
Whole-Exome Sequencing Revealed New Candidate Genes for Human Dilated Cardiomyopathy
 , ,
, ,  , ,
, ,  , , , , , ,
, , , , , ,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients’ Clinical Characteristics
2.2. Whole-Exome Sequencing (WES)
2.3. Bioinformatics Analysis
3. Results
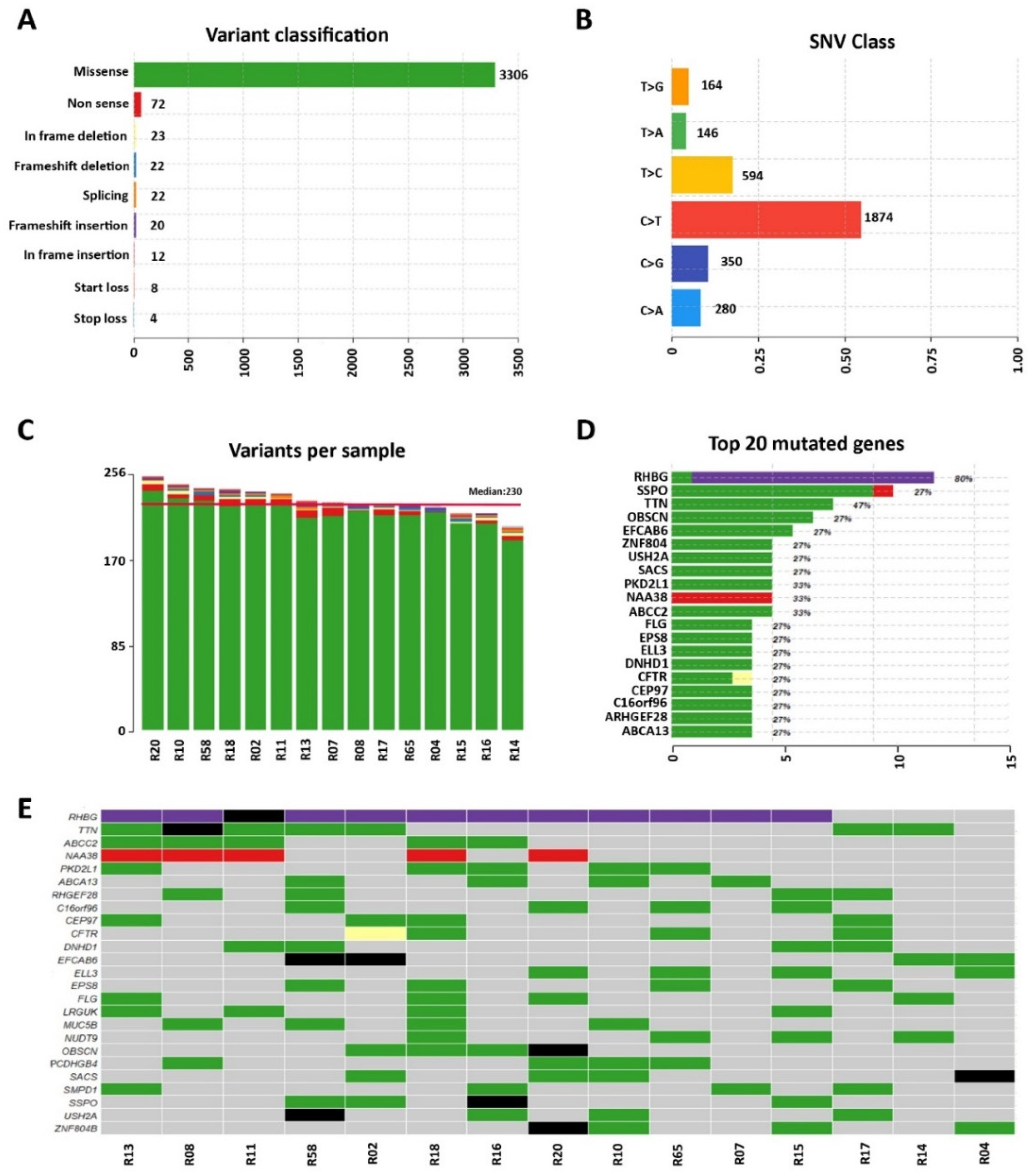
3.1. Whole-Exome Sequencing of Sporadic DCM Patients Revealed Variants in Genes Known for Their Association with the Disease
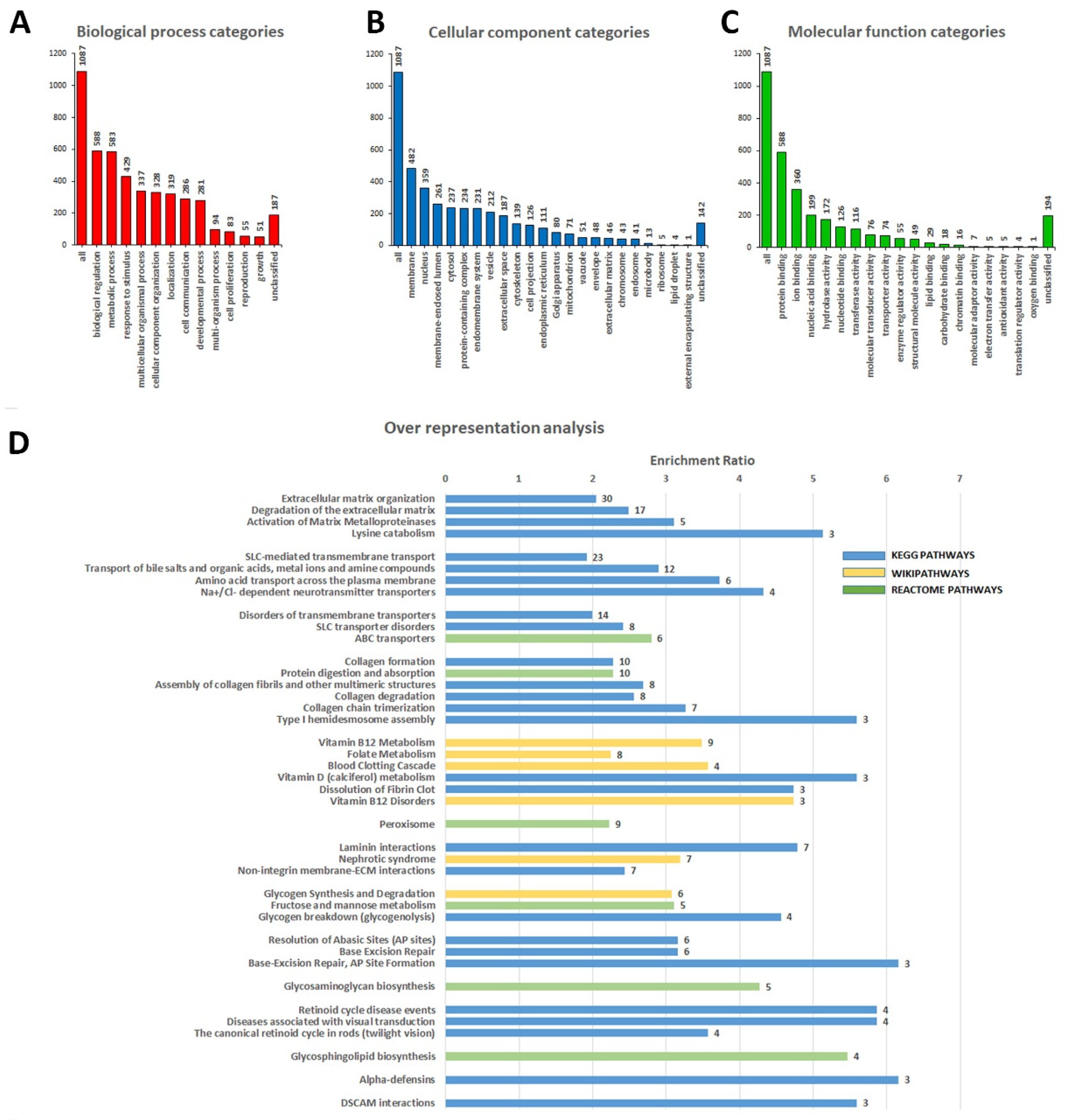
3.2. Identification of Variants in Genes Not Previously Associated with DCM
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Elliott, P.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P.; Dubourg, O.; Kuhl, U.; Maisch, B.; McKenna, W.J.; et al. Classification of the cardiomyopathies: A position statement from the european society of cardiology working group on myocardial and pericardial diseases. Eur. Hear. J. 2007, 29, 270–276. [Google Scholar] [CrossRef]
- Jefferies, J.L.; Towbin, J.A. Dilated cardiomyopathy. Lancet 2010, 375, 752–762. [Google Scholar] [CrossRef]
- Bozkurt, B.; Colvin, M.; Cook, J.; Cooper, L.; Deswal, A.; Fonarow, G.; Francis, G.S.; Lenihan, D.; Lewis, E.F.; McNamara, D.M.; et al. Current Diagnostic and Treatment Strategies for Specific Dilated Cardiomyopathies: A Scientific Statement From the American Heart Association. Circulation 2016, 134, e579–e646. [Google Scholar] [CrossRef]
- Schultheiss, H.P.; Fairweather, D.; Caforio, A.L.P.; Escher, F.; Hershberger, R.E.; Lipshultz, S.E.; Liu, P.P.; Matsumori, A.; Mazzanti, A.; McMurray, J.; et al. Dilated cardiomyopathy. Nat. Rev. Dis. Primers 2019, 5, 32. [Google Scholar] [CrossRef]
- Reichart, D.; Magnussen, C.; Zeller, T.; Blankenberg, S. Dilated cardiomyopathy: From epidemiologic to genetic phenotypes: A translational review of current literature. J. Intern. Med. 2019, 286, 362–372. [Google Scholar] [CrossRef]
- Mestroni, L.; Milasin, J.; Vatta, M.; Pinamonti, B.; Sinagra, G.; Rocco, C.; Matulic, M.; Falaschi, A.; Giacca, M.; Camerini, F. Genetic factors in dilated cardiomyopathy. Arch. des Mal. du Coeur et des Vaiss. 1996, 89, 15–20. [Google Scholar]
- McNally, E.M.; Mestroni, L. Dilated Cardiomyopathy: Genetic Determinants and Mechanisms. Circ. Res. 2017, 121, 731–748. [Google Scholar] [CrossRef] [PubMed]
