
Small RNA-Sequencing: Approaches and Considerations for miRNA Analysis


Abstract
1. Introduction
2. Small RNA-seq Technology Overview
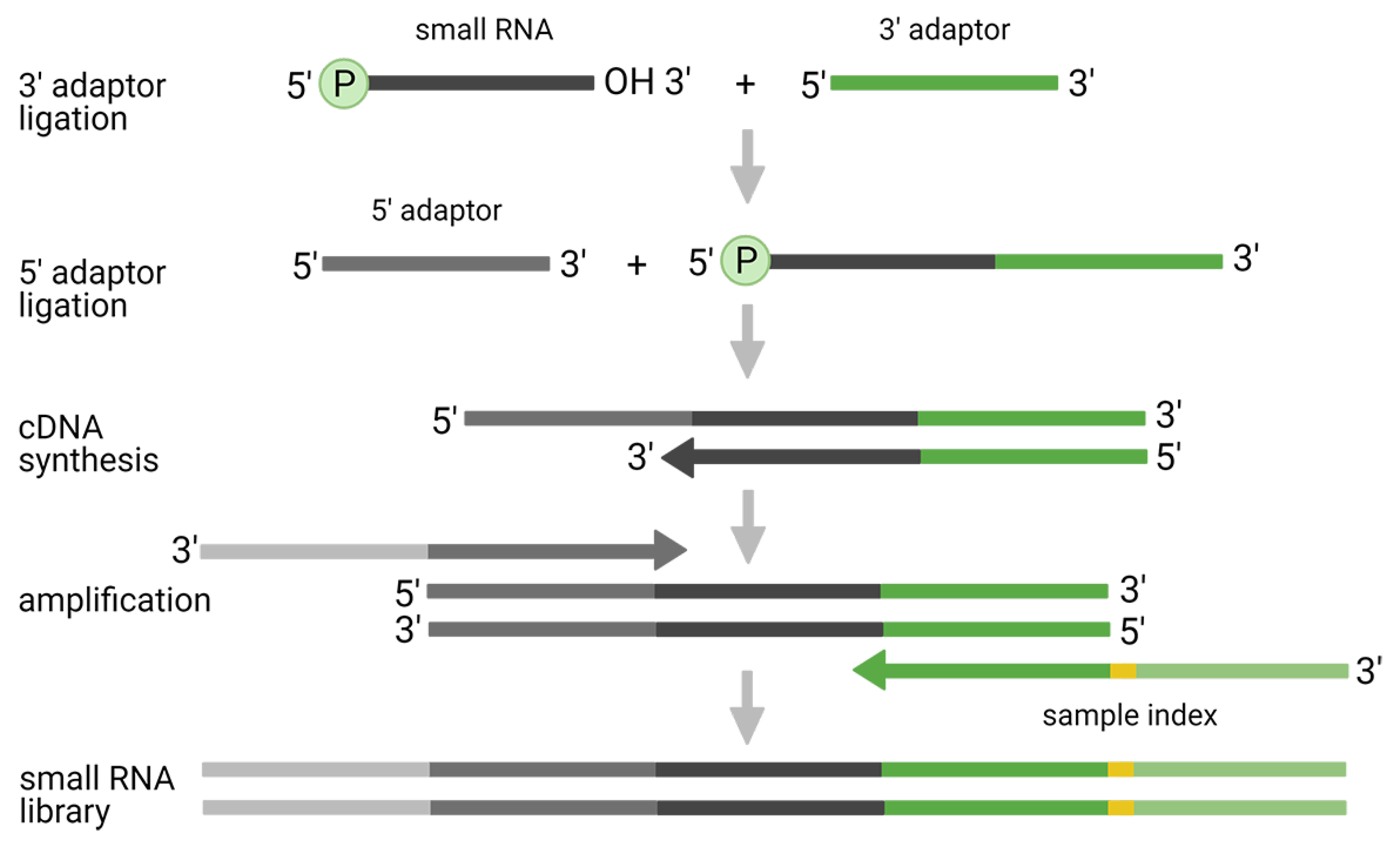
2.1. Two-Adaptor Ligation-Based Methods
2.2. Improved Two-Adaptor Ligation-Based Methods
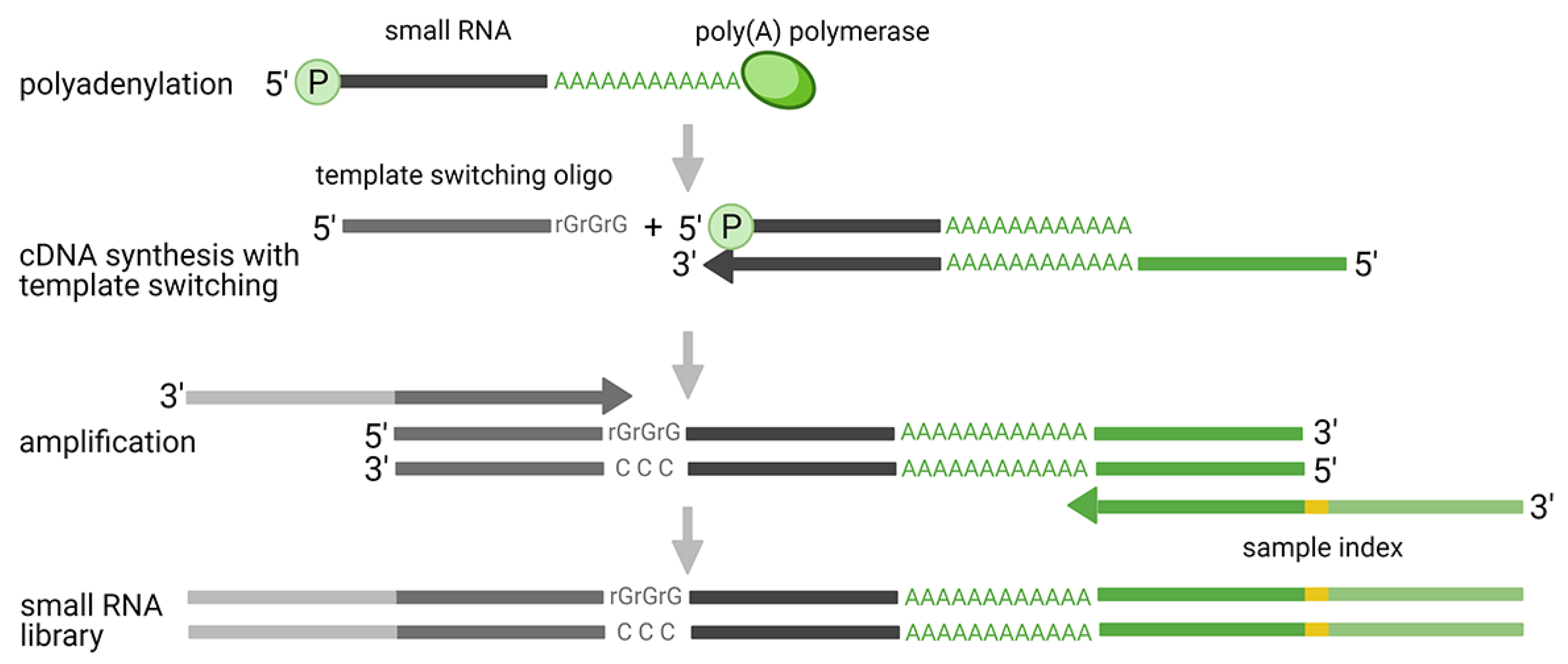
2.3. Ligation Free Methods
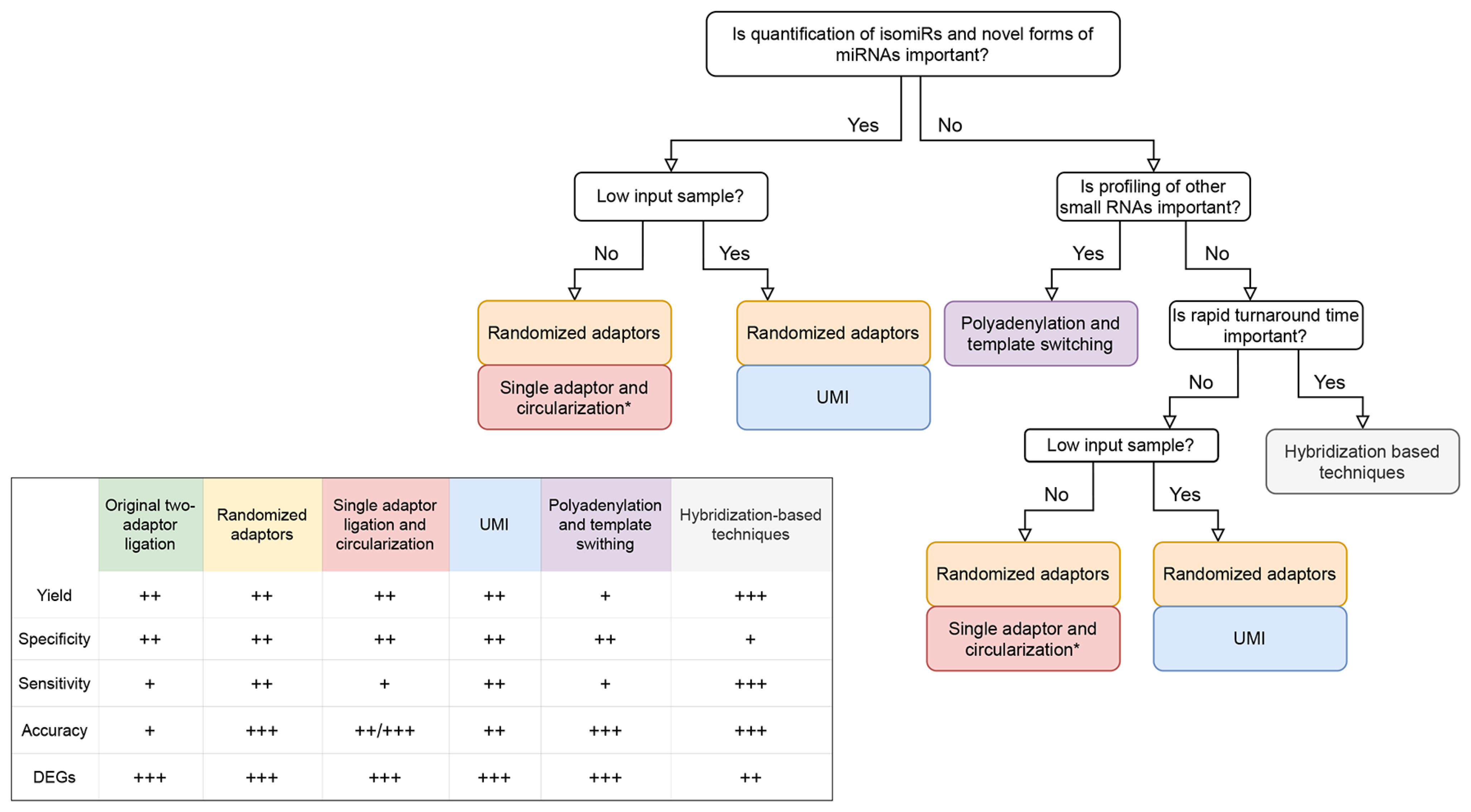
3. Benchmarking Studies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Benchmarking Study | Included Approaches | Commercial Protocols Included | Sample Types | Conclusion |
|---|---|---|---|---|
| Dard-Dascot, C. et al. BMC Genomics 19, 1–16 (2018) [58]. | Original two-adaptor ligation | TruSeq Small RNA Library Prep Kit (Illumina) + in house modifications | Synthetic RNAs | Protocol utilizing randomized adaptors performed best |
| Randomized adaptors | NEXTflex (Bioo Scientific) + in house modifications | Arabidopsis thaliana total RNA | on both human and plant miRNAs. Addition of polyethylene glycol (PEG) | |
| Polyadenylation and template switching | SMARTer (Clontech) and CATS (Diagenode) | Oilseed rape total RNA | and usage of chimeric DNA-RNA 5′ adaptor with random nucleotides | |
| HeLa cells total RNA | led to improved performance. | |||
| Coenen-Stass, A. M. L. et al. RNA Biol. 15, 1133–1145 (2018) [78]. | Original two-adaptor ligation | NEBNext Multiplex Small RNA Library Prep Set (New England Biolabs) | Human plasma miRNAs | Protocols utilizing randomized adaptors and UMIs performed well across |
| Randomized adaptors | NEXTflex (Bioo Scientific) | Human serum miRNAs | all measured characteristics and showed the least sequence bias. | |
| Polyadenylation and template switching | SMARTer (Clontech) | miRXplore universal reference | ||
| UMI | QIAseq miRNA library Kit (Qiagen) | |||
| Giraldez, M. D. et al. Nat. Biotechnol. 36, 746–757 (2018) [59]. | Original two-adaptor ligation | NEBNext Multiplex Small RNA Library Prep Set (New England Biolabs) | Human plasma total RNA | In-house adjusted protocol with randomized adaptors showed the least bias. |
| Randomized adaptors | NEXTflex (Bioo Scientific) + in house modifications | Equimolar and ratiometric pool of synthetic RNAs | Optimization of ligation temperature and PEG helped to reduce bias. | |
| Yeri, A. et al. BMC Genomics 19, 1–15 (2018) [76]. | Original two-adaptor ligation | TruSeq Small RNA Library Prep Kit (Illumina) | Human brain total RNA | Protocol utilizing randomized adaptors detected the highest number of miRNAs |
| NEBNext Multiplex Small RNA Library Prep Set (New England Biolabs) | Human liver total RNA | and had the highest correlation with results from ligation free methods, but | ||
| Randomized adaptors | NEXTflex (Bioo Scientific) + in house modifications | Human placenta total RNA | it required user experience to be performed consistently. | |
| Sequencing of hybridization probes | EdgeSeq (HTG Molecular Diagnostics) | Human plasma total RNA | ||
| Hybridization-based technique | FirePlex (Abcam) | |||
| Barberán-Soler, S. et al. Genome Biol. 19, 105 (2018) [25]. | Original two-adaptor ligation | NEBNext Multiplex Small RNA Library Prep Set (New England Biolabs) | Human brain total RNA | Single adaptor ligation and circularization protocol detected the |
| UMI | QIAseq miRNA library Kit (Qiagen) | miRXplore universal reference | the largest spectrum of miRNAs and showed the least bias. | |
| Randomized adaptors | NEXTflex (Bioo Scientific) | |||
| Polyadenylation and template switching | SMARTer smRNA-seq Kit (Takara Bio) | |||
| Single adaptor ligation and circularization | Beta version of RealSeq-AC Kit (Somagenics) | |||
