
| GO:0019958 | C-X-C chemokine binding | 5/1656 | 2.3133 × 10−5 | 0.009907 |
| GO:0030545 | receptor regulator activity | 85/1656 | 2.1418 × 10−6 | 0.002111 |
| GO:0032501 | multicellular organismal process | 802/1685 | 2.1589 × 10−6 | 0.002132 |
| GO:0042221 | response to chemical | 513/1685 | 0.0001 | 0.018405 |
| GO:0042246 | tissue regeneration | 17/1685 | 0.0002 | 0.027754 |
| GO:0042692 | muscle cell differentiation | 55/1685 | 0.0003 | 0.037022 |
| GO:0043403 | skeletal muscle tissue regeneration | 11/1685 | 0.0003 | 0.044909 |
| GO:0043503 | skeletal muscle fiber adaptation | 4/1685 | 4.4531 × 10−5 | 0.011269 |
| GO:0045165 | cell fate commitment | 46/1685 | 1.9621 × 10−5 | 0.006707 |
| GO:0048018 | receptor ligand activity | 79/1656 | 2.4643 × 10−6 | 0.002111 |
| GO:0051272 | positive regulation of cellular component movement | 84/1685 | 7.8914 × 10−6 | 0.003597 |
| GO:0051493 | regulation of cytoskeleton organization | 73/1685 | 0.00012 | 0.020485 |
| GO:1902903 | regulation of supramolecular fiber organization | 51/1685 | 0.0004 | 0.047617 |
| GO:1904888 | cranial skeletal system development | 15/1685 | 0.0003 | 0.044566 |
| GO:2001046 | positive regulation of integrin-mediated signaling | 5/1685 | 6.6284 × 10−5 | 0.014261 |
| Cases vs. Control_Smokers: | ||||
| GO:0003729 | mRNA binding | 23/81 | 1.0069 × 10−5 | 0.001057 |
| GO:0010608 | posttranscriptional regulation of gene expression | 23/81 | 3.0028 × 10−10 | 4.69 × 10−8 |
| GO:0016441 | posttranscriptional gene silencing | 22/84 | 4.4483 × 10−15 | 1.62 × 10−12 |
| GO:0016442 | RISC complex | 23/81 | 7.5451 × 10−17 | 6.64 × 10−15 |
| GO:0016458 | gene silencing | 23/81 | 1.5066 × 10−13 | 3.29 × 10−11 |
| GO:0031047 | gene silencing by RNA | 22/84 | 1.2005 × 10−14 | 3.28 × 10−12 |
| GO:0031332 | RNAi effector complex | 23/81 | 7.5451 × 10−17 | 6.64 × 10−15 |
| GO:0035194 | posttranscriptional gene silencing by RNA | 23/81 | 4.3205 × 10−14 | 1.62 × 10−12 |
| GO:0035195 | gene silencing by miRNA | 23/81 | 3.2204 × 10−15 | 1.62 × 10−12 |
| GO:0040029 | regulation of gene expression, epigenetic | 23/81 | 7.5803 × 10−13 | 1.38 × 10−10 |
| GO:1903231 | mRNA binding involved in posttranscriptional gene silencing | 23/84 | 2.5252 × 10−8 | 5.3 × 10−6 |
| GO:1990904 | ribonucleoprotein complex | 23/84 | 1.9793 × 10−8 | 1.16 × 10−6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seneviratne, C.; Shetty, A.C.; Geng, X.; McCracken, C.; Cornell, J.; Mullins, K.; Jiang, F.; Stass, S. A Pilot Analysis of Circulating cfRNA Transcripts for the Detection of Lung Cancer. Diagnostics 2022, 12, 2897. https://doi.org/10.3390/diagnostics12122897
Seneviratne C, Shetty AC, Geng X, McCracken C, Cornell J, Mullins K, Jiang F, Stass S. A Pilot Analysis of Circulating cfRNA Transcripts for the Detection of Lung Cancer. Diagnostics. 2022; 12(12):2897. https://doi.org/10.3390/diagnostics12122897
Chicago/Turabian StyleSeneviratne, Chamindi, Amol Carl Shetty, Xinyan Geng, Carrie McCracken, Jessica Cornell, Kristin Mullins, Feng Jiang, and Sanford Stass. 2022. "A Pilot Analysis of Circulating cfRNA Transcripts for the Detection of Lung Cancer" Diagnostics 12, no. 12: 2897. https://doi.org/10.3390/diagnostics12122897
APA StyleSeneviratne, C., Shetty, A. C., Geng, X., McCracken, C., Cornell, J., Mullins, K., Jiang, F., & Stass, S. (2022). A Pilot Analysis of Circulating cfRNA Transcripts for the Detection of Lung Cancer. Diagnostics, 12(12), 2897. https://doi.org/10.3390/diagnostics12122897