- Orphanou, N.; Papatheodorou, E.; Anastasakis, A. Dilated cardiomyopathy in the era of precision medicine: Latest concepts and developments. Hear. Fail. Rev. 2021, 27, 1173–1191. [Google Scholar] [CrossRef]
- Mestroni, L.; Maisch, B.; McKenna, W.J.; Schwartz, K.; Charron, P.; Rocco, C.; Tesson, F.; Richter, A.; Wilke, A.; Komajda, M. Guidelines for the study of familial dilated cardiomyopathies. Collaborative Research Group of the European Human and Capital Mobility Project on Familial Dilated Cardiomyopathy. Eur. Heart J. 1999, 20, 93–102. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Hedges, D.J.; Morales, A. Dilated cardiomyopathy: The complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 2013, 10, 531–547. [Google Scholar] [CrossRef]
- McNally, E.M.; Golbus, J.; Puckelwartz, M.J. Genetic mutations and mechanisms in dilated cardiomyopathy. J. Clin. Investig. 2013, 123, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Pugh, T.J.; Kelly, M.A.; Gowrisankar, S.; Hynes, E.; Seidman, M.A.; Baxter, S.M.; Bowser, M.; Harrison, B.; Aaron, D.; Mahanta, L.M.; et al. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet. Med. 2014, 16, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, J.; van Tintelen, J.P.; Fokstuen, S.; Elliott, P.; van Langen, I.M.; Meder, B.; Richard, P.; Syrris, P.; Caforio, A.L.; Adler, Y.; et al. The current role of next-generation DNA sequencing in routine care of patients with hereditary cardiovascular conditions: A viewpoint paper of the European Society of Cardiology working group on myocardial and pericardial diseases and members of the European Society of Human Genetics. Eur. Hear. J. 2015, 36, 1367–1370. [Google Scholar] [CrossRef]
- Ciarambino, T.; Menna, G.; Sansone, G.; Giordano, M. Cardiomyopathies: An Overview. Int. J. Mol. Sci. 2021, 22, 7722. [Google Scholar] [CrossRef] [PubMed]
- Gilissen, C.; Hoischen, A.; Brunner, H.G.; Veltman, J. Disease gene identification strategies for exome sequencing. Eur. J. Hum. Genet. 2012, 20, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, A.N.; Agre, K.E.; Pereira, N.L. Genetics of dilated cardiomyopathy: Practical implications for heart failure management. Nat. Rev. Cardiol. 2019, 17, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Henry, W.L.; Gardin, J.M.; Ware, J.H. Echocardiographic measurements in normal subjects from infancy to old age. Circulation 1980, 62, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Van Der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Aguilera, M.A.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef] [PubMed]