| Godoy, P. M. et al. Cell Rep. 29, 4212–4222.e5 (2019) [75]. | Randomized adaptors | TruSeq Small RNA Library Prep Kit (Illumina) + in house modifications | Equimolar and ratio metric pool of synthetic RNAs | Small RNA-seq showed better specificity, ability to detect expected differential |
| Sequencing of hybridization probes | EdgeSeq (HTG Molecular Diagnostics) | Human plasma total RNA | expression, but higher level of bias. | |
| Hybridization based techniques | FirePlex (Abcam) + nCounter (NanoString) | |||
| Wong, R. K. Y. et al. BMC Genomics 20, 1–12 (2019) [36]. | Original two-adaptor ligation | CleanTag Small RNA Library Prep Kit (TriLink BioTechnologies) | Human plasma total RNA | Protocol utilizing UMIs detected the highest number of miRNAs and correlated |
| Randomized adaptors | NEXTflex (Bioo Scientific) | most closely to RT-qPCR validation data. | ||
| UMI | QIAseq miRNA library Kit (Qiagen) | |||
| Wright, C. et al. BMC Genomics 20, 513 (2019) [60]. | Original two-adaptor ligation | TruSeq Small RNA Library Prep Kit (Illumina) | Human brain total RNA | Protocol utilizing randomized adaptors performed the best. Authors suggested |
| Randomized adaptors | NEXTflex (Bioo Scientific) | miRXplore universal reference | usage of random nucleotides as UMIs to reduce PCR bias. | |
| Polyadenylation and template switching | SMARTer (Clontech) | |||
| Heinicke, F. et al. RNA Biol. 17, 75–86 (2020) [66]. | Original two-adaptor ligation | Small RNA-Seq Library Prep Kit (Lexogen) + TailorMix miRNA Sample Preparation Kit (SeqMatic) | Equimolar and ratio metric pool of synthetic RNAs | Best performance was shown by protocol utilizing UMIs. Polyadenylation |
| CleanTag Small RNA Library Prep Kit (TriLink BioTechnologies) | Human CD8+ T cells total RNA | and circularization protocols showed a poor yield of miRNAs reads | ||
| Single adaptor ligation and circularization | SMARTer microRNA-seq Kit (Takara Bio) | and were not considered for further analysis. | ||
| Polyadenylation and template switching | CATS (Diagenode) | |||
| UMI | QIAseq miRNA library Kit (Qiagen) | |||
| Herbert, Z. T. et al. J. Biomol. Tech. 31, 47–56 (2020) [65]. | All available approaches to small RNA-seq | Small RNA-Seq Library Prep Kit (Lexogen) + TruSeq Small RNA Library Prep Kit (Illumina) | miRXplore universal reference | All methods showed high reproducibility and there was no protocol |
| NEBNext Multiplex Small RNA Library Prep Set (New England Biolabs) | Mesenchymal stem cells total RNA | outperforming others across all the metrics. | ||
| CleanTag Small RNA Library Prep Kit (TriLink BioTechnologies) | ||||
| NEXTflex (PerkinElmer) | ||||
| SMARTer smRNA-seq Kit + CATS (Diagenode) | ||||
| RealSeq-AC Kit (Somagenics) | ||||
| QIAseq miRNA library Kit (Qiagen) | ||||
| Hybridization-based method | nCounter (NanoString) | |||
| Baldrich, P. et al. bioRxiv (2020) [79] | Original two-adaptor ligation | NEBNext Multiplex Small RNA Library Prep Set (New England Biolabs) | Maize anthers total RNA | Protocols using randomized adaptors and two-adaptor ligation generated |
| TruSeq Small RNA Library Prep Kit (Illumina) | highest number of reads mapping to miRNAs and phasiRNAs. | |||
| CleanTag Small RNA Library Prep Kit (TriLink BioTechnologies) | ||||
| Randomized adaptors | NEXTflex v2, NEXTflex v3 (Bioo Scientific) | |||
| Single adaptor ligation and circularization | RealSeq-AC Kit (Somagenics) | |||
| Polyadenylation and template switching | SMARTer smRNA-seq Kit (Takara Bio) | |||
| Androvic, P. et al. bioRxiv (2021) [61]. | All available approaches to small RNA-seq | Small RNA-Seq Library Prep Kit (Lexogen) + Small RNA Library Prep Kit (Norgen) | Human plasma total RNA | Protocols utilizing randomized adaptors and UMIs had the best overall |
