
Deep Learning of Histopathological Features for the Prediction of Tumour Molecular Genetics


Abstract
:1. Introduction
1.1. Deep Learning
1.1.1. Convolutional Neural Networks
1.1.2. Generative Adversarial Networks
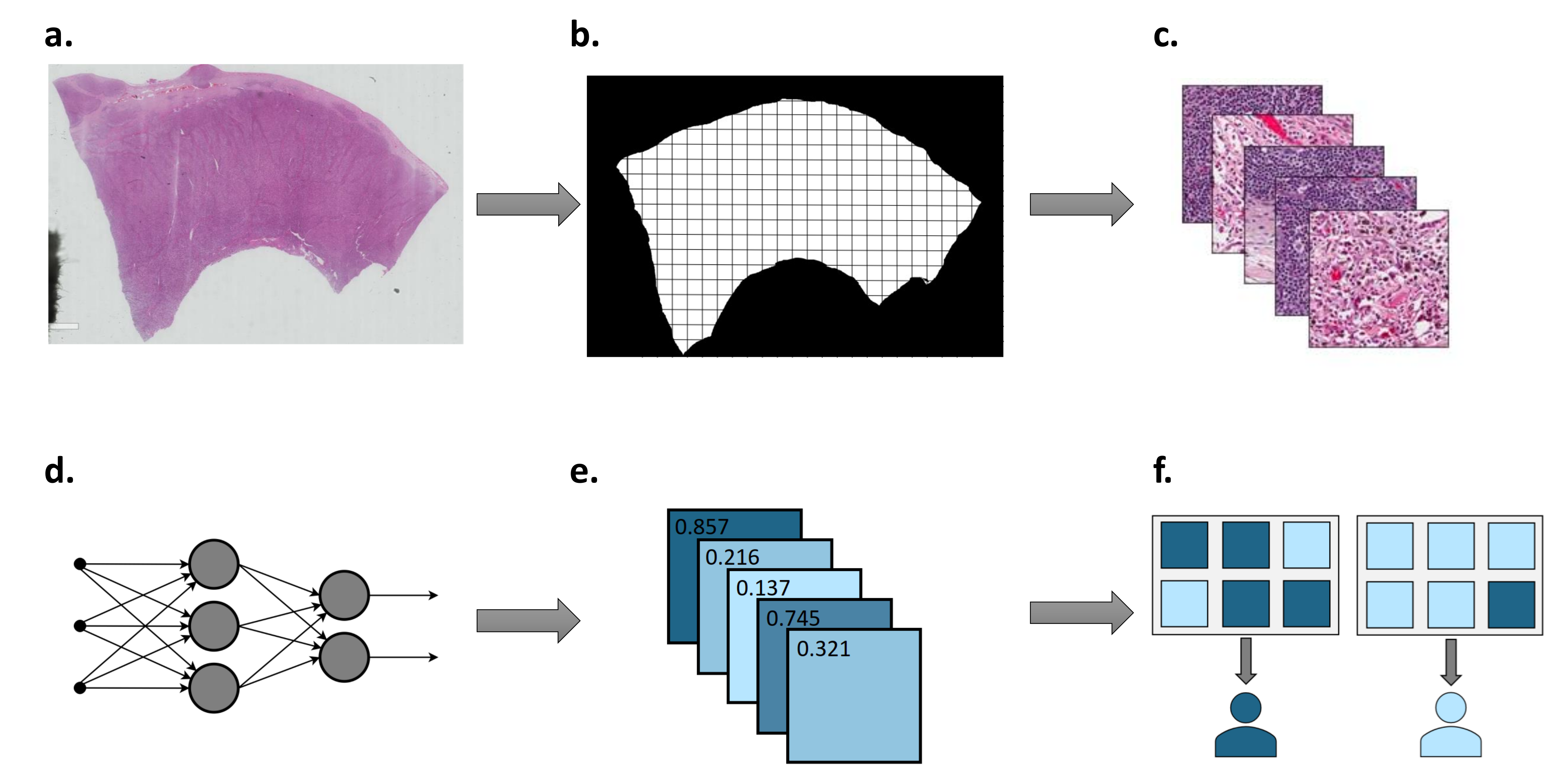
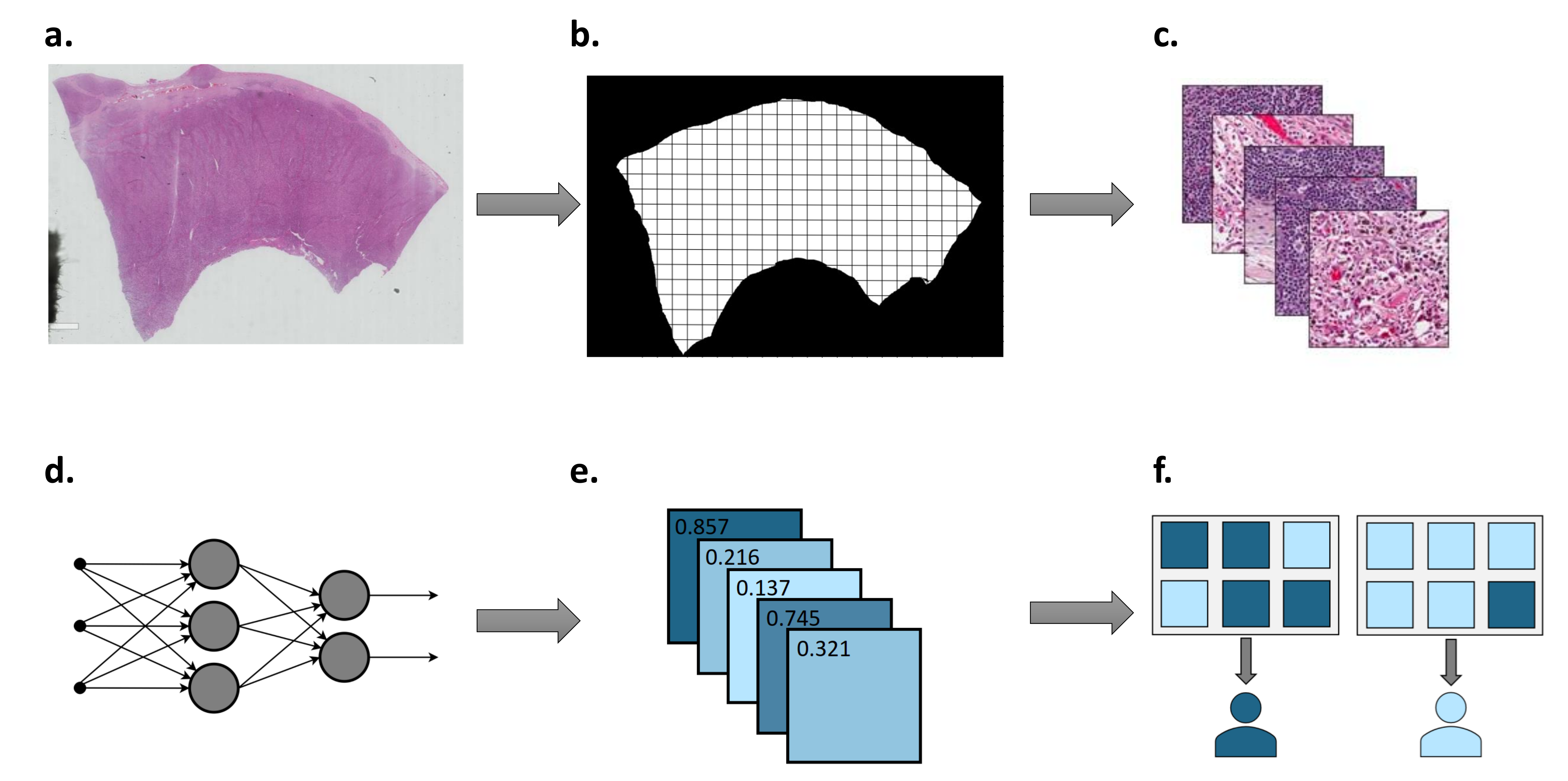
1.2. Deep Learning Workflow in Digital Pathology
1.2.1. Sample Preparation and Annotation
1.2.2. Colour Normalisation and Augmentation
1.2.3. Transfer Learning and Tile Aggregation
1.2.4. Model Interpretation
2. Materials and Methods
- Full-text available in English.
- Not a review article, commentary or editorial.
- Digitised whole-slide images are used as an input to a neural network.
- The neural network is used to predict the presence of a molecular feature, namely, mutations, mutated genes, gene expression, hormone receptor status, TMB, or MSI.
3. Results
3.1. Predicting Mutations
3.2. Predicting Gene Expression and Hormone Receptor Status
3.3. Predicting Tumour Mutational Burden
3.4. Predicting Microsatellite Instability
4. Discussion
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| EGFR | Epidermal growth factor receptor |
| NSCLC | Non-small cell lung cancer |
| TMB | Tumour mutational burden |
| MSI | Microsatellite instability |
| MIL | Multiple-instance learning |
| CNN | Convolutional neural network |
| WSI | Whole-slide image |
| FFPE | Formalin-fixed paraffin-embedded |
| H&E | Hematoxylin and eosin |
| AUC | Area under the ROC curve |
| TCGA | The Cancer Genome Atlas |
| WGD | Whole-genome duplication |
| GAN | Generative adversarial network |
| IHC | Immunohistochemistry |
| PR | Progesterone receptor |
| HER2 | Human epidermal growth factor receptor 2 |
| FDA | US Food and Drug Administration |
| BAP1 | BRCA1-associated protein 1 |
| ABCTB | Australian Breast Cancer Tissue Bank |
| ER | Estrogen rceptor |
| WES | Whole-exome sequencing |
| STAD | Atomach adenomcarcinoma |
| COAD | Colon adenocarcinoma |
| TIL | Tumour-infiltrating lymphocytes |
| MMR | Mismatch repair |
References
- Sequist, L.V.; Lynch, T.J. EGFR Tyrosine Kinase Inhibitors in Lung Cancer: An Evolving Story. Annu. Rev. Med. 2008, 59, 429–442. [Google Scholar] [CrossRef]
- Bollag, G.; Hirth, P.; Tsai, J.; Zhang, J.; Ibrahim, P.N.; Cho, H.; Spevak, W.; Zhang, C.; Zhang, Y.; Habets, G.; et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 2010, 467, 596–599. [Google Scholar] [CrossRef]
- Tsao, M.S.; Aviel-Ronen, S.; Ding, K.; Lau, D.; Liu, N.; Sakurada, A.; Whitehead, M.; Zhu, C.Q.; Livingston, R.; Johnson, D.H.; et al. Prognostic and predictive importance of p53 and RAS for adjuvant chemotherapy in non-small-cell lung cancer. J. Clin. Oncol. 2007, 25, 5240–5247. [Google Scholar] [CrossRef]
- Davis, A.A.; Patel, V.G. The role of PD-L1 expression as a predictive biomarker: An analysis of all US food and drug administration (FDA) approvals of immune checkpoint inhibitors. J. Immunother. Cancer 2019, 7, 278. [Google Scholar] [CrossRef]
- Kim, H.; Chung, J.H. PD-L1 Testing in Non-Small Cell Lung Cancer: Past, Present, and Future. J. Pathol. Transl. Med. 2019, 53, 199–206. [Google Scholar] [CrossRef] [Green Version]
- Hellmann, M.D.; Ciuleanu, T.E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Li, L.; Jiang, X.; Li, Q. Mismatch repair deficiency/microsatellite instability-high as a predictor for anti-PD-1/PD-L1 immunotherapy efficacy. J. Hematol. Oncol. 2019, 12, 54. [Google Scholar] [CrossRef] [PubMed]
- Viros, A.; Fridlyand, J.; Bauer, J.; Lasithiotakis, K.; Garbe, C.; Pinkel, D.; Bastian, B.C. Improving melanoma classification by integrating genetic and morphologic features. PLoS Med. 2008, 5, e120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ninomiya, H.; Hiramatsu, M.; Inamura, K.; Nomura, K.; Okui, M.; Miyoshi, T.; Okumura, S.; Satoh, Y.; Nakagawa, K.; Nishio, M.; et al. Correlation between morphology and EGFR mutations in lung adenocarcinomas. Significance of the micropapillary pattern and the hobnail cell type. Lung Cancer 2009, 63, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Brockmoeller, S.; Young, C.; Lee, J.; Arends, M.J.; Wilkins, B.S.; Thomas, G.J.; Oien, K.A.; Jones, L.; Hunter, K.D. Survey of UK histopathology consultants’ attitudes towards academic and molecular pathology. J. Clin. Pathol. 2019, 72, 399–405. [Google Scholar] [CrossRef] [Green Version]
- Märkl, B.; Füzesi, L.; Huss, R.; Bauer, S.; Schaller, T. Number of pathologists in Germany: Comparison with European countries, USA, and Canada. Virchows Arch. 2021, 478, 335–341. [Google Scholar] [CrossRef]
- Benjamens, S.; Dhunnoo, P.; Meskó, B. The state of artificial intelligence-based FDA-approved medical devices and algorithms: An online database. NPJ Digit. Med. 2020, 3, 118. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Dong, W.; Socher, R.; Li, L.J.; Li, K.; Li, F.-F. ImageNet: A Large-Scale Hierarchical Image Database. In Proceedings of the 2009 IEEE Conference on Computer Vision and Pattern Recognition, Miami, FL, USA, 20–25 June 2009; pp. 248–255. [Google Scholar] [CrossRef] [Green Version]
- LeCun, Y.A.; Bottou, L.; Orr, G.B.; Müller, K.R. Efficient BackProp. In Lecture Notes in Computer Science (Including Subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics); Springer: Berlin/Heidelberg, Germany, 2012; pp. 9–48. [Google Scholar] [CrossRef]
- Bishop, C.M. Pattern Recognition and Machine Learning; Springer: New York, NY, USA, 2006; p. 738. [Google Scholar]
- LeCun, Y.; Bengio, Y. Convolutional networks for images, speech, and time series. In The Handbook of Brain Theory and Neural Networks; MIT Press: Cambridge, MA, USA, 1995; Volume 3361. [Google Scholar]
- Arvaniti, E.; Fricker, K.S.; Moret, M.; Rupp, N.; Hermanns, T.; Fankhauser, C.; Wey, N.; Wild, P.J.; Rüschoff, J.H.; Claassen, M. Automated Gleason grading of prostate cancer tissue microarrays via deep learning. Sci. Rep. 2018, 8, 12054. [Google Scholar] [CrossRef]
- Kather, J.N.; Krisam, J.; Charoentong, P.; Luedde, T.; Herpel, E.; Weis, C.A.; Gaiser, T.; Marx, A.; Valous, N.A.; Ferber, D.; et al. Predicting survival from colorectal cancer histology slides using deep learning: A retrospective multicenter study. PLoS Med. 2019, 16, e1002730. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.H.; Li, J.; Gong, H.F.; Yu, G.Y.; Liu, P.; Hao, L.Q.; Liu, L.J.; Bai, C.G.; Zhang, W. Comparison of Fresh Frozen Tissue With Formalin-Fixed Paraffin-Embedded Tissue for Mutation Analysis Using a Multi-Gene Panel in Patients With Colorectal Cancer. Front. Oncol. 2020, 10, 310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khened, M.; Kori, A.; Rajkumar, H.; Krishnamurthi, G.; Srinivasan, B. A generalized deep learning framework for whole-slide image segmentation and analysis. Sci. Rep. 2021, 11, 11579. [Google Scholar] [CrossRef]
- Priego-Torres, B.M.; Sanchez-Morillo, D.; Fernandez-Granero, M.A.; Garcia-Rojo, M. Automatic segmentation of whole-slide H&E stained breast histopathology images using a deep convolutional neural network architecture. Expert Syst. Appl. 2020, 151, 113387. [Google Scholar] [CrossRef]
- Castiglioni, I.; Rundo, L.; Codari, M.; Di Leo, G.; Salvatore, C.; Interlenghi, M.; Gallivanone, F.; Cozzi, A.; D’Amico, N.C.; Sardanelli, F. AI applications to medical images: From machine learning to deep learning. Phys. Med. 2021, 83, 9–24. [Google Scholar] [CrossRef]
- Howard, F.M.; Dolezal, J.; Kochanny, S.; Schulte, J.; Chen, H.; Heij, L.; Huo, D.; Nanda, R.; Olopade, O.I.; Kather, J.N.; et al. The Impact of Digital Histopathology Batch Effect on Deep Learning Model Accuracy and Bias. bioRxiv 2020. [Google Scholar] [CrossRef]
- Tellez, D.; Litjens, G.; Bándi, P.; Bulten, W.; Bokhorst, J.M.; Ciompi, F.; van der Laak, J. Quantifying the effects of data augmentation and stain color normalization in convolutional neural networks for computational pathology. Med. Image Anal. 2019, 58. [Google Scholar] [CrossRef] [Green Version]
- Russakovsky, O.; Deng, J.; Su, H.; Krause, J.; Satheesh, S.; Ma, S.; Huang, Z.; Karpathy, A.; Khosla, A.; Bernstein, M.; et al. ImageNet Large Scale Visual Recognition Challenge. Int. J. Comput. Vis. 2014, 115, 211–252. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.; Sun, F.; Kong, T.; Zhang, W.; Yang, C.; Liu, C. A Survey on Deep Transfer Learning. In Proceedings of the 27th International Conference on Artificial Neural Networks, Rhodes, Greece, 4–7 October 2018; Springer: Cham, Switzerland, 2018; pp. 270–279. [Google Scholar] [CrossRef] [Green Version]
- Krause, J.; Grabsch, H.I.; Kloor, M.; Jendrusch, M.; Echle, A.; Buelow, R.D.; Boor, P.; Luedde, T.; Brinker, T.J.; Trautwein, C.; et al. Deep learning detects genetic alterations in cancer histology generated by adversarial networks. J. Pathol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Yang, F.; Ma, S.C.; Liu, L.; Zhao, Y.; Li, Y.; Wu, D.H.; Wang, T.; Lu, W.J.; Cai, W.J.; et al. Development and interpretation of a pathomics-based model for the prediction of microsatellite instability in Colorectal Cancer. Theranostics 2020, 10, 11080–11091. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Khosla, A.; Lapedriza, A.; Oliva, A.; Torralba, A. Learning Deep Features for Discriminative Localization. In Proceedings of the IEEE Computer Society Conference on Computer Vision and Pattern Recognition, Las Vegas, NV, USA, 27–30 June 2016; IEEE Computer Society: Washington, DC, USA, 2016; pp. 2921–2929. [Google Scholar] [CrossRef] [Green Version]
- Bach, S.; Binder, A.; Montavon, G.; Klauschen, F.; Müller, K.R.; Samek, W. On pixel-wise explanations for non-linear classifier decisions by layer-wise relevance propagation. PLoS ONE 2015, 10, e0130140. [Google Scholar] [CrossRef] [Green Version]
- Simonyan, K.; Vedaldi, A.; Zisserman, A. Deep Inside Convolutional Networks: Visualising Image Classification Models and Saliency Maps. In Proceedings of the 2nd International Conference on Learning Representations, ICLR 2014—Workshop Track Proceedings, Banff, AB, Canada, 14–16 April 2014. [Google Scholar]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [Green Version]
- Kumari, N.; Singh, S.; Haloi, D.; Mishra, S.K.; Krishnani, N.; Nath, A.; Neyaz, Z. Epidermal Growth Factor Receptor Mutation Frequency in Squamous Cell Carcinoma and Its Diagnostic Performance in Cytological Samples: A Molecular and Immunohistochemical Study. World J. Oncol. 2019, 10, 142–150. [Google Scholar] [CrossRef]
- Coudray, N.; Ocampo, P.S.; Sakellaropoulos, T.; Narula, N.; Snuderl, M.; Fenyö, D.; Moreira, A.L.; Razavian, N.; Tsirigos, A. Classification and mutation prediction from non–small cell lung cancer histopathology images using deep learning. Nat. Med. 2018, 24, 1559–1567. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhang, B.; Topatana, W.; Cao, J.; Zhu, H.; Juengpanich, S.; Mao, Q.; Yu, H.; Cai, X. Classification and mutation prediction based on histopathology H&E images in liver cancer using deep learning. NPJ Precis. Oncol. 2020, 4, 1–7. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, J.; Liang, Y.; Liao, B.; Zhu, W.; Mo, X.; Huang, K. Identification and Validation of Efficacy of Immunological Therapy for Lung Cancer From Histopathological Images Based on Deep Learning. Front. Genet. 2021, 12, 642981. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Huang, X.; Huang, S.; Ding, X.; Wang, L. Direct Prediction of BRAFV600E Mutation from Histopathological Images in Papillary Thyroid Carcinoma with a Deep Learning Workflow. In Proceedings of the 4th International Conference on Computer Science and Artificial Intelligence, Zhuhai, China, 11–13 December 2020; Association for Computing Machinery: New York, NY, USA, 2020; pp. 146–151. [Google Scholar] [CrossRef]
- Loeffler, C.M.L.; Ortiz Bruechle, N.; Jung, M.; Seillier, L.; Rose, M.; Laleh, N.G.; Knuechel, R.; Brinker, T.J.; Trautwein, C.; Gaisa, N.T.; et al. Artificial Intelligence–based Detection of FGFR3 Mutational Status Directly from Routine Histology in Bladder Cancer: A Possible Preselection for Molecular Testing? Eur. Urol. Focus 2021, 7, 497–668. [Google Scholar] [CrossRef]
- Jang, H.J.; Lee, A.; Kang, J.; Song, I.H.; Lee, S.H. Prediction of clinically actionable genetic alterations from colorectal cancer histopathology images using deep learning. World J. Gastroenterol. 2020, 26, 6207–6223. [Google Scholar] [CrossRef]
- Tsou, P.; Wu, C.J. Mapping Driver Mutations to Histopathological Subtypes in Papillary Thyroid Carcinoma: Applying a Deep Convolutional Neural Network. J. Clin. Med. 2019, 8, 1675. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Shah, Z.; Sav, A.; Russo, C.; Berkovsky, S.; Qian, Y.; Coiera, E.; Di Ieva, A. Isocitrate dehydrogenase (IDH) status prediction in histopathology images of gliomas using deep learning. Sci. Rep. 2020, 10, 7733. [Google Scholar] [CrossRef]
- Fu, Y.; Jung, A.W.; Torne, R.V.; Gonzalez, S.; Vöhringer, H.; Shmatko, A.; Yates, L.R.; Jimenez-Linan, M.; Moore, L.; Gerstung, M. Pan-cancer computational histopathology reveals mutations, tumor composition and prognosis. Nat. Cancer 2020, 1, 800–810. [Google Scholar] [CrossRef]
- Kather, J.N.; Heij, L.R.; Grabsch, H.I.; Loeffler, C.; Echle, A.; Muti, H.S.; Krause, J.; Niehues, J.M.; Sommer, K.A.; Bankhead, P.; et al. Pan-cancer image-based detection of clinically actionable genetic alterations. Nat. Cancer 2020, 1, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Noorbakhsh, J.; Farahmand, S.; Foroughi Pour, A.; Namburi, S.; Caruana, D.; Rimm, D.; Soltanieh-ha, M.; Zarringhalam, K.; Chuang, J.H. Deep learning-based cross-classifications reveal conserved spatial behaviors within tumor histological images. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, L.M.; Lopiano, K.K.; Morse, A.M.; Amin, V.; Oberg, A.L.; Young, L.J.; Nuzhdin, S.V. RNA-seq: Technical variability and sampling. BMC Genom. 2011, 12, 293. [Google Scholar] [CrossRef] [Green Version]
- Dowsett, M.; Houghton, J.; Iden, C.; Salter, J.; Farndon, J.; A’Hern, R.; Sainsbury, R.; Baum, M. Benefit from adjuvant tamoxifen therapy in primary breast cancer patients according oestrogen receptor, progesterone receptor, EGF receptor and HER2 status. Ann. Oncol. 2006, 17, 818–826. [Google Scholar] [CrossRef] [PubMed]
- Hudis, C.A. Trastuzumab—Mechanism of Action and Use in Clinical Practice. N. Engl. J. Med. 2007, 357, 39–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sha, L.; Osinski, B.; Ho, I.; Tan, T.; Willis, C.; Weiss, H.; Beaubier, N.; Mahon, B.; Taxter, T.; Yip, S. Multi-field-of-view deep learning model predicts nonsmall cell lung cancer programmed death-ligand 1 status from whole-slide hematoxylin and eosin images. J. Pathol. Inform. 2019, 10, 24. [Google Scholar] [CrossRef] [PubMed]
- Griewank, K.G.; Van De Nes, J.; Schilling, B.; Moll, I.; Sucker, A.; Kakavand, H.; Haydu, L.E.; Asher, M.; Zimmer, L.; Hillen, U.; et al. Genetic and clinico-pathologic analysis of metastatic uveal melanoma. Mod. Pathol. 2014, 27, 175–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, M.; Zhou, W.; Qi, X.; Zhang, G.; Girnita, L.; Seregard, S.; Grossniklaus, H.E.; Yao, Z.; Zhou, X.; Stålhammar, G. Prediction of BAP1 expression in uveal melanoma using densely-connected deep classification networks. Cancers 2019, 11, 1579. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Kalirai, H.; Acha-Sagredo, A.; Yang, X.; Zheng, Y.; Coupland, S.E. Piloting a deep learning model for predicting nuclear BAP1 immunohistochemical expression of uveal melanoma from hematoxylin-and-eosin sections. Transl. Vis. Sci. Technol. 2020, 9, 1–13. [Google Scholar] [CrossRef]
- Schmauch, B.; Romagnoni, A.; Pronier, E.; Saillard, C.; Maillé, P.; Calderaro, J.; Kamoun, A.; Sefta, M.; Toldo, S.; Zaslavskiy, M.; et al. A deep learning model to predict RNA-Seq expression of tumours from whole slide images. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Bergenstråhle, L.; Stenbeck, L.; Abid, A.; Andersson, A.; Borg, Å.; Maaskola, J.; Lundeberg, J.; Zou, J. Integrating spatial gene expression and breast tumour morphology via deep learning. Nat. Biomed. Eng. 2020, 4, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Levy-Jurgenson, A.; Tekpli, X.; Kristensen, V.N.; Yakhini, Z. Spatial transcriptomics inferred from pathology whole-slide images links tumor heterogeneity to survival in breast and lung cancer. Sci. Rep. 2020, 10, 18802. [Google Scholar] [CrossRef] [PubMed]
- Jaber, M.I.; Song, B.; Taylor, C.; Vaske, C.J.; Benz, S.C.; Rabizadeh, S.; Soon-Shiong, P.; Szeto, C.W. A deep learning image-based intrinsic molecular subtype classifier of breast tumors reveals tumor heterogeneity that may affect survival. Breast Cancer Res. 2020, 22, 12. [Google Scholar] [CrossRef] [Green Version]
- Naik, N.; Madani, A.; Esteva, A.; Keskar, N.S.; Press, M.F.; Ruderman, D.; Agus, D.B.; Socher, R. Deep learning-enabled breast cancer hormonal receptor status determination from base-level H&E stains. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef]
- Anand, D.; Kurian, N.; Dhage, S.; Kumar, N.; Rane, S.; Gann, P.; Sethi, A. Deep learning to estimate human epidermal growth factor receptor 2 status from hematoxylin and eosin-stained breast tissue images. J. Pathol. Inform. 2020, 11, 19. [Google Scholar] [CrossRef]
- Rawat, R.R.; Ortega, I.; Roy, P.; Sha, F.; Shibata, D.; Ruderman, D.; Agus, D.B. Deep learned tissue “fingerprints” classify breast cancers by ER/PR/Her2 status from H&E images. Sci. Rep. 2020, 10, 7275. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.A.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Johnson, B.A.; Lutz, E.R.; Laheru, D.A.; Jaffee, E.M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 2017, 17, 209–222. [Google Scholar] [CrossRef]
- Bersanelli, M. Tumour mutational burden as a driver for treatment choice in resistant tumours (and beyond). Lancet Oncol. 2020, 21, 1255–1257. [Google Scholar] [CrossRef]
- Tang, Y.; Li, Y.; Wang, W.; Lizaso, A.; Hou, T.; Jiang, L.; Huang, M. Tumor mutation burden derived from small next generation sequencing targeted gene panel as an initial screening method. Transl. Lung Cancer Res. 2020, 9, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.S.; Massoud, T.F. Predicting tumour mutational burden from histopathological images using multiscale deep learning. Nat. Mach. Intell. 2020, 2, 356–362. [Google Scholar] [CrossRef]
- Wang, L.; Jiao, Y.; Qiao, Y.; Zeng, N.; Yu, R. A novel approach combined transfer learning and deep learning to predict TMB from histology image. Pattern Recognit. Lett. 2020, 135, 244–248. [Google Scholar] [CrossRef]
- Shimada, Y.; Okuda, S.; Watanabe, Y.; Tajima, Y.; Nagahashi, M.; Ichikawa, H.; Nakano, M.; Sakata, J.; Takii, Y.; Kawasaki, T.; et al. Histopathological characteristics and artificial intelligence for predicting tumor mutational burden-high colorectal cancer. J. Gastroenterol. 2021, 56, 547–559. [Google Scholar] [CrossRef]
- Kawakami, H.; Zaanan, A.; Sinicrope, F.A. Microsatellite Instability Testing and Its Role in the Management of Colorectal Cancer. Curr. Treat. Opt. Oncol. 2015, 16, 30. [Google Scholar] [CrossRef]
- Boland, C.R.; Goel, A. Microsatellite Instability in Colorectal Cancer. Gastroenterology 2010, 138, 2073–2087.e3. [Google Scholar] [CrossRef]
- Lemery, S.; Keegan, P.; Pazdur, R. First FDA Approval Agnostic of Cancer Site—When a Biomarker Defines the Indication. N. Engl. J. Med. 2017, 377, 1409–1412. [Google Scholar] [CrossRef]
- Kather, J.N.; Halama, N.; Jaeger, D. Genomics and emerging biomarkers for immunotherapy of colorectal cancer. Semin. Cancer Biol. 2018, 52, 189–197. [Google Scholar] [CrossRef] [PubMed]
- De Smedt, L.; Lemahieu, J.; Palmans, S.; Govaere, O.; Tousseyn, T.; Van Cutsem, E.; Prenen, H.; Tejpar, S.; Spaepen, M.; Matthijs, G.; et al. Microsatellite instable vs stable colon carcinomas: Analysis of tumour heterogeneity, inflammation and angiogenesis. Br. J. Cancer 2015, 113, 500–509. [Google Scholar] [CrossRef] [Green Version]
- Kather, J.N.; Pearson, A.T.; Halama, N.; Jäger, D.; Krause, J.; Loosen, S.H.; Marx, A.; Boor, P.; Tacke, F.; Neumann, U.P.; et al. Deep learning can predict microsatellite instability directly from histology in gastrointestinal cancer. Nat. Med. 2019, 25, 1054–1056. [Google Scholar] [CrossRef]
- Ke, J.; Shen, Y.; Guo, Y.; Wright, J.D.; Liang, X. A Prediction Model of Microsatellite Status from Histology Images. In Proceedings of the ICBET 2020: 2020 10th International Conference on Biomedical Engineering and Technology, Tokyo, Japan, 15–18 September 2020; Association for Computing Machinery: New York, NY, USA, 2020; pp. 334–338. [Google Scholar] [CrossRef]
- Echle, A.; Grabsch, H.I.; Quirke, P.; van den Brandt, P.A.; West, N.P.; Hutchins, G.G.; Heij, L.R.; Tan, X.; Richman, S.D.; Krause, J.; et al. Clinical-Grade Detection of Microsatellite Instability in Colorectal Tumors by Deep Learning. Gastroenterology 2020, 159, 1406–1416.e11. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, R.; Long, J.; Longacre, T.; Peng, L.; Berry, G.; Martin, B.; Higgins, J.; Rubin, D.L.; Shen, J. Deep learning model for the prediction of microsatellite instability in colorectal cancer: A diagnostic study. Lancet Oncol. 2021, 22, 132–141. [Google Scholar] [CrossRef]
- Lee, S.H.; Song, I.H.; Jang, H. Feasibility of deep learning-based fully automated classification of microsatellite instability in tissue slides of colorectal cancer. Int. J. Cancer 2021, 149, 728–740. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Reference | Description | Cohorts | Num. of Samples (ext. Validation) | External Validation | Neural Network | Outcome Measures |
|---|---|---|---|---|---|---|
| Coudray, 2018 [34] | Predicted the mutation status of 6 of 10 selected genes in lung cancer. | TCGA-LUAD | 567 (1) | Yes | Inception-v3 | AUC range 0.73 (KRAS) to 0.85 (STK11) |
| Tsou, 2019 [40] | Predicted BRAF V600E or RAS mutant in papillary thyroid carcinoma. | TCGA-PTC | 103 | No | Inception-v3 | AUC 0.951 |
| Liu, 2020 [41] | GAN-based data augmentation to improve prediction of IDH mutation status in glioma. | TCGA-GBM, -LGG, internal data set | 266 | No | ResNet50 | AUC 0.927 |
| Chen, 2020 [35] | Predicted the mutation status of 4 of 10 selected genes in liver cancer. | TCGA-HCC, internal data set | 387 (101) | Yes | Inception-v3 | AUC range 0.71 (ZFX4) to 0.89 (CTNNB1) |
| Fu, 2020 [42] | Predicted a number of mutations across 28 cancers. | TCGA, METABRIC, BASIS | 17,355 (622) | Yes | Inception-v4 | AUC 0.73 (WGD) |
| Kather, 2020 [43] | Predicted mutations and gene expression signatures across 14 cancers. | TCGA, DACHS | >5000 (408) | Yes | ShuffleNet | AUC 0.77 (BRAF in CRC); AUC 0.66 (CIMP-high in CRC) |
| Jang, 2020 [39] | Predicted the mutation status of 5 genes in colorectal cancer. | TCGA-READ, -COAD, internal data set | 629 (142) | Yes | Inception-v3 | AUC range 0.693 (SMAD4) to 0.809 (TP53) on frozen; AUC range 0.645 (KRAS) to 0.783 (TP53) on FFPE |
| Noorbakhsh, 2020 [44] | Predicted TP53 mutation status across cancer types. | TCGA-BRCA, -LUAD, -STAD, -COAD, -BLCA | 27,815 (2115) | Yes | Inception-v3 | AUC range 0.65 to 0.80 |
| Wu, 2020 [37] | Predicted BRAF V600E status in papillary thyroid carcinoma. | Internal data set, PCam-BRCA, PAIP-LIHC | 439 | Yes | DenseNet-121 | AUC 0.884 |
| Yang, 2021 [36] | Predicted mutation status of immune-related genes in lung cancer. | TCGA-LUAD, -LUSC | 180 | No | ResNet | AUC range 0.71 to 0.87 |
| Loeffler, 2021 [38] | Predicted mutation status of FGFR3 in bladder cancer. | TCGA-BLCA, Aachen cohort | 327 (182) | Yes | ShuffleNet | AUC 0.70 (TCGA-BLCA); AUC 0.62 (ext. validation) |
| Reference | Description | Cohorts | Num. of Samples (ext. Validation) | External Validation | Neural Network | Outcome Measures |
|---|---|---|---|---|---|---|
| Sha, 2019 [48] | Predicted PD-L1 expression status in lung cancer. | Internal data set | 130 | No | ResNet18 | AUC 0.80 |
| Sun, 2019 [50] | Predicted BAP1 expression status in uveal melanoma. | Internal data sets | 17 (30) | Yes | DenseNet-121 | AUC-positive 0.99; AUC-negative 0.98 |
| Jaber, 2020 [55] | Predicted PAM50 gene expression- based subtypes in breast cancer. | TCGA-BRCA | 1142 | No | Inception-v3 | AUC 0.82 (Basal/non-Basal) |
| Rawat, 2020 [58] | Predicted hormone receptor status in breast cancer. | TCGA-BRCA, ABCTB | 939 (2351) | Yes | ResNet34 | AUC 0.89 (ER); AUC 0.81 (PR); AUC 0.79 (HER2) |
| He, 2020 [53] | Predicted spatial gene expression profiles in breast cancer. | Internal data set, 10x Genomics BC, TCGA-BRCA | 23 (1094) | Yes | DenseNet-121 | Pearson’s R range for top 5 genes 0.43–0.54 |
| Anand, 2020 [57] | Predicted HER2 overexpression in breast cancer. | Warwick, TCGA-BRCA | 52 (45) | Yes | U-Net and custom architecture | AUC 0.76 |
| Schmauch, 2020 [52] | Predicted gene expression of multiple genes across cancers. | TCGA | 8725 (369) | Yes | ResNet50 | Pearson’s R 0.47 (MKI67 in LIHC), 0.43 (CD3D in COAD) |
| Zhang, 2020 [51] | Predicted BAP1 expression status in uveal melanoma. | Internal data set | 184 | No | ResNet18 | AUC 0.93 |
| Levy-Jurgenson, 2020 [54] | Predicted spatially resolved transctiptomics from bulk mRNA and miRNA expression. | TCGA-BRCA, -LUAD | 761 BRCA, 469 LUAD | No | Inception-v3 | AUC 0.95 (miR-17) |
| Naik, 2020 [56] | Predicted hormone receptor status in breast cancer. | ABCTB, TCGA-BRCA | 2535 ABCTB, 1014 BRCA | Yes | ResNet50 | AUC 0.92 |
| Reference | Description | Cohorts | Num. of Samples (ext. Validation) | External Validation | Neural Network | Outcome Measures |
|---|---|---|---|---|---|---|
| Jain, 2020 [63] | Predicted TMB in lung adenocarcinoma. | TCGA-LUAD | 760 | No | Inception-v3 | AUPRC 0.92 |
| Wang, 2020 [64] | Predicted TMB in gastrointestinal cancer. | TCGA-STAD, -COAD, -READ | 644 | No | Resnet50, Googlenet, Inception v3, Alexnet, VGG19, Squeezenet, Densenet | AUC 0.75 STAD (Googlenet); AUC 0.82 COAD (VGG19) |
| Shimada, 2021 [65] | Predicted TMB in colorectal cancer. | TCGA-CRC, Japanese-CRC | 278 | No | Inception-v3 | AUPRC 0.91 |
| Reference | Description | Cohorts | Num. of Samples (ext. Validation) | External Validation | Neural Network | Outcome Measures |
|---|---|---|---|---|---|---|
| Kather, 2019 [71] | Predicted MSI status in gastointestinal cancer. | TCGA-STAD, -CRC, DACHS | 315 STAD, 747 CRC, (378) | Yes | ResNet18 | AUC 0.81 TCGA-STAD; AUC 0.84 TCGA-CRC-KR; AUC 0.77 TCGA-CRC-DX |
| Ke, 2019 [72] | Predicted MSI status in colorectal cancer. | TCGA-CRC | 387 CRC-KR, 360 CRC-DX | No | AlexNet, ResNet, VGG, Inception-v3 | Accuracy 98.8% (AlexNet) |
| Cao, 2020 [28] | Predicted MSI status in colorectal cancer. | TCCG-COAD, internal data set | 429 COAD, 785 internal | Yes | ResNet18 | AUC 0.88 (TCGA-COAD); AUC 0.85 (internal) |
| Echle, 2020 [73] | Predicted MSI status in colorectal cancer using a large, multicenter cohort. | MSIDETECT | 6406 (771) | Yes | ShuffleNet | AUC 0.96 |
| Yamashita, 2021 [74] | Predicted MSI status in colorectal cancer. | Internal data set, TCGA-CRC | 100 (484) | Yes | MobileNetV2 | AUC 0.779 |
| Krause, 2021 [27] | Used GAN-generated images to improve prediction of MSI in colorectal cancer. | TCGA-CRC, NLCS | 256 CRC, 1457 NLCS | No | Conditional GAN, ShuffleNet | AUC 0.742 (TCGA-CRC); AUC 0.757 (NLCS); AUC 0.777 (real and synthetic data) |
| Lee, 2021 [75] | Predicted MSI status in colorectal cancer. | TCGA-CRC, SMH | 1920 (365) | Yes | Inception-v3 | AUC 0.972 (TCGA-CRC); AUC 0.787 (SMH) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murchan, P.; Ó’Brien, C.; O’Connell, S.; McNevin, C.S.; Baird, A.-M.; Sheils, O.; Ó Broin, P.; Finn, S.P. Deep Learning of Histopathological Features for the Prediction of Tumour Molecular Genetics. Diagnostics 2021, 11, 1406. https://doi.org/10.3390/diagnostics11081406
Murchan P, Ó’Brien C, O’Connell S, McNevin CS, Baird A-M, Sheils O, Ó Broin P, Finn SP. Deep Learning of Histopathological Features for the Prediction of Tumour Molecular Genetics. Diagnostics. 2021; 11(8):1406. https://doi.org/10.3390/diagnostics11081406
Chicago/Turabian StyleMurchan, Pierre, Cathal Ó’Brien, Shane O’Connell, Ciara S. McNevin, Anne-Marie Baird, Orla Sheils, Pilib Ó Broin, and Stephen P. Finn. 2021. "Deep Learning of Histopathological Features for the Prediction of Tumour Molecular Genetics" Diagnostics 11, no. 8: 1406. https://doi.org/10.3390/diagnostics11081406
APA StyleMurchan, P., Ó’Brien, C., O’Connell, S., McNevin, C. S., Baird, A.-M., Sheils, O., Ó Broin, P., & Finn, S. P. (2021). Deep Learning of Histopathological Features for the Prediction of Tumour Molecular Genetics. Diagnostics, 11(8), 1406. https://doi.org/10.3390/diagnostics11081406