- Gobron, S.; Monnerie, H.; Meiniel, R.; Creveaux, I.; Lehmann, W.; Lamalle, D.; Dastugue, B. SCO-spondin: A new member of the thrombospondin family secreted by the subcommissural organ is a candidate in the modulation of neuronal aggregation. J. Cell Sci. 1996, 109, 1053–1061. [Google Scholar] [CrossRef] [PubMed]
- Granzier, H.; Irving, T. Passive tension in cardiac muscle: Contribution of collagen, titin, microtubules, and intermediate filaments. Biophys. J. 1995, 68, 1027–1044. [Google Scholar] [CrossRef]
- Grogan, A.; Kontrogianni-Konstantopoulos, A. Unraveling obscurins in heart disease. Pflugers Arch. 2018, 471, 735–743. [Google Scholar] [CrossRef]
- Fukuzawa, A.; Lange, S.; Holt, M.; Vihola, A.; Carmignac, V.; Ferreiro, A.; Udd, B.; Gautel, M. Interactions with titin and myomesin target obscurin and obscurin-like 1 to the M-band—Implications for hereditary myopathies. J. Cell Sci. 2008, 121, 1841–1851. [Google Scholar] [CrossRef]
- Grabowski, K.; Riemenschneider, M.; Schulte, L.; Witten, A.; Schulz, A.; Stoll, M.; Kreutz, R. Fetal-Adult Cardiac Transcriptome Analysis in Rats with Contrasting Left Ventricular Mass Reveals New Candidates for Cardiac Hypertrophy. PLoS ONE 2015, 10, e0116807. [Google Scholar] [CrossRef]
- Toualbi, L.; Toms, M.; Moosajee, M. USH2A-retinopathy: From genetics to therapeutics. Exp. Eye Res. 2020, 201, 108330. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488. [Google Scholar] [CrossRef]
- Zha, W. Transporter-mediated natural product–drug interactions for the treatment of cardiovascular diseases. J. Food Drug Anal. 2018, 26, S32–S44. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Magida, J.A.; Leinwand, L.A. Metabolic crosstalk between the heart and liver impacts familial hypertrophic cardiomyopathy. EMBO Mol. Med. 2014, 6, 482–495. [Google Scholar] [CrossRef]
- Kim, T.T.; Dyck, J.R. The role of CD36 in the regulation of myocardial lipid metabolism. Biochim. et Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2016, 1861, 1450–1460. [Google Scholar] [CrossRef]
- Tanaka, T.; Sohmiya, K.; Kawamura, K. Is CD36 deficiency an etiology of hereditary hypertrophic cardiomyopathy? J. Mol. Cell Cardiol. 1997, 29, 121–127. [Google Scholar] [CrossRef]
- Hirooka, K.; Yasumura, Y.; Ishida, Y.; Komamura, K.; Hanatani, A.; Nakatani, S.; Yamagishi, M.; Miyatake, K. Improvement in Cardiac Function and Free Fatty Acid Metabolism in a Case of Dilated Cardiomyopathy With CD36 Deficiency. Jpn. Circ. J. 2000, 64, 731–735. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tanaka, T.; Nakata, T.; Oka, T.; Ogawa, T.; Okamoto, F.; Kusaka, Y.; Sohmiya, K.; Shimamoto, K.; Itakura, K. Defect in human myocardial long-chain fatty acid uptake is caused by FAT/CD36 mutations. J. Lipid Res. 2001, 42, 751–759. [Google Scholar] [CrossRef]
- Tkatchenko, T.V.; Moreno-Rodriguez, R.A.; Conway, S.J.; Molkentin, J.D.; Markwald, R.R.; Tkatchenko, A.V. Lack of periostin leads to suppression of Notch1 signaling and calcific aortic valve disease. Physiol. Genom. 2009, 39, 160–168. [Google Scholar] [CrossRef]
- Katsuragi, N.; Morishita, R.; Nakamura, N.; Ochiai, T.; Taniyama, Y.; Hasegawa, Y.; Kawashima, K.; Kaneda, Y.; Ogihara, T.; Sugimura, K. Periostin as a Novel Factor Responsible for Ventricular Dilation. Circulation 2004, 110, 1806–1813. [Google Scholar] [CrossRef] [PubMed]
- Komlosi, K.; Claris, O.; Collardeau-Frachon, S.; Kopp, J.; Hausser, I.; Mazereeuw-Hautier, J.; Jonca, N.; Zimmer, A.D.; Sanlaville, D.; Fischer, J. Fatal Neonatal DOLK-CDG as a Rare Form of Syndromic Ichthyosis. Front. Genet. 2021, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lefeber, D.J.; de Brouwer, A.P.M.; Morava, E.; Riemersma, M.; Schuurs-Hoeijmakers, J.H.M.; Absmanner, B.; Verrijp, K.; Akker, W.M.R.V.D.; Huijben, K.; Steenbergen, G.; et al. Autosomal Recessive Dilated Cardiomyopathy due to DOLK Mutations Results from Abnormal Dystroglycan O-Mannosylation. PLoS Genet. 2011, 7, e1002427. [Google Scholar] [CrossRef]
- Joshi, S.K.; Keck, J.M.; Eide, C.A.; Bottomly, D.; Traer, E.; Tyner, J.W.; McWeeney, S.K.; Tognon, C.E.; Druker, B.J. ERBB2/HER2 mutations are transforming and therapeutically targetable in leukemia. Leukemia 2020, 34, 2798–2804. [Google Scholar] [CrossRef] [PubMed]
- Crone, S.; Zhao, Y.-Y.; Fan, L.; Gu, Y.; Minamisawa, S.; Liu, Y.; Peterson, K.L.; Chen, J.; Kahn, R.; Condorelli, G.; et al. ErbB2 is essential in the prevention of dilated cardiomyopathy. Nat. Med. 2002, 8, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Özcelik, C.; Erdmann, B.; Pilz, B.; Wettschureck, N.; Britsch, S.; Hübner, N.; Chien, K.R.; Birchmeier, C.; Garratt, A.N. Conditional mutation of the ErbB2 (HER2) receptor in cardiomyocytes leads to dilated cardiomyopathy. Proc. Natl. Acad. Sci. USA 2002, 99, 8880–8885. [Google Scholar] [CrossRef]
- Reischauer, S.; Arnaout, R.; Ramadass, R.; Stainier, D.Y.R. Actin Binding GFP Allows 4D In Vivo Imaging of Myofilament Dynamics in the Zebrafish Heart and the Identification of Erbb2 Signaling as a Remodeling Factor of Myofibril Architecture. Circ. Res. 2014, 115, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Opreanu, M.; Tikhonenko, M.; Bozack, S.; Lydic, T.A.; Reid, G.E.; McSorley, K.M.; Sochacki, A.; Perez, G.I.; Esselman, W.J.; Kern, T.; et al. The Unconventional Role of Acid Sphingomyelinase in Regulation of Retinal Microangiopathy in Diabetic Human and Animal Models. Diabetes 2011, 60, 2370–2378. [Google Scholar] [CrossRef]
- Panigrahi, I.; Dhanorkar, M.; Suthar, R.; Kumar, C.; Baalaaji, M.; Thapa, B.R.; Kalra, J. Niemann-Pick Disease: An Underdiagnosed Lysosomal Storage Disorder. Case Rep. Genet. 2019, 2019, 3108093. [Google Scholar] [CrossRef]
- Drevinge, C.; Karlsson, L.O.; Ståhlman, M.; Larsson, T.; Perman Sundelin, J.; Grip, L.; Andersson, L.; Borén, J.; Levin, M.C. Cholesteryl Esters Accumulate in the Heart in a Porcine Model of Ischemia and Reperfusion. PLoS ONE 2013, 8, e61942. [Google Scholar] [CrossRef]
- Raucci Jr, F.J.; Wijesinghe, D.S.; Chalfant, C.E.; Baumgarten, C.M. Exogenous and endogenous ceramides elicit volume-sensitive chloride current in ventricular myocytes. Cardiovasc. Res. 2010, 86, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Park, T.-S.; Hu, Y.; Noh, H.-L.; Drosatos, K.; Okajima, K.; Buchanan, J.; Tuinei, J.; Homma, S.; Jiang, X.-C.; Abel, E.D. Ceramide is a cardiotoxin in lipotoxic cardiomyopathys. J. Lipid. Res. 2008, 49, 2101–2112. [Google Scholar] [CrossRef]
- Alsarah, A.; Alsara, O.; Laird-Fick, H.S. Cardiac manifestations of Familial Mediterranean fever. Avicenna J. Med. 2017, 7, 158–163. [Google Scholar] [CrossRef]
- Ueda, K.; Valdivia, C.; Medeiros-Domingo, A.; Tester, D.J.; Vatta, M.; Farrugia, G.; Ackerman, M.J.; Makielski, J.C. Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. Proc. Natl. Acad. Sci. USA 2008, 105, 9355–9360. [Google Scholar] [CrossRef] [PubMed]
- Akcay, A.; Acar, G.; Sayarlioglu, M.; Sokmen, A.; Kaya, H.; Ispiroglu, M.; Koroglu, S. QT dispersion and transmural dispersion of repolarization in patients with familial Mediterranean fever. Mod. Rheumatol. 2009, 19, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Koca, B.; Kasapçopur, Ö.; Bakari, S.; Sönmez, E.; Öztunç, F.; Eroğlu, A.G.; Saltik, L.; Calay, Ö. QT dispersion and cardiac involvement in children with Familial Mediterranean fever. Cardiol. Young 2011, 22, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Celik, M.M.; Buyukkaya, E.; Ustun, N.; Nacar, A.B.; Kurt, M.; Karakas, M.F.; Bilen, P.; Duru, M.; Şen, N.; Akcay, A.B. Relation of fragmented QRS to tissue Doppler-derived parametersin patients with familial Mediterranean fever. Wien. Klin. Wochenschr. 2015, 127, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Parnell, E.; Shapiro, L.P.; Voorn, R.A.; Forrest, M.P.; Jalloul, H.A.; Loizzo, D.D.; Penzes, P. KALRN: A central regulator of synaptic function and synaptopathies. Gene 2020, 768, 145306. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.-M. Kalirin-7 is a Key Player in the Formation of Excitatory Synapses in Hippocampal Neurons. Sci. World J. 2010, 10, 1655–1666. [Google Scholar] [CrossRef]
- Molina-Navarro, M.M.; Roselló-Lletí, E.; Tarazón, E.; Ortega, A.; Sánchez-Izquierdo, D.; Lago, F.; González-Juanatey, J.R.; García-Pavía, P.; Salvador, A.; Montero, J.A.; et al. Heart failure entails significant changes in human nucleocytoplasmic transport gene expression. Int. J. Cardiol. 2013, 168, 2837–2843. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.; Schinde, V.; Bateman, A.C.; Gallagher, P.J. Interstitial fibrosis in the dilated non-ischaemic myocardium. Heart 2003, 89, 1255–1256. [Google Scholar] [CrossRef]
- Kapelko, V.I. Extracellular matrix alterations in cardiomyopathy: The possible crucial role in the dilative form. Exp. Clin. Cardiol. 2001, 6, 41–49. [Google Scholar]
- Heneghan, M.A.; Malone, D.; Dervan, P.A. Myocardial collagen network in dilated cardiomyopathy. Ir. J. Med Sci. 1991, 160, 399–401. [Google Scholar] [CrossRef] [PubMed]
- Rasi, K.; Piuhola, J.; Czabanka, M.; Sormunen, R.; Ilves, M.; Leskinen, H.; Rysä, J.; Kerkelä, R.; Janmey, P.; Heljasvaara, R.; et al. Collagen XV Is Necessary for Modeling of the Extracellular Matrix and Its Deficiency Predisposes to Cardiomyopathy. Circ. Res. 2010, 107, 1241–1252. [Google Scholar] [CrossRef]
- Louzao-Martinez, L.; Vink, A.; Harakalova, M.; Asselbergs, F.W.; Verhaar, M.C.; Cheng, C. Characteristic adaptations of the extracellular matrix in dilated cardiomyopathy. Int. J. Cardiol. 2016, 220, 634–646. [Google Scholar] [CrossRef]
- Zha, Y.; Li, Y.; Ge, Z.; Wang, J.; Jiao, Y.; Zhang, J.; Zhang, S. ADAMTS8 Promotes Cardiac Fibrosis Partly Through Activating EGFR Dependent Pathway. Front. Cardiovasc. Med. 2022, 9, 1–18. [Google Scholar] [CrossRef]
- Glavinas, H.; Krajcsi, P.; Cserepes, J.; Sarkadi, B. The Role of ABC Transporters in Drug Resistance, Metabolism and Toxicity. Curr. Drug Deliv. 2004, 1, 27–42. [Google Scholar] [CrossRef]
- Schumacher, T.; Benndorf, R.A. ABC Transport Proteins in Cardiovascular Disease—A Brief Summary. Molecules 2017, 22, 589. [Google Scholar] [CrossRef]
- Quinton, P.M. Physiological Basis of Cystic Fibrosis: A Historical Perspective. Physiol. Rev. 1999, 79, S3–S22. [Google Scholar] [CrossRef]
- Labombarda, F.; Saloux, E.; Brouard, J.; Bergot, E.; Milliez, P. Heart involvement in cystic fibrosis: A specific cystic fibrosis-related myocardial changes? Respir. Med. 2016, 118, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lin, Z.; Liu, M.; Liao, H.; Chen, Y.; Zhang, X.; Chan, H.C.; Zhou, B.; Rao, L.; Sun, H. CFTR deficiency causes cardiac dysplasia during zebrafish embryogenesis and is associated with dilated cardiomyopathy. Mech. Dev. 2020, 163, 103627. [Google Scholar] [CrossRef]
- Hausner, E.A.; Elmore, S.A.; Yang, X. Overview of the Components of Cardiac Metabolism. Drug Metab. Dispos. 2019, 47, 673–688. [Google Scholar] [CrossRef] [PubMed]
- Nhiri, R.; ElOuafi, N. An Unusual Treatable Cause of Reversible Cardiomyopathy: Vitamin B12 Deficiency. ARC J. Cardiol. 2020, 6, 14–17. [Google Scholar]
- Yamamoto, K.; Ikeda, U.; Furuhashi, K.; Irokawa, M.; Nakayama, T.; Shimada, K. The coagulation system is activated in idiopathic cardiomyopathy. J. Am. Coll. Cardiol. 1995, 25, 1634–1640. [Google Scholar] [CrossRef]
- Ito, K.; Hongo, K.; Date, T.; Ikegami, M.; Hano, H.; Owada, M.; Morimoto, S.; Kashiwagi, Y.; Katoh, D.; Yoshino, T.; et al. Tissue thrombin is associated with the pathogenesis of dilated cardiomyopathy. Int. J. Cardiol. 2016, 228, 821–827. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Variables | Value |
|---|---|
| Age, years (mean ± SD) | 56 ± 6 |
| BMI (Kg/m2) | 30.3 ± 7.6 |
| Male, sex n (%) | 10 (66.7) |
| NYHA class | |
| I | none |
| II | 6 |
| III | 9 |
| IV | none |
| Medical history, n (%) | |
| Diabetes mellitus | 5 (33.3) |
| Hypertension | 8 (53) |
| Dyslipidemia | 5 (31) |
| Current smoker n (%) | 2 (12.5) |
| eGFR | 76.3 ± 29.8 |
| Hemoglobin g/dL | 13.4 ± 1.2 |
| Echocardiography parameters | |
| EF (%) | 37.4 ± 9.2 |
| VTD (mL) | 206.8 ± 80 |
| EDV (mL) | 135.8 ± 67.8 |
| E/E’ | 11.4 ± 5.9 |
| TAPSE (mm) | 18.4 ± 4.8 |
| RVS’ (cm/s) | 11.5 ± 1.6 |
| PASP (mmHg) | 28.8 ± 4.5 |
| Medication baseline, n (%) | |
| ß-blocker | 13 (86.7) |
| ACEi/ARB | 7 (46.7) |
| Aldosterone antagonist | 9 (60) |
| Diuretic | 10 (66.7) |
| Sacubitril + Valsartan | 4 (26.6) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Agostino, Y.; Palumbo, D.; Rusciano, M.R.; Strianese, O.; Amabile, S.; Di Rosa, D.; De Angelis, E.; Visco, V.; Russo, F.; Alexandrova, E.; et al. Whole-Exome Sequencing Revealed New Candidate Genes for Human Dilated Cardiomyopathy. Diagnostics 2022, 12, 2411. https://doi.org/10.3390/diagnostics12102411
D’Agostino Y, Palumbo D, Rusciano MR, Strianese O, Amabile S, Di Rosa D, De Angelis E, Visco V, Russo F, Alexandrova E, et al. Whole-Exome Sequencing Revealed New Candidate Genes for Human Dilated Cardiomyopathy. Diagnostics. 2022; 12(10):2411. https://doi.org/10.3390/diagnostics12102411
Chicago/Turabian StyleD’Agostino, Ylenia, Domenico Palumbo, Maria Rosaria Rusciano, Oriana Strianese, Sonia Amabile, Domenico Di Rosa, Elena De Angelis, Valeria Visco, Fabio Russo, Elena Alexandrova, and et al. 2022. "Whole-Exome Sequencing Revealed New Candidate Genes for Human Dilated Cardiomyopathy" Diagnostics 12, no. 10: 2411. https://doi.org/10.3390/diagnostics12102411
APA StyleD’Agostino, Y., Palumbo, D., Rusciano, M. R., Strianese, O., Amabile, S., Di Rosa, D., De Angelis, E., Visco, V., Russo, F., Alexandrova, E., Salvati, A., Giurato, G., Nassa, G., Tarallo, R., Galasso, G., Ciccarelli, M., Weisz, A., & Rizzo, F. (2022). Whole-Exome Sequencing Revealed New Candidate Genes for Human Dilated Cardiomyopathy. Diagnostics, 12(10), 2411. https://doi.org/10.3390/diagnostics12102411