| QIAseq miRNA library Kit (Qiagen) | miRXplore universal reference | performance. Hybridization-based methods showed the highest sensitivity and | ||
| NEXTflex (Bioo Scientific) | low specificity. | |||
| SMARTer smRNA-seq Kit (Takara Bio) | ||||
| RealSeq-Biofluids Kit (Somagenics) | ||||
| Hybridization-based method | EdgeSeq (HTG Molecular Diagnostics) |
4. Considerations and Future Prospects
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Vasudevan, S.; Tong, Y.; Steitz, J.A. Switching from Repression to Activation: microRNAs Can Up-Regulate Translation. Science 2007, 318, 1931–1934. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, S. Posttranscriptional Upregulation by MicroRNAs. Wiley Interdiscip. Rev. RNA 2012, 3, 311–330. [Google Scholar] [CrossRef]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of microRNA biogenesis, mechanisms of actions, and circulation. Front. Endocrinol. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Catalanotto, C.; Cogoni, C.; Zardo, G. MicroRNA in control of gene expression: An overview of nuclear functions. Int. J. Mol. Sci. 2016, 17, 1721. [Google Scholar] [CrossRef] [PubMed]
- Kim, V.N.; Nam, J.W. Genomics of microRNA. Trends Genet. 2006, 22, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Gebert, L.F.; MacRae, I.J. Regulation of microRNA function in animals. Nat. Rev. Mol. Cell Biol. 2019, 20, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Neilsen, C.T.; Goodall, G.J.; Bracken, C.P. IsomiRs—The overlooked repertoire in the dynamic microRNAome. Trends Genet. 2012, 28, 544–549. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. MiRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Friedman, R.C.; Farh, K.K.H.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef]
- Ruan, K.; Fang, X.; Ouyang, G. MicroRNAs: Novel regulators in the hallmarks of human cancer. Cancer Lett. 2009, 285, 116–126. [Google Scholar] [CrossRef]
- Anfossi, S.; Babayan, A.; Pantel, K.; Calin, G.A. Clinical utility of circulating non-coding RNAs—An update. Nat. Rev. Clin. Oncol. 2018, 15, 541–563. [Google Scholar] [CrossRef] [PubMed]
- Cortez, M.A.; Bueso-Ramos, C.; Ferdin, J.; Lopez-Berestein, G.; Sood, A.K.; Calin, G.A. MicroRNAs in body fluids-the mix of hormones and biomarkers. Nat. Rev. Clin. Oncol. 2011, 8, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, C.C.; Cheng, H.H.; Tewari, M. MicroRNA profiling: Approaches and considerations. Nat. Rev. Genet. 2012, 13, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Koppers-Lalic, D.; Hackenberg, M.; Menezes, R.D. Non invasive prostate cancer detection by measuring miRNA variants (isomiRs) in urine extracellular vesicles. Oncotarget 2016, 7, 22566. [Google Scholar] [CrossRef]
- Telonis, A.G.; Loher, P.; Jing, Y.; Londin, E.; Rigoutsos, I. Beyond the one-locus-one-miRNA paradigm: MicroRNA isoforms enable deeper insights into breast cancer heterogeneity. Nucleic Acids Res. 2015, 43, 9158–9175. [Google Scholar] [CrossRef]
- Telonis, A.G.; Magee, R.; Loher, P.; Chervoneva, I.; Londin, E.; Rigoutsos, I. Knowledge about the presence or absence of miRNA isoforms (isomiRs) can successfully discriminate amongst 32 TCGA cancer types. Nucleic Acids Res. 2017, 45, 2973–2985. [Google Scholar] [CrossRef]
- Ono, S.; Lam, S.; Nagahara, M.; Hoon, D. Circulating microRNA Biomarkers as Liquid Biopsy for Cancer Patients: Pros and Cons of Current Assays. J. Clin. Med. 2015, 4, 1890–1907. [Google Scholar] [CrossRef]
- Valihrach, L.; Androvic, P.; Kubista, M. Circulating miRNA analysis for cancer diagnostics and therapy. Mol. Asp. Med. 2020, 72, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Shore, S.; Henderson, J.M.; Lebedev, A.; Salcedo, M.P.; Zon, G.; McCaffrey, A.P.; Paul, N.; Hogrefe, R.I. Small RNA library preparation method for next-generation sequencing using chemical modifications to prevent adapter dimer formation. PLoS ONE 2016, 11, e0167009. [Google Scholar] [CrossRef] [PubMed]
- Pease, J. Small-RNA sequencing libraries with greatly reduced adaptor-dimer background. Nat. Methods 2011, 8, iii–iv. [Google Scholar] [CrossRef]
- Baran-Gale, J.; Lisa Kurtz, C.; Erdos, M.R.; Sison, C.; Young, A.; Fannin, E.E.; Chines, P.S.; Sethupathy, P. Addressing bias in small RNA library preparation for sequencing: A new protocol recovers microRNAs that evade capture by current methods. Front. Genet. 2015, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Barberán-Soler, S.; Vo, J.M.; Hogans, R.E.; Dallas, A.; Johnston, B.H.; Kazakov, S.A. Decreasing miRNA sequencing bias using a single adapter and circularization approach. Genome Biol. 2018, 19, 105. [Google Scholar] [CrossRef]
- Dennis, D.; Rhodes, M.; Maclean, K. Targeted miRNA Discovery and Validation—Using the nCounter® Platform; NanoString Technologies—WHITE PAPER—nCounter PanCancer Immune Profiling Panel; NanoString Technologies, Inc.: Seattle, WA, USA, 2015; pp. 1–8. [Google Scholar]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics in Western Equatoria State. Nat. Rev. Genet. 2009, 10, 57. [Google Scholar] [CrossRef]
- Lu, C.; Tej, S.S.; Luo, S.; Haudenschild, C.D.; Meyers, B.C.; Green, P.J. Genetics: Elucidation of the small RNA component of the transcriptome. Science 2005, 309, 1567–1569. [Google Scholar] [CrossRef]
- Ruby, J.G.; Jan, C.; Player, C.; Axtell, M.J.; Lee, W.; Nusbaum, C.; Ge, H.; Bartel, D.P. Large-Scale Sequencing Reveals 21U-RNAs and Additional MicroRNAs and Endogenous siRNAs in C. elegans. Cell 2006, 127, 1193–1207. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]
- Chakraborty, C.; Bhattacharya, M.; Agoramoorthy, G. Single-cell sequencing of miRNAs: A modified technology. Cell Biol. Int. 2020, 44, 1773–1780. [Google Scholar] [CrossRef]
- Hafner, M.; Landgraf, P.; Ludwig, J.; Rice, A.; Ojo, T.; Lin, C.; Holoch, D.; Lim, C.; Tuschl, T. Identification of microRNAs and other small regulatory RNAs using cDNA library sequencing. Methods 2008, 44, 3–12. [Google Scholar] [CrossRef]
- Redshaw, N.; Wilkes, T.; Whale, A.; Cowen, S.; Huggett, J.; Foy, C.A. A comparison of miRNA isolation and RT-qPCR technologies and their effects on quantification accuracy and repeatability. BioTechniques 2013, 54, 155–164. [Google Scholar] [CrossRef]
- Guo, Y.; Vickers, K.; Xiong, Y.; Zhao, S.; Sheng, Q.; Zhang, P.; Zhou, W.; Flynn, C.R. Comprehensive evaluation of extracellular small RNA isolation methods from serum in high throughput sequencing. BMC Genom. 2017, 18, 1–9. [Google Scholar] [CrossRef]
- Srinivasan, S.; Yeri, A.; Cheah, P.S.; Chung, A.; Dehoff, P.; Filant, J.; Laurent, C.D.; Laurent, L.D.; Magee, R.; Moeller, C.; et al. Small RNA sequencing across diverse biofluids identifies optimal methods for exRNA isolation. Cell 2019, 177, 446–462. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.K.; MacMahon, M.; Woodside, J.V.; Simpson, D.A. A comparison of RNA extraction and sequencing protocols for detection of small RNAs in plasma. BMC Genom. 2019, 20, 1–12. [Google Scholar] [CrossRef]
- Raabe, C.A.; Tang, T.H.; Brosius, J.; Rozhdestvensky, T.S. Biases in small RNA deep sequencing data. Nucleic Acids Res. 2014, 42, 1414–1426. [Google Scholar] [CrossRef]
- Fuchs, R.T.; Sun, Z.; Zhuang, F.; Robb, G.B. Bias in ligation-based small RNA sequencing library construction is determined by adaptor and RNA structure. PLoS ONE 2015, 10, e0126049. [Google Scholar] [CrossRef] [PubMed]
- Kalle, E.; Kubista, M.; Rensing, C. Multi-template polymerase chain reaction. Biomol. Detect. Quantif. 2014, 2, 11–29. [Google Scholar] [CrossRef] [PubMed]
- Kivioja, T.; Vähärautio, A.; Karlsson, K.; Bonke, M.; Enge, M.; Linnarsson, S.; Taipale, J. Counting absolute numbers of molecules using unique molecular identifiers. Nat. Methods 2012, 9, 72–74. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Wu, P.H.; Beane, T.; Zamore, P.D.; Weng, Z. Elimination of PCR duplicates in RNA-seq and small RNA-seq using unique molecular identifiers. BMC Genom. 2018, 19, 531. [Google Scholar] [CrossRef] [PubMed]
- Buschmann, D.; Haberberger, A.; Kirchner, B.; Spornraft, M.; Riedmaier, I.; Schelling, G.; Pfaffl, M.W. Toward reliable biomarker signatures in the age of liquid biopsies—How to standardize the small RNA-Seq workflow. Nucleic Acids Res. 2016, 44, 5995–6018. [Google Scholar] [CrossRef] [PubMed]
- Hafner, M.; Renwick, N.; Brown, M.; Mihailović, A.; Holoch, D.; Lin, C.; Pena, J.T.; Nusbaum, J.D.; Morozov, P.; Ludwig, J.; et al. RNA-ligase-dependent biases in miRNA representation in deep-sequenced small RNA cDNA libraries. RNA 2011, 17, 1697–1712. [Google Scholar] [CrossRef] [PubMed]
- Sorefan, K.; Pais, H.; Hall, A.E.; Kozomara, A.; Griffiths-Jones, S.; Moulton, V.; Dalmay, T. Reducing ligation bias of small RNAs in libraries for next generation sequencing. Silence 2012, 3, 1–11. [Google Scholar] [CrossRef]
- Zhuang, F.; Fuchs, R.T.; Robb, G.B. Small RNA expression profiling by high-throughput sequencing: Implications of enzymatic manipulation. J. Nucleic Acids 2012, 2012, 360358. [Google Scholar] [CrossRef]
- Baroin-Tourancheau, A.; Jaszczyszyn, Y.; Benigni, X.; Amar, L. Evaluating and Correcting Inherent Bias of microRNA Expression in Illumina Sequencing Analysis. Front. Mol. Biosci. 2019, 6, 17. [Google Scholar] [CrossRef]
- Xu, P.; Billmeier, M.; Mohorianu, I.; Green, D.; Fraser, W.D.; Dalmay, T. An improved protocol for small RNA library construction using High Definition adapters. Methods Next Gener. Seq. 2015, 2, 1–10. [Google Scholar] [CrossRef]
- Pak, J.; Fire, A. Distinct Populations of Primary and Secondary Effectors During RNAi in C. elegans. Science 2007, 315, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Kugelberg, U.; Nätt, D.; Skog, S.; Kutter, C.; Öst, A. 5′XP sRNA-seq: Efficient identification of transcripts with and without 5′ phosphorylation reveals evolutionary conserved small RNA. RNA Biol. 2021, 1–12. [Google Scholar] [CrossRef]
- Giraldez, M.D.; Spengler, R.M.; Etheridge, A.; Goicochea, A.J.; Tuck, M.; Choi, S.W.; Galas, D.J.; Tewari, M. Phospho-RNA-seq: A modified small RNA-seq method that reveals circulating mRNA and lncRNA fragments as potential biomarkers in human plasma. EMBO J. 2019, 38, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Munafó, D.B.; Robb, G.B. Optimization of enzymatic reaction conditions for generating representative pools of cDNA from small RNA. RNA 2010, 16, 2537–2552. [Google Scholar] [CrossRef]
- Head, S.R.; Kiyomi Komori, H.; LaMere, S.A.; Whisenant, T.; Van Nieuwerburgh, F.; Salomon, D.R.; Ordoukhanian, P. Library construction for next-generation sequencing: Overviews and challenges. BioTechniques 2014, 56, 61–77. [Google Scholar] [CrossRef] [PubMed]
- Vigneault, F.; Ter-Ovanesyan, D.; Alon, S.; Eminaga, S.; Christodoulou, D.C.; Seidman, J.G.; Eisenberg, E.; Church, G.M. High-throughput multiplex sequencing of miRNA. Curr. Protoc. Hum. Genet. 2012, 73, 1–10. [Google Scholar] [CrossRef]
- Kawano, M.; Kawazu, C.; Lizio, M.; Kawaji, H.; Carninci, P.; Suzuki, H.; Hayashizaki, Y. Reduction of non-insert sequence reads by dimer eliminator LNA oligonucleotide for small RNA deep sequencing. BioTechniques 2010, 49, 751–754. [Google Scholar] [CrossRef]
- Hardigan, A.A.; Roberts, B.S.; Moore, D.E.; Ramaker, R.C.; Jones, A.L.; Myers, R.M. CRISPR/Cas9-targeted removal of unwanted sequences from small-RNA sequencing libraries. Nucleic Acids Res. 2019, 47, e84. [Google Scholar] [CrossRef] [PubMed]
- Wickersheim, M.L.; Blumenstiel, J.P. Terminator oligo blocking efficiently eliminates rRNA from Drosophila small RNA sequencing libraries. Biotechniques 2013, 55, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Roberts, B.S.; Hardigan, A.A.; Kirby, M.K.; Fitz-Gerald, M.B.; Wilcox, C.M.; Kimberly, R.P.; Myers, R.M. Blocking of targeted microRNAs from next-generation sequencing libraries. Nucleic Acids Res. 2015, 43, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Dard-Dascot, C.; Naquin, D.; D’Aubenton-Carafa, Y.; Alix, K.; Thermes, C.; van Dijk, E. Systematic comparison of small RNA library preparation protocols for next-generation sequencing. BMC Genom. 2018, 19, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Giraldez, M.D.; Spengler, R.M.; Etheridge, A.; Godoy, P.M.; Barczak, A.J.; Srinivasan, S.; De Hoff, P.L.; Tanriverdi, K.; Courtright, A.; Lu, S.; et al. Comprehensive multi-center assessment of small RNA-seq methods for quantitative miRNA profiling. Nat. Biotechnol. 2018, 36, 746–757. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.; Rajpurohit, A.; Burke, E.E.; Williams, C.; Collado-Torres, L.; Kimos, M.; Brandon, N.J.; Cross, A.J.; Jaffe, A.E.; Weinberger, D.R.; et al. Comprehensive assessment of multiple biases in small RNA sequencing reveals significant differences in the performance of widely used methods. BMC Genom. 2019, 20, 513. [Google Scholar] [CrossRef]
- Androvic, P.; Benesova, S.; Rohlova, E.; Kubista, M.; Valihrach, L. Small RNA-sequencing for Analysis of Circulating miRNAs: Benchmark Study. bioRxiv 2021. [Google Scholar] [CrossRef]
- Zhuang, F.; Fuchs, R.T.; Sun, Z.; Zheng, Y.; Robb, G.B. Structural bias in T4 RNA ligase-mediated 3′-adapter ligation. Nucleic Acids Res. 2012, 40, e54. [Google Scholar] [CrossRef] [PubMed]
- Belair, C.D.; Hu, T.; Chu, B.; Freimer, J.W.; Cooperberg, M.R.; Blelloch, R.H. High-throughput, Efficient, and Unbiased Capture of Small RNAs from Low-input Samples for Sequencing. Sci. Rep. 2019, 9, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Saunders, K.; Bert, A.G.; Dredge, B.K.; Toubia, J.; Gregory, P.A.; Pillman, K.A.; Goodall, G.J.; Bracken, C.P. Insufficiently complex unique-molecular identifiers (UMIs) distort small RNA sequencing. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Herbert, Z.T.; Thimmapuram, J.; Xie, S.; Kershner, J.P.; Kolling, F.W.; Ringelberg, C.S.; Leclerc, A.; Alekseyev, Y.O.; Fan, J.; Podnar, J.W.; et al. Multisite evaluation of next-generation methods for small RNA quantification. J. Biomol. Tech. 2020, 31, 47–56. [Google Scholar] [CrossRef]
- Heinicke, F.; Zhong, X.; Zucknick, M.; Breidenbach, J.; Sundaram, A.Y.; Flåm, S.T.; Leithaug, M.; Dalland, M.; Farmer, A.; Henderson, J.M.; et al. Systematic assessment of commercially available low-input miRNA library preparation kits. RNA Biol. 2020, 17, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Jayaprakash, A.D.; Jabado, O.; Brown, B.D.; Sachidanandam, R. Identification and remediation of biases in the activity of RNA ligases in small-RNA deep sequencing. Nucleic Acids Res. 2011, 39, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lipps, C.; Northe, P.; Figueiredo, R.; Rohde, M.; Brahmer, A.; Krämer-Albers, E.M.; Liebetrau, C.; Wiedenroth, C.B.; Mayer, E.; Kriechbaum, S.D.; et al. Non-invasive approach for evaluation of pulmonary hypertension using extracellular vesicle-associated small non-coding RNA. Biomolecules 2019, 9, 666. [Google Scholar] [CrossRef]
- Berezikov, E.; Van Tetering, G.; Verheul, M.; Van De Belt, J.; Van Laake, L.; Vos, J.; Verloop, R.; Van De Wetering, M.; Guryev, V.; Takada, S.; et al. Many novel mammalian microRNA candidates identified by extensive cloning and RAKE analysis. Genome Res. 2006, 16, 1289–1298. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.Y.; Machleder, E.M.; Chenchik, A.; Li, R.; Siebert, P.D. Reverse transcriptase template switching: A SMART™ approach for full-length cDNA library construction. BioTechniques 2001, 30, 892–897. [Google Scholar] [CrossRef]
- Geiss, G.K.; Bumgarner, R.E.; Birditt, B.; Dahl, T.; Dowidar, N.; Dunaway, D.L.; Fell, H.P.; Ferree, S.; George, R.D.; Grogan, T.; et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat. Biotechnol. 2008, 26, 317–325. [Google Scholar] [CrossRef]
- Chapin, S.C.; Appleyard, D.C.; Pregibon, D.C.; Doyle, P.S. Rapid microRNA profiling on encoded gel microparticles. Angew. Chem. Int. Ed. 2011, 50, 2289–2293. [Google Scholar] [CrossRef]
- El-Khoury, V.; Pierson, S.; Kaoma, T.; Bernardin, F.; Berchem, G. Assessing cellular and circulating miRNA recovery: The impact of the RNA isolation method and the quantity of input material. Sci. Rep. 2016, 6, 1–14. [Google Scholar] [CrossRef]
- Girard, L.; Rodriguez-Canales, J.; Behrens, C.; Thompson, D.M.; Botros, I.W.; Tang, H.; Xie, Y.; Rekhtman, N.; Travis, W.D.; Wistuba, I.I.; et al. An Expression Signature as an Aid to the Histologic Classification of Non-Small Cell Lung Cancer. Clin. Cancer Res. 2016, 22, 4880–4889. [Google Scholar] [CrossRef]
- Godoy, P.M.; Barczak, A.J.; DeHoff, P.; Srinivasan, S.; Etheridge, A.; Galas, D.; Das, S.; Erle, D.J.; Laurent, L.C. Comparison of Reproducibility, Accuracy, Sensitivity, and Specificity of miRNA Quantification Platforms. Cell Rep. 2019, 29, 4212–4222.e5. [Google Scholar] [CrossRef] [PubMed]
- Yeri, A.; Courtright, A.; Danielson, K.; Hutchins, E.; Alsop, E.; Carlson, E.; Hsieh, M.; Ziegler, O.; Das, A.; Shah, R.V.; et al. Evaluation of commercially available small RNASeq library preparation kits using low input RNA. BMC Genom. 2018, 19, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Abdel-Mageed, A.B.; Adamidi, C.; Adelson, P.D.; Akat, K.M.; Alsop, E.; Ansel, K.M.; Arango, J.; Aronin, N.; Avsaroglu, S.K.; et al. The Extracellular RNA Communication Consortium: Establishing Foundational Knowledge and Technologies for Extracellular RNA Research. Cell 2019, 177, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Coenen-Stass, A.M.L.; Magen, I.; Brooks, T.; Ben-Dov, I.Z.; Greensmith, L.; Hornstein, E.; Fratta, P. Evaluation of methodologies for microRNA biomarker detection by next generation sequencing. RNA Biol. 2018, 15, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Baldrich, P.; Tamim, S.; Mathioni, S.; Meyers, B. Ligation bias is a major contributor to nonstoichiometric abundances of secondary siRNAs and impacts analyses of microRNAs. bioRxiv 2020. [Google Scholar] [CrossRef]
- Maguire, S.; Lohman, G.J.; Guan, S. A low-bias and sensitive small RNA library preparation method using randomized splint ligation. Nucleic Acids Res. 2020, 48, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, J.; Kim, K.; Chang, H.; You, K.V.; Kim, N. Bias-minimized quantification of microRNA reveals widespread alternative processing and 3 end modification. Nucleic Acids Res. 2019, 47, 2630–2640. [Google Scholar] [CrossRef]
- Raine, A.; Manlig, E.; Wahlberg, P.; Syvänen, A.C.; Nordlund, J. SPlinted Ligation Adapter Tagging (SPLAT), a novel library preparation method for whole genome bisulphite sequencing. Nucleic Acids Res. 2017, 45. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Dai, W.; Wu, L.; Wang, J. SALP, a new single-stranded DNA library preparation method especially useful for the high-throughput characterization of chromatin openness states. BMC Genom. 2018, 19, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Persson, H.; Søkilde, R.; Pirona, A.C.; Rovira, C. Preparation of highly multiplexed small RNA sequencing libraries. BioTechniques 2017, 63, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Heikkinen, L.; Wang, C.L.; Yang, Y.; Knott, K.E.; Wong, G. MiRToolsGallery: A tag-based and rankable microRNA bioinformatics resources database portal. Database 2018, 2018, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Heikkinen, L.; Wang, C.; Yang, Y.; Sun, H.; Wong, G. Trends in the development of miRNA bioinformatics tools. Brief. Bioinform. 2019, 20, 1836–1852. [Google Scholar] [CrossRef] [PubMed]




| Technology | Product Name | Company | Reference |
|---|---|---|---|
| Original two-adaptor ligation | Small RNA-Seq Library Prep Kit | Lexogen GmbH, Vienna, Austria, | not available |
| Small RNA Library Prep Kit | Norgen Biotek Corp., Thorold, ON, Canada | not available | |
| TruSeq Small RNA Library Prep Kit | Illumina, San Diego, CA, USA | not available | |
| TailorMix miRNA Sample Preparation Kit | SeqMatic, Fremont, CA, USA | not available | |
| NEBNext Multiplex Small RNA Library Prep Set | New England Biolabs, Ipswich, MA, USA | not available | |
| CleanTag Small RNA Library Prep Kit | TriLink BioTechnologies, Inc., San Diego, CA, USA | [22] | |
| ScriptMiner Library preparation Technology for Small RNA | Cambio Ltd., Cambridge, UK | [23] | |
| Randomized adaptors | NEXTflex Small RNA Sequencing Kit | PerkinElmer, Waltham, MA, USA | [24] |
| Single adaptor ligation and circularization | RealSeq-AC Kit | Somagenics, Santa Cruz, CA, USA | [25] |
| RealSeq-biofluids Kit | Somagenics, Santa Cruz, CA, USA | not available | |
| SMARTer microRNA-Seq Kit | Takara Bio, Shiga, Japan | not available | |
| UMI | TrueQuant SmallRNA Seq Kit for Ultra Low Input | GenXPro GmbH, Frankfurt Main, Germany | not available |
| QIAseq miRNA Library Kit (12bp UMIs) | Qiagen, Hilden, Germany | not available | |
| Polyadenylation and template switching | SMARTer smRNA-seq Kit | Takara Bio, Shiga, Japan | not available |
| CATS Small RNA-seq Kit | Diagenode, Liege, Belgium | not available | |
| Sequencing of hybridization probes | HTG EdgeSeq miRNA Whole Transcriptome Assay | HTG Molecular Diagnostics, Inc., Tuscon, AZ, USA | not available |
| Hybridization based techniques without NGS readout | FirePlex miRNA assays | Abcam, Cambridge, UK | not available |
| nCounter miRNA Expression Panels | NanoString, Seattle, WA, USA | [26] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benesova, S.; Kubista, M.; Valihrach, L. Small RNA-Sequencing: Approaches and Considerations for miRNA Analysis. Diagnostics 2021, 11, 964. https://doi.org/10.3390/diagnostics11060964
Benesova S, Kubista M, Valihrach L. Small RNA-Sequencing: Approaches and Considerations for miRNA Analysis. Diagnostics. 2021; 11(6):964. https://doi.org/10.3390/diagnostics11060964
Chicago/Turabian StyleBenesova, Sarka, Mikael Kubista, and Lukas Valihrach. 2021. "Small RNA-Sequencing: Approaches and Considerations for miRNA Analysis" Diagnostics 11, no. 6: 964. https://doi.org/10.3390/diagnostics11060964
APA StyleBenesova, S., Kubista, M., & Valihrach, L. (2021). Small RNA-Sequencing: Approaches and Considerations for miRNA Analysis. Diagnostics, 11(6), 964. https://doi.org/10.3390/diagnostics11060964