
Medulloblastoma Associated with Down Syndrome: From a Rare Event Leading to a Pathogenic Hypothesis

 , , , ,
, , , ,  , , ,
, , ,  ,
,  and
and 
Abstract
1. Introduction
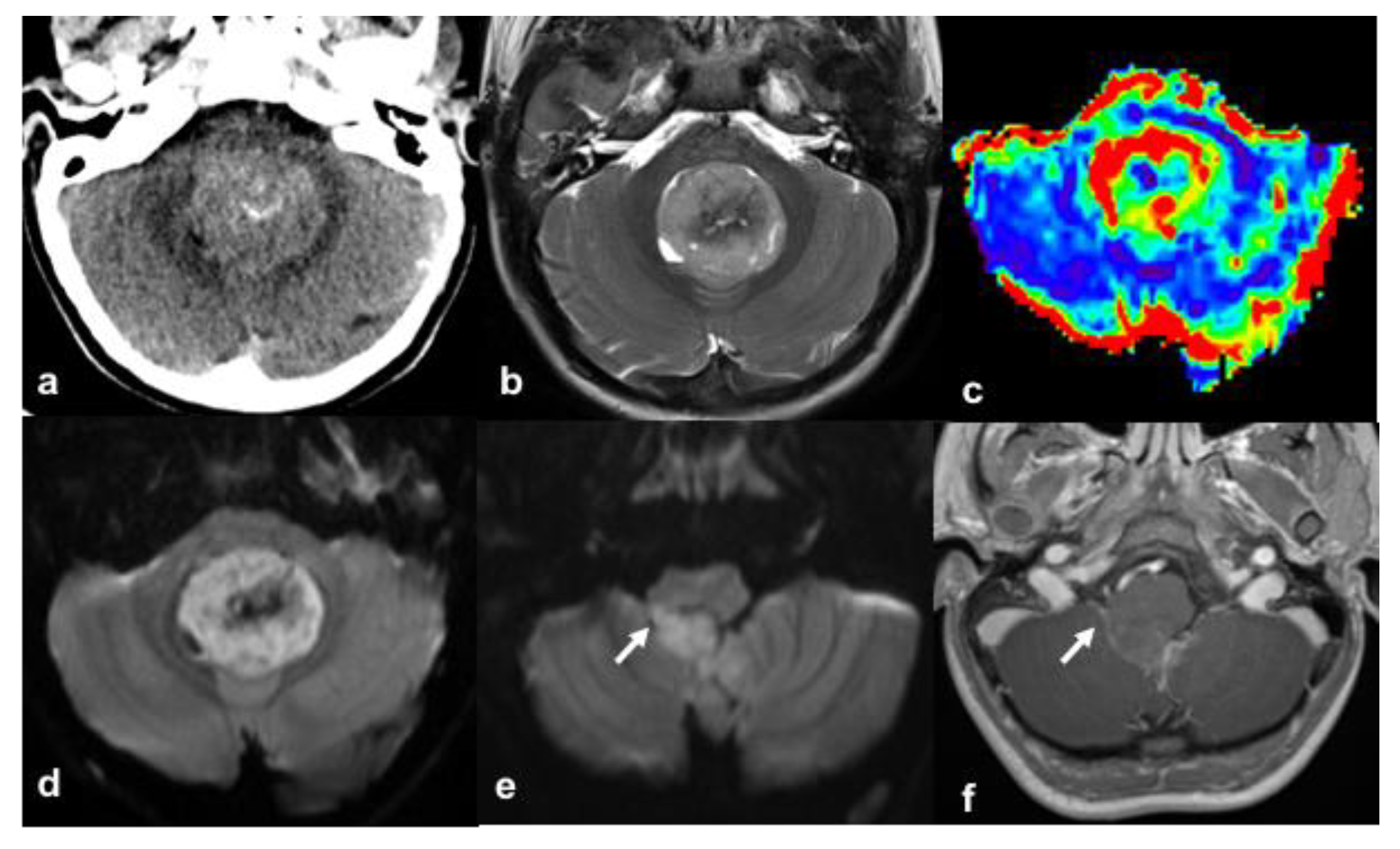
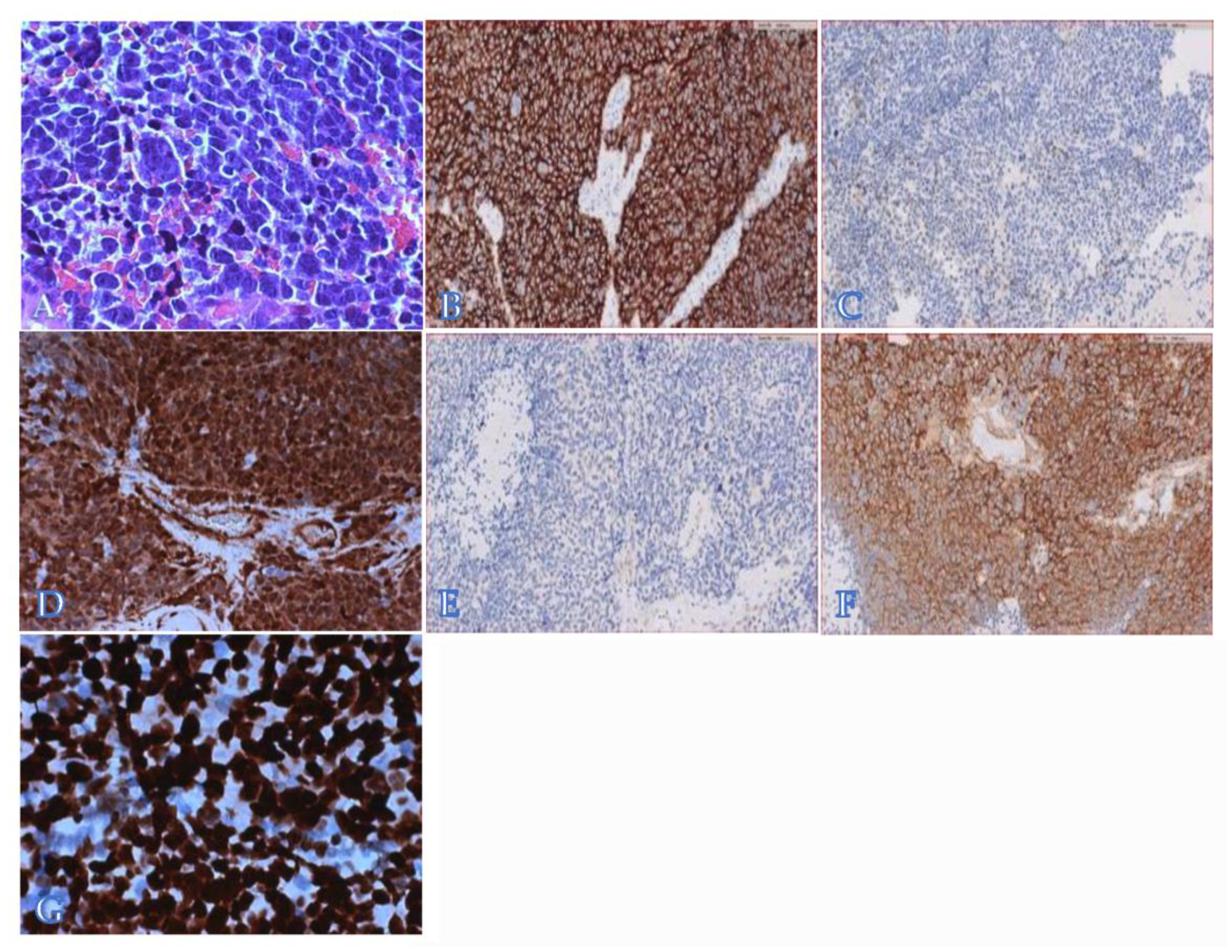
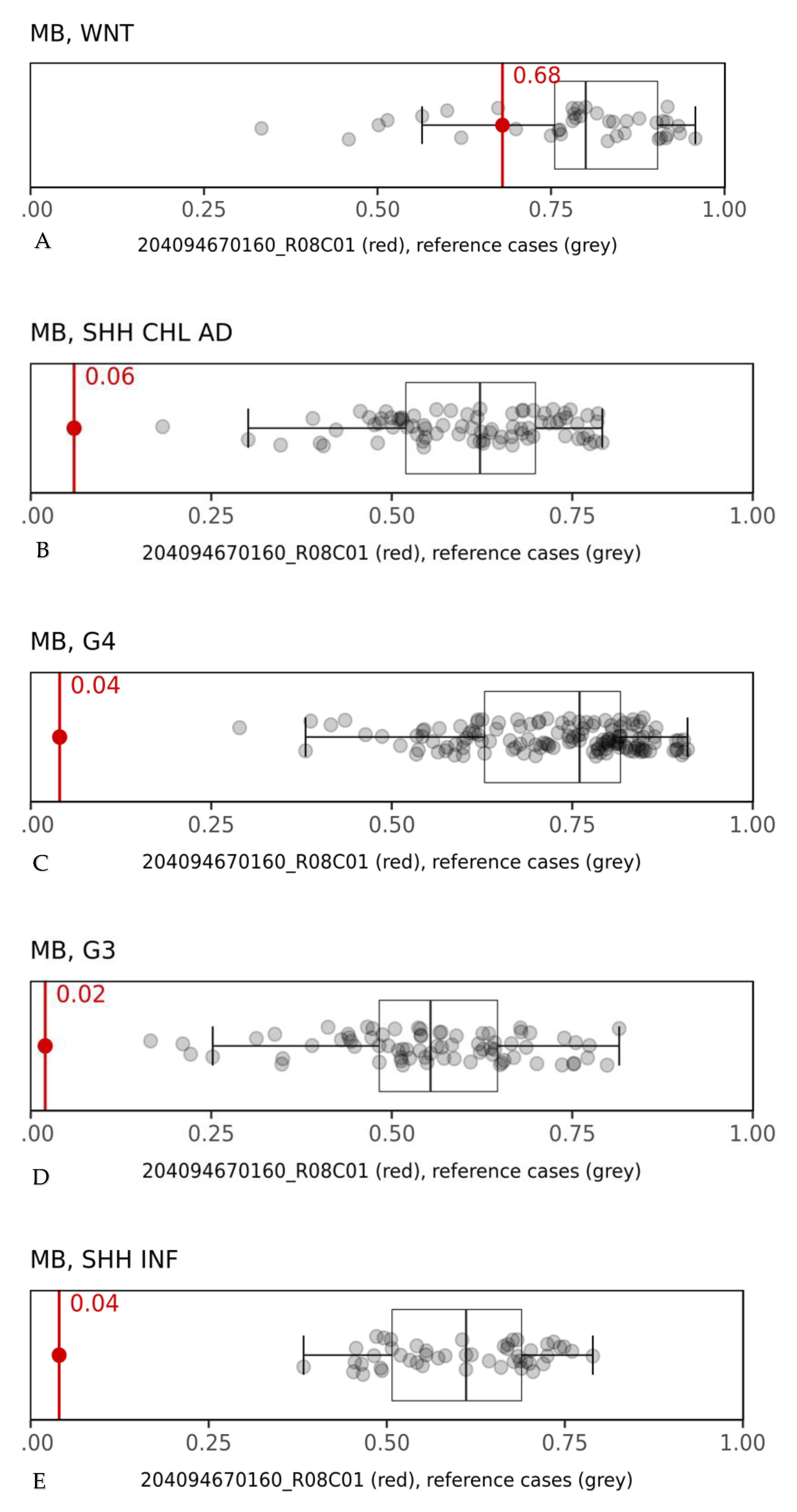
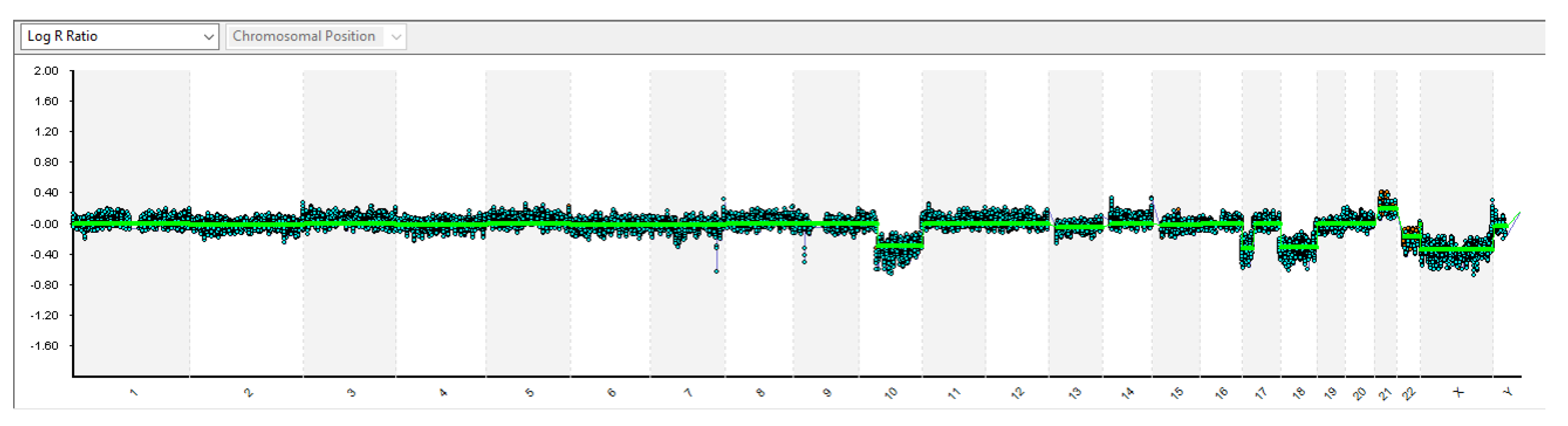
2. Case Presentation
3. Discussion
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DS | Down syndrome |
| MB | medulloblastoma |
| WNT | wingless |
| CNS | central nervous system |
| CPS | cancer predisposition syndrome |
| CT | computed tomography |
| MRI | magnetic resonance imaging |
| SNP | single nucleotide polymorphism |
| NGS | next-generation sequencing |
| CES | clinical exome sequencing |
| CH | congenital hypothyroidism |
| ABD | adaptor-binding domain |
| CNS-PNET | central nervous system primitive neuroectodermal tumors |
| SHH | Sonic hedgehog |
| CGNPs | cerebellar granule neuron progenitor cells |
References
- Asim, A.; Kumar, A.; Muthuswamy, S.; Jain, S.; Agarwal, S. Down syndrome: An insight of the disease. J. Biomed. Sci. 2015, 22, 41. [Google Scholar] [CrossRef]
- Hasle, H.; Clemmensen, I.H.; Mikkelsen, M. Risks of leukaemia and solid tumours in individuals with Down’s syndrome. Lancet 2000, 355, 165–169. [Google Scholar] [CrossRef]
- Rabin, K.R.; Whitlock, J.A. Malignancy in Children with Trisomy 21. Oncologist 2009, 14, 164–173. [Google Scholar] [CrossRef]
- Millard, N.E.; De Braganca, K.C. Medulloblastoma. J. Child Neurol. 2016, 31, 1341–1353. [Google Scholar] [CrossRef]
- Raffel, C. Medulloblastoma: Molecular Genetics and Animal Models. Neoplasia 2004, 6, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.G.; Farndon, P.A.; Burnell, L.D.; Gattamaneni, H.R.; Birch, J.M. The incidence of Gorlin syndrome in 173 consecutive cases of medulloblastoma. Br. J. Cancer 1991, 64, 959–961. [Google Scholar] [CrossRef] [PubMed]
- Waszak, S.M.; A Northcott, P.; Buchhalter, I.; Robinson, G.W.; Sutter, C.; Groebner, S.; Grund, K.B.; Brugières, L.; Jones, D.T.W.; Pajtler, K.W.; et al. Spectrum and prevalence of genetic predisposition in medulloblastoma: A retrospective genetic study and prospective validation in a clinical trial cohort. Lancet Oncol. 2018, 19, 785–798. [Google Scholar] [CrossRef]
- Carta, R.; Del Baldo, G.; Miele, E.; Po, A.; Besharat, Z.M.; Nazio, F.; Colafati, G.S.; Piccirilli, E.; Agolini, E.; Rinelli, M.; et al. Cancer Predisposition Syndromes and Medulloblastoma in the Molecular Era. Front. Oncol. 2020, 10, 566822. [Google Scholar] [CrossRef] [PubMed]
- Benesch, M.; Moser, A.; Sovinz, P.; Lackner, H.; Schwinger, W.; Eder, H.; Urban, C. Medulloblastoma in a child with down syndrome: Long-term remission with multimodality treatment. Pediatr. Blood Cancer 2009, 53, 1150–1151. [Google Scholar] [CrossRef] [PubMed]
- Mangum, R.; Varga, E.; Boué, D.R.; Capper, D.; Benesch, M.; Leonard, J.; Osorio, D.S.; Pierson, C.R.; Zumberge, N.; Sahm, F.; et al. SHH desmoplastic/nodular medulloblastoma and Gorlin syndrome in the setting of Down syndrome: Case report, molecular profiling, and review of the literature. Child Nerv. Syst. 2016, 32, 2439–2446. [Google Scholar] [CrossRef] [PubMed]
- Patay, Z.; Desain, L.A.; Hwang, S.N.; Coan, A.; Li, Y.; Ellison, D.W. MR Imaging Characteristics of Wingless-Type-Subgroup Pediatric Medulloblastoma. Am. J. Neuroradiol. 2015, 36, 2386–2393. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, V.; Remke, M.; Bouffet, E.; Bailey, S.; Clifford, S.C.; Doz, F.; Kool, M.; Dufour, C.; Vassal, G.; Milde, T.; et al. Risk stratification of childhood medulloblastoma in the molecular era: The current consensus. Acta Neuropathol. 2016, 131, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Ballabio, C.; Anderle, M.; Gianesello, M.; Lago, C.; Miele, E.; Cardano, M.; Aiello, G.; Piazza, S.; Caron, D.; Gianno, F.; et al. Modeling medulloblastoma in vivo and with human cerebellar organoids. Nat. Commun. 2020, 11, 1–18. [Google Scholar] [CrossRef]
- Miele, E.; De Vito, R.; Andrea, C.; Pedace, L.; Russo, I.; De Pasquale, M.D.; Di Giannatale, A.; Crocoli, A.; De Angelis, B.; Tartaglia, M.; et al. DNA Methylation Profiling for Diagnosing Undifferentiated Sarcoma with Capicua Transcriptional Receptor (CIC) Alterations. Int. J. Mol. Sci. 2020, 21, 1818. [Google Scholar] [CrossRef]
- Capper, D.; Stichel, D.; Sahm, F.; Jones, D.T.W.; Schrimpf, D.; Sill, M.; Schmid, S.; Hovestadt, V.; Reuss, D.E.; Koelsche, C.; et al. Practical implementation of DNA methylation and copy-number-based CNS tumor diagnostics: The Heidelberg experience. Acta Neuropathol. 2018, 136, 181–210. [Google Scholar] [CrossRef]
- Ligresti, G.; Militello, L.; Steelman, L.S.; Cavallaro, A.; Basile, F.; Nicoletti, F.; Stivala, F.; McCubrey, J.A.; Libra, M. PIK3CA mutations in human solid tumors: Role in sensitivity to various therapeutic approaches. Cell Cycle 2009, 8, 1352–1358. [Google Scholar] [CrossRef] [PubMed]
- Fernando, T.M.; Piskol, R.; Bainer, R.; Sokol, E.S.; Trabucco, S.E.; Zhang, Q.; Trinh, H.; Maund, S.; Kschonsak, M.; Chaudhuri, S.; et al. Functional characterization of SMARCA4 variants identified by targeted exome-sequencing of 131,668 cancer patients. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Takeshima, H.; Niwa, T.; Takahashi, T.; Wakabayashi, M.; Yamashita, S.; Ando, T.; Inagawa, Y.; Taniguchi, H.; Katai, H.; Sugiyama, T.; et al. Frequent involvement of chromatin remodeler alterations in gastric field cancerization. Cancer Lett. 2015, 357, 328–338. [Google Scholar] [CrossRef]
- Kim, Y.; Cho, M.-Y.; Kim, J.; Kim, S.N.; Oh, S.C.; Lee, K.-A. Profiling cancer-associated genetic alterations and molecular classification of cancer in Korean gastric cancer patients. Oncotarget 2017, 8, 69888–69905. [Google Scholar] [CrossRef]
- Peters, C.; Nicholas, A.K.; Schoenmakers, E.; Lyons, G.; Langham, S.; Serra, E.G.; Sebire, N.J.; Muzza, M.; Fugazzola, L.; Schoenmakers, N. DUOX2/DUOXA2 Mutations Frequently Cause Congenital Hypothyroidism that Evades Detection on Newborn Screening in the United Kingdom. Thyroid 2019, 29, 790–801. [Google Scholar] [CrossRef]
- Bull, M.J. Down Syndrome. N. Engl. J. Med. 2020, 382, 2344–2352. [Google Scholar] [CrossRef]
- Cairney, C.J.; Sanguinetti, G.; Ranghini, E.; Chantry, A.D.; Nostro, M.C.; Bhattacharyya, A.; Svendsen, C.; Keith, W.N.; Bellantuono, I. A systems biology approach to Down syndrome: Identification of Notch/Wnt dysregulation in a model of stem cells aging. Biochim. Biophys. Acta 2009, 1792, 353–363. [Google Scholar] [CrossRef]
- Satgé, D.; Seidel, M.G. The Pattern of Malignancies in Down Syndrome and Its Potential Context with the Immune System. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Satgé, D.; Sasco, A.J.; Carlsen, N.L.; A Stiller, C.; Rubie, H.; Hero, B.; De Bernardi, B.; De Kraker, J.; Coze, C.; Kogner, P.; et al. A lack of neuroblastoma in Down syndrome: A study from 11 European countries. Cancer Res. 1998, 58, 448–452. [Google Scholar] [PubMed]
- Satgé, D.; Sasco, A.J.; Chompret, A.; Orbach, D.; Méchinaud, F.; Lacour, B.; Roullet, B.; Martelli, H.; Bergeron, C.; Bertrand, Y.; et al. A 22-year French experience with solid tumors in children with Down syndrome. Pediatr. Hematol. Oncol. 2003, 20, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Satgé, D.; A Stiller, C.; Rutkowski, S.; Von Bueren, A.O.; Lacour, B.; Sommelet, D.; Nishi, M.; Massimino, M.; Garrè, M.L.; Moreno, F.; et al. A very rare cancer in Down syndrome: Medulloblastoma. Epidemiological data from 13 countries. J. Neuro-Oncol. 2013, 112, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Taub, J.W.; Berman, J.N.; Hitzler, J.K.; Sorrell, A.D.; Lacayo, N.J.; Mast, K.; Head, D.; Raimondi, S.; Hirsch, B.; Ge, Y.; et al. Improved outcomes for myeloid leukemia of Down syndrome: A report from the Children’s Oncology Group AAML0431 trial. Blood 2017, 129, 3304–3313. [Google Scholar] [CrossRef] [PubMed]
- Bruwier, A.; Chantrain, C.F. Hematological disorders and leukemia in children with Down syndrome. Eur. J. Nucl. Med. Mol. Imaging 2011, 171, 1301–1307. [Google Scholar] [CrossRef]
- Baxter, L.L.; Moran, T.H.; Richtsmeier, J.T.; Troncoso, J.; Reeves, R.H. Discovery and genetic localization of Down syndrome cerebellar phenotypes using the Ts65Dn mouse. Hum. Mol. Genet. 2000, 9, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.D.; Northcott, P.A.; Korshunov, A.; Remke, M.; Cho, Y.-J.; Clifford, S.C.; Eberhart, C.G.; Parsons, D.W.; Rutkowski, S.; Gajjar, A.; et al. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol. 2011, 123, 465–472. [Google Scholar] [CrossRef]
- Clifford, S.C.; Lusher, M.E.; Lindsey, J.C.; Langdon, J.A.; Gilbertson, R.J.; Straughton, D.; Ellison, D.W. Wnt/Wingless Pathway Activation and Chromosome 6 Loss Characterise a Distinct Molecular Sub-Group of Medulloblastomas Associated with a Favourable Prognosis. Cell Cycle 2006, 5, 2666–2670. [Google Scholar] [CrossRef] [PubMed]
- Ellison, D.W.; Dalton, J.; Kocak, M.; Nicholson, S.L.; Fraga, C.; Neale, G.; Kenney, A.M.; Brat, D.J.; Perry, A.; Yong, W.H.; et al. Medulloblastoma: Clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta Neuropathol. 2011, 121, 381–396. [Google Scholar] [CrossRef]
- Zhukova, N.; Ramaswamy, V.; Remke, M.; Pfaff, E.; Shih, D.J.; Martin, D.C.; Castelo-Branco, P.; Baskin, B.; Ray, P.N.; Bouffet, E.; et al. Subgroup-Specific Prognostic Implications of TP53 Mutation in Medulloblastoma. J. Clin. Oncol. 2013, 31, 2927–2935. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, J.; Hill, R.M.; Megahed, H.; Lusher, M.E.; Schwalbe, E.C.; Cole, M.; Hogg, T.L.; Gilbertson, R.J.; Ellison, D.W.; Bailey, S.; et al. TP53 Mutations in Favorable-Risk Wnt/Wingless-Subtype Medulloblastomas. J. Clin. Oncol. 2011, 29, e344–e346. [Google Scholar] [CrossRef] [PubMed]
- Pfaff, E.; Remke, M.; Sturm, D.; Benner, A.; Witt, H.; Milde, T.; Von Bueren, A.O.; Wittmann, A.; Schöttler, A.; Jorch, N.; et al. TP53 Mutation is frequently associated with CTNNB1 mutation or MYCN amplification and is compatible with long-term survival in medulloblastoma. J. Clin. Oncol. 2010, 28, 5188–5196. [Google Scholar] [CrossRef]
- Surun, A.; Varlet, P.; Brugières, L.; Lacour, B.; Faure-Conter, C.; Leblond, P.; Bertozzi-Salomon, A.-I.; Berger, C.; André, N.; Sariban, E.; et al. Medulloblastomas associated with an APC germline pathogenic variant share the good prognosis of CTNNB1-mutated medulloblastomas. Neuro Oncol. 2020, 22, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Gibson, P.; Tong, Y.; Robinson, G.W.; Thompson, M.C.; Currle, D.S.; Eden, C.; Kranenburg, T.A.; Hogg, T.L.; Poppleton, H.; Martin, J.; et al. Subtypes of medulloblastoma have distinct developmental origins. Nat. Cell Biol. 2010, 468, 1095–1099. [Google Scholar] [CrossRef] [PubMed]
- Colafati, G.S.; Voicu, