
Molecular Autopsy of Sudden Cardiac Death in the Genomics Era

 , ,
, , 
Abstract
1. Introduction
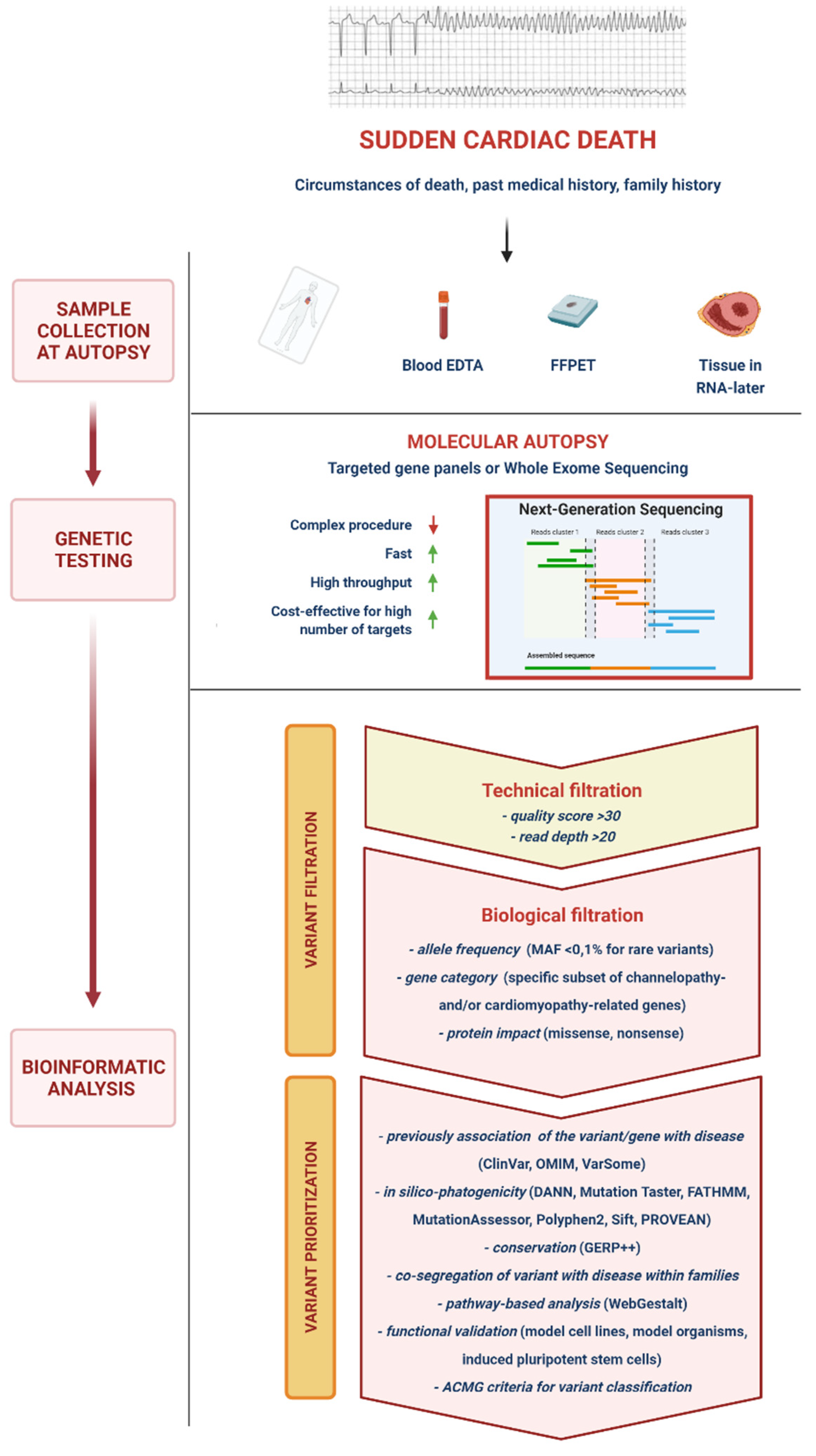
2. Methods
3. Molecular Autopsy
4. Sanger Sequencing
Sanger Sequencing in Sudden Cardiac Death
5. Next-Generation Sequencing
5.1. Variant Calling, Filtering, Prioritization and Interpretation
5.2. Challenges and Technical Issues
5.2.1. Sample Collection
5.2.2. Sequencing-Related Issues
5.2.3. Variants of Unknown Significance
5.3. Next-Generation Sequencing in Sudden Cardiac Death
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Priori, S.G.; Blomstrom-Lundqvist, C.; Mazzanti, A.; Bloma, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. 2015 ESC Guidelines for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death the Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the Europea. Eur. Heart J. 2015, 36, 2793–2867. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.X.; Brown, A.; Lau, D.H.; Chugh, S.S.; Albert, C.M.; Kalman, J.M.; Sanders, P. Epidemiology of Sudden Cardiac Death: Global and Regional Perspectives. Heart Lung Circ. 2019, 28, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Eckart, R.E.; Shry, E.A.; Burke, A.P.; McNear, J.A.; Appel, D.A.; Castillo-Rojas, L.M.; Avedissian, L.; Pearse, L.A.; Potter, R.N.; Tremaine, L.; et al. Sudden Death in Young Adults: An Autopsy-Based Series of a Population Undergoing Active Surveillance. J. Am. Coll. Cardiol. 2011, 58, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, M.J.; Priori, S.G.; Willems, S.; Berul, C.; Brugada, R.; Calkins, H.; Camm, A.J.; Ellinor, P.T.; Gollob, M.; Hamilton, R.; et al. HRS/EHRA Expert Consensus Statement on the State of Genetic Testing for the Channelopathies and Cardiomyopathies: This Document Was Developed as a Partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm 2011, 8, 1308–1339. [Google Scholar] [CrossRef]
- Buja, L.M.; Ottaviani, G.; Mitchell, R.N. Pathobiology of Cardiovascular Diseases: An Update. Cardiovasc. Pathol. 2019, 42, 44–53. [Google Scholar] [CrossRef]
- Priori, S.G.; Wilde, A.A.; Horie, M.; Cho, Y.; Behr, E.R.; Berul, C.; Blom, N.; Brugada, J.; Chiang, C.-E.; Huikuri, H.; et al. HRS/EHRA/APHRS Expert Consensus Statement on the Diagnosis and Management of Patients with Inherited Primary Arrhythmia Syndromes: Document Endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm 2013, 10, 1932–1963. [Google Scholar] [CrossRef]
- Ottaviani, G.; Buja, L.M. Anatomopathological Changes of the Cardiac Conduction System in Sudden Cardiac Death, Particularly in Infants: Advances over the Last 25 Years. Cardiovasc. Pathol. 2016, 25, 489–499. [Google Scholar] [CrossRef]
- Bagnall, R.D.; Weintraub, R.G.; Ingles, J.; Duflou, J.; Yeates, L.; Lam, L.; Davis, A.M.; Thompson, T.; Connell, V.; Wallace, J.; et al. A Prospective Study of Sudden Cardiac Death among Children and Young Adults. N. Engl. J. Med. 2016, 374, 2441–2452. [Google Scholar] [CrossRef] [PubMed]
- Haïssaguerre, M.; Duchateau, J.; Dubois, R.; Hocini, M.; Cheniti, G.; Sacher, F.; Lavergne, T.; Probst, V.; Surget, E.; Vigmond, E.; et al. Idiopathic Ventricular Fibrillation: Role of Purkinje System and Microstructural Myocardial Abnormalities. JACC Clin. Electrophysiol. 2020, 6, 591–608. [Google Scholar] [CrossRef]
- Orland, K.M.; Anderson, K.B. Molecular Autopsy for Sudden Cardiac Death: Current State and Considerations. Curr. Genet. Med. Rep. 2019, 7, 145–152. [Google Scholar] [CrossRef]
- Bagnall, R.D.; Singer, E.S.; Tfelt-Hansen, J. Sudden Cardiac Death in the Young. Heart Lung Circ. 2020, 29, 498–504. [Google Scholar] [CrossRef]
- Brohus, M.; Arsov, T.; Wallace, D.A.; Jensen, H.H.; Nyegaard, M.; Crotti, L.; Adamski, M.; Zhang, Y.; Field, M.A.; Athanasopoulos, V.; et al. Infanticide vs. Inherited Cardiac Arrhythmias. EP Eur. 2021, 23, 441–450. [Google Scholar] [CrossRef]
- Grassi, S.; Vidal, M.C.; Campuzano, O.; Arena, V.; Alfonsetti, A.; Rossi, S.S.; Scarnicci, F.; Iglesias, A.; Brugada, R.; Oliva, A. Sudden Death without a Clear Cause after Comprehensive Investigation: An Example of Forensic Approach to Atypical/Uncertain Findings. Diagnostics 2021, 11, 886. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of Age: Ten Years of next-Generation Sequencing Technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef] [PubMed]
- International Human Genome Sequencing Consortium. Finishing the Euchromatic Sequence of the Human Genome. Nature 2004, 431, 931–945. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, M.J.; Tester, D.J.; Porter, C.J.; Edwards, W.D. Molecular Diagnosis of the Inherited Long-QT Syndrome in a Woman Who Died after near-Drowning. N. Engl. J. Med. 1999, 341, 1121–1125. [Google Scholar] [CrossRef]
- Chugh, S.S.; Senashova, O.; Watts, A.; Tran, P.T.; Zhou, Z.; Gong, Q.; Titus, J.L.; Hayflick, S.J. Postmortem Molecular Screening in Unexplained Sudden Death. J. Am. Coll. Cardiol. 2004, 43, 1625–1629. [Google Scholar] [CrossRef]
- Di Paolo, M.; Luchini, D.; Bloise, R.; Priori, S.G. Postmortem Molecular Analysis in Victims of Sudden Unexplained Death. Am. J. Forensic Med. Pathol. 2004, 25, 182–184. [Google Scholar] [CrossRef]
- Skinner, J.R.; Crawford, J.; Smith, W.; Aitken, A.; Heaven, D.; Evans, C.-A.; Hayes, I.; Neas, K.R.; Stables, S.; Koelmeyer, T.; et al. Prospective, Population-Based Long QT Molecular Autopsy Study of Postmortem Negative Sudden Death in 1 to 40 Year Olds. Heart Rhythm 2011, 8, 412–419. [Google Scholar] [CrossRef]
- Winkel, B.G.; Holst, A.G.; Theilade, J.; Kristensen, I.B.; Thomsen, J.L.; Ottesen, G.L.; Bundgaard, H.; Svendsen, J.H.; Haunsø, S.; Tfelt-Hansen, J. Nationwide Study of Sudden Cardiac Death in Persons Aged 1-35 Years. Eur. Heart J. 2011, 32, 983–990. [Google Scholar] [CrossRef]
- Tester, D.J.; Medeiros-Domingo, A.; Will, M.L.; Haglund, C.M.; Ackerman, M.J. Cardiac Channel Molecular Autopsy: Insights from 173 Consecutive Cases of Autopsy-Negative Sudden Unexplained Death Referred for Postmortem Genetic Testing. Mayo Clin. Proc. 2012, 87, 524–539. [Google Scholar] [CrossRef] [PubMed]
- Priest, J.R. A Primer to Clinical Genome Sequencing. Curr. Opin. Pediatr. 2017, 29, 513–519. [Google Scholar] [CrossRef]
- Adams, D.R.; Eng, C.M. Next-Generation Sequencing to Diagnose Suspected Genetic Disorders. N. Engl. J. Med. 2018, 379, 1353–1362. [Google Scholar] [CrossRef] [PubMed]
- Beck, T.F.; Mullikin, J.C.; NISC Comparative Sequencing Program; Biesecker, L.G. Systematic Evaluation of Sanger Validation of Next-Generation Sequencing Variants. Clin. Chem. 2016, 62, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Bowdin, S.; Gilbert, A.; Bedoukian, E.; Carew, C.; Adam, M.P.; Belmont, J.; Bernhardt, B.; Biesecker, L.; Bjornsson, H.T.; Blitzer, M.; et al. Recommendations for the Integration of Genomics into Clinical Practice. Genet. Med. Off. J. Am. Coll. Med. Genet. 2016, 18, 1075–1084. [Google Scholar] [CrossRef] [PubMed]
- Lahrouchi, N.; Raju, H.; Lodder, E.M.; Papatheodorou, E.; Ware, J.S.; Papadakis, M.; Tadros, R.; Cole, D.; Skinner, J.R.; Crawford, J.; et al. Utility of Post-Mortem Genetic Testing in Cases of Sudden Arrhythmic Death Syndrome. J. Am. Coll. Cardiol. 2017, 69, 2134–2145. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A Revolutionary Tool for Transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Miles, C.J.; Behr, E.R. The Role of Genetic Testing in Unexplained Sudden Death. Transl. Res. J. Lab. Clin. Med. 2016, 168, 59–73. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Biesecker, L.G.; Harrison, S.M.; the ClinGen Sequence Variant Interpretation Working Group. The ACMG/AMP Reputable Source Criteria for the Interpretation of Sequence Variants. Genet. Med. Off. J. Am. Coll. Med. Genet. 2018, 20, 1687–1688. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, O.; Sarquella-Brugada, G.; Fernandez-Falgueras, A.; Coll, M.; Iglesias, A.; Ferrer-Costa, C.; Cesar, S.; Arbelo, E.; García-Álvarez, A.; Jordà, P.; et al. Reanalysis and Reclassification of Rare Genetic Variants Associated with Inherited Arrhythmogenic Syndromes. EBioMedicine 2020, 54, 102732. [Google Scholar] [CrossRef]
- Middleton, O.; Baxter, S.; Demo, E.; Honeywell, C.; Jentzen, J.; Miller, F.; Pinckard, J.K.; Reichard, R.R.; Rutberg, J.; Stacy, C.; et al. National Association of Medical Examiners Position Paper: Retaining Postmortem Samples for Genetic Testing. Acad. Forensic Pathol. 2013, 3, 191–194. [Google Scholar] [CrossRef]
- Stiles, M.K.; Wilde, A.A.M.; Abrams, D.J.; Ackerman, M.J.; Albert, C.M.; Behr, E.R.; Chugh, S.S.; Cornel, M.C.; Gardner, K.; Ingles, J.; et al. 2020 APHRS/HRS Expert Consensus Statement on the Investigation of Decedents with Sudden Unexplained Death and Patients with Sudden Cardiac Arrest, and of Their Families. Heart Rhythm 2021, 18, e1–e50. [Google Scholar] [CrossRef]
- Srinivasan, M.; Sedmak, D.; Jewell, S. Effect of Fixatives and Tissue Processing on the Content and Integrity of Nucleic Acids. Am. J. Pathol. 2002, 161, 1961–1971. [Google Scholar] [CrossRef]
- Baudhuin, L.M.; Leduc, C.; Train, L.J.; Avula, R.; Kluge, M.L.; Kotzer, K.E.; Lin, P.T.; Ackerman, M.J.; Maleszewski, J.J. Technical Advances for the Clinical Genomic Evaluation of Sudden Cardiac Death. Circ. Cardiovasc. Genet. 2017, 10, e001844. [Google Scholar] [CrossRef]
- Bagnall, R.D.; Ingles, J.; Yeates, L.; Berkovic, S.F.; Semsarian, C. Exome Sequencing–Based Molecular Autopsy of Formalin-Fixed Paraffin-Embedded Tissue after Sudden Death. Genet. Med. 2017, 19, 1127–1133. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Gryazeva, T.; Wang, D.; Zhou, B.; Um, S.Y.; Eng, L.S.; Ruiter, K.; Rojas, L.; Williams, N.; Sampson, B.A.; et al. Using Postmortem Formalin Fixed Paraffin-Embedded Tissues for Molecular Testing of Sudden Cardiac Death: A Cautionary Tale of Utility and Limitations. Forensic Sci. Int. 2020, 308, 110177. [Google Scholar] [CrossRef] [PubMed]
- Bagnall, R.D.; K, J.D.; Duflou, J.; Semsarian, C. Exome Analysis–Based Molecular Autopsy in Cases of Sudden Unexplained Death in the Young. Heart Rhythm 2014, 11, 655–662. [Google Scholar] [CrossRef]
- Hata, Y.; Kinoshita, K.; Mizumaki, K.; Yamaguchi, Y.; Hirono, K.; Ichida, F.; Takasaki, A.; Mori, H.; Nishida, N. Postmortem Genetic Analysis of Sudden Unexplained Death Syndrome under 50 Years of Age: A next-Generation Sequencing Study. Heart Rhythm 2016, 13, 1544–1551. [Google Scholar] [CrossRef]
- Modena, M.; Castiglione, V.; Aretini, P.; Mazzanti, C.M.; Chiti, E.; Giannoni, A.; Emdin, M.; Di Paolo, M. Unveiling a Sudden Unexplained Death Case by Whole Exome Sequencing and Bioinformatic Analysis. Mol. Genet. Genomic Med. 2020, 8, e1182. [Google Scholar] [CrossRef]
- Nunn, L.M.; Lopes, L.R.; Syrris, P.; Murphy, C.; Plagnol, V.; Firman, E.; Dalageorgou, C.; Zorio, E.; Domingo, D.; Murday, V.; et al. Diagnostic Yield of Molecular Autopsy in Patients with Sudden Arrhythmic Death Syndrome Using Targeted Exome Sequencing. EP Eur. 2016, 18, 888–896. [Google Scholar] [CrossRef] [PubMed]
- Hertz, C.L.; Christiansen, S.L.; Ferrero-Miliani, L.; Dahl, M.; Weeke, P.E.; Ottesen, G.L.; Frank-Hansen, R.; Bundgaard, H.; Morling, N. Next-Generation Sequencing of 100 Candidate Genes in Young Victims of Suspected Sudden Cardiac Death with Structural Abnormalities of the Heart. Int. J. Leg. Med. 2016, 130, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Ripoll-Vera, T.; Pérez Luengo, C.; Borondo Alcázar, J.C.; García Ruiz, A.B.; Sánchez Del Valle, N.; Barceló Martín, B.; Poncela García, J.L.; Gutiérrez Buitrago, G.; Dasi Martínez, C.; Canós Villena, J.C.; et al. Sudden Cardiac Death in Persons Aged 50 Years or Younger: Diagnostic Yield of a Regional Molecular Autopsy Program Using Massive Sequencing. Rev. Esp. Cardiol. Engl. Ed. 2021, 74, 402–413. [Google Scholar] [CrossRef]
- Dewar, L.J.; Alcaide, M.; Fornika, D.; D’Amato, L.; Shafaatalab, S.; Stevens, C.M.; Balachandra, T.; Phillips, S.M.; Sanatani, S.; Morin, R.D.; et al. Investigating the Genetic Causes of Sudden Unexpected Death in Children Through Targeted Next-Generation Sequencing Analysis. Circ. Cardiovasc. Genet. 2017, 10, e001738. [Google Scholar] [CrossRef]
- Farrugia, A.; Keyser, C.; Hollard, C.; Raul, J.S.; Muller, J.; Ludes, B. Targeted next Generation Sequencing Application in Cardiac Channelopathies: Analysis of a Cohort of Autopsy-Negative Sudden Unexplained Deaths. Forensic Sci. Int. 2015, 254, 5–11. [Google Scholar] [CrossRef]
- Anderson, J.H.; Tester, D.J.; Will, M.L.; Ackerman, M.J. Whole-Exome Molecular Autopsy After Exertion-Related Sudden Unexplained Death in the Young. Circ. Cardiovasc. Genet. 2016, 9, 259–265. [Google Scholar] [CrossRef]
- Rueda, M.; Wagner, J.L.; Phillips, T.C.; Topol, S.E.; Muse, E.D.; Lucas, J.R.; Wagner, G.N.; Topol, E.J.; Torkamani, A. Molecular Autopsy for Sudden Death in the Young: Is Data Aggregation the Key? Front. Cardiovasc. Med. 2017, 4, 72. [Google Scholar] [CrossRef]
- Buscemi, L.; Alessandrini, F.; Perna, G.; Tagliabracci, A. Next-Generation Sequencing of 68 Genes in Sudden Unexplained Death of Young Individuals in Forensics. Forensic Sci. Int. Genet. Suppl. Ser. 2015, 5, e138–e140. [Google Scholar] [CrossRef][Green Version]
- Brion, M.; Blanco-Verea, A.; Sobrino, B.; Santori, M.; Gil, R.; Ramos-Luis, E.; Martinez, M.; Amigo, J.; Carracedo, A. Next Generation Sequencing Challenges in the Analysis of Cardiac Sudden Death Due to Arrhythmogenic Disorders. Electrophoresis 2014, 35, 3111–3116. [Google Scholar] [CrossRef]
- Narula, N.; Tester, D.J.; Paulmichl, A.; Maleszewski, J.J.; Ackerman, M.J. Post-Mortem Whole Exome Sequencing with Gene-Specific Analysis for Autopsy Negative Sudden Unexplained Death in the Young: A Case Series. Pediatr. Cardiol. 2015, 36, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Santori, M.; Blanco-Verea, A.; Gil, R.; Cortis, J.; Becker, K.; Schneider, P.M.; Carracedo, A.; Brion, M. Broad-Based Molecular Autopsy: A Potential Tool to Investigate the Involvement of Subtle Cardiac Conditions in Sudden Unexpected Death in Infancy and Early Childhood. Arch. Dis. Child. 2015, 100, 952–956. [Google Scholar] [CrossRef]
- Christiansen, S.L.; Hertz, C.L.; Ferrero-Miliani, L.; Dahl, M.; Weeke, P.E.; LuCamp; Ottesen, G.L.; Frank-Hansen, R.; Bundgaard, H.; Morling, N. Genetic Investigation of 100 Heart Genes in Sudden Unexplained Death Victims in a Forensic Setting. Eur. J. Hum. Genet. 2016, 24, 1797–1802. [Google Scholar] [CrossRef]
- Hellenthal, N.; Gaertner-Rommel, A.; Klauke, B.; Paluszkiewicz, L.; Stuhr, M.; Kerner, T.; Farr, M.; Püschel, K.; Milting, H. Molecular Autopsy of Sudden Unexplained Deaths Reveals Genetic Predispositions for Cardiac Diseases among Young Forensic Cases. EP Eur. 2017, 19, 1881–1890. [Google Scholar] [CrossRef]
- Neubauer, J.; Lecca, M.R.; Russo, G.; Bartsch, C.; Medeiros-Domingo, A.; Berger, W.; Haas, C. Exome Analysis in 34 Sudden Unexplained Death (SUD) Victims Mainly Identified Variants in Channelopathy-Associated Genes. Int. J. Leg. Med. 2018, 132, 1057–1065. [Google Scholar] [CrossRef]
- Sanchez, O.; Campuzano, O.; Fernández-Falgueras, A.; Sarquella-Brugada, G.; Cesar, S.; Mademont, I.; Mates, J.; Pérez-Serra, A.; Coll, M.; Pico, F.; et al. Natural and Undetermined Sudden Death: Value of Post-Mortem Genetic Investigation. PLoS ONE 2016, 11, e0167358. [Google Scholar] [CrossRef]
- Campuzano, O.; Beltramo, P.; Fernandez, A.; Iglesias, A.; García, L.; Allegue, C.; Sarquella-Brugada, G.; Coll, M.; Perez-Serra, A.; Mademont-Soler, I.; et al. Molecular Autopsy in a Cohort of Infants Died Suddenly at Rest. Forensic Sci. Int. Genet. 2018, 37, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Shanks, G.W.; Tester, D.J.; Ackerman, J.P.; Simpson, M.A.; Behr, E.R.; White, S.M.; Ackerman, M.J. Importance of Variant Interpretation in Whole-Exome Molecular Autopsy. Circulation 2018, 137, 2705–2715. [Google Scholar] [CrossRef] [PubMed]
- Mak, C.M.; Mok, N.S.; Shum, H.C.; Siu, W.K.; Chong, Y.K.; Lee, H.H.C.; Fong, N.C.; Tong, S.F.; Lee, K.W.; Ching, C.K.; et al. Sudden Arrhythmia Death Syndrome in Young Victims: A Five-Year Retrospective Review and Two-Year Prospective Molecular Autopsy Study by next-Generation Sequencing and Clinical Evaluation of Their First-Degree Relatives. Hong Kong Med. J. 2019, 25, 21–29. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Features | Sanger Sequencing | Next-Generation Sequencing |
|---|---|---|
| Approach | Irreversible chain termination method using dideoxynucleotides (ddNTPs) | Different NGS platform uses different principles (e.g., Illumina—reversible chain termination) |
| Throughput | Low | High |
| Sequencing samples | Clones, PCR | DNA libraries |
| Preparation steps | Few; simple procedure | Many; complex procedure |
| Speed | Slow | Fast (WGS in 2 days) |
| Read length | 600–800 bp | 35–20,000 bp (depends on the platform) |
| Data | 1 read | Millions of reads |
| Data accuracy | 99% | 99.999% |
| Run cost | Cost-effective for low number of targets | Cost-effective for high number of targets |
| Data interpretation | Electrophoresis gel | Bioinformatic analysis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castiglione, V.; Modena, M.; Aimo, A.; Chiti, E.; Botto, N.; Vittorini, S.; Guidi, B.; Vergaro, G.; Barison, A.; Rossi, A.; et al. Molecular Autopsy of Sudden Cardiac Death in the Genomics Era. Diagnostics 2021, 11, 1378. https://doi.org/10.3390/diagnostics11081378
Castiglione V, Modena M, Aimo A, Chiti E, Botto N, Vittorini S, Guidi B, Vergaro G, Barison A, Rossi A, et al. Molecular Autopsy of Sudden Cardiac Death in the Genomics Era. Diagnostics. 2021; 11(8):1378. https://doi.org/10.3390/diagnostics11081378
Chicago/Turabian StyleCastiglione, Vincenzo, Martina Modena, Alberto Aimo, Enrica Chiti, Nicoletta Botto, Simona Vittorini, Benedetta Guidi, Giuseppe Vergaro, Andrea Barison, Andrea Rossi, and et al. 2021. "Molecular Autopsy of Sudden Cardiac Death in the Genomics Era" Diagnostics 11, no. 8: 1378. https://doi.org/10.3390/diagnostics11081378
APA StyleCastiglione, V., Modena, M., Aimo, A., Chiti, E., Botto, N., Vittorini, S., Guidi, B., Vergaro, G., Barison, A., Rossi, A., Passino, C., Giannoni, A., Di Paolo, M., & Emdin, M. (2021). Molecular Autopsy of Sudden Cardiac Death in the Genomics Era. Diagnostics, 11(8), 1378. https://doi.org/10.3390/diagnostics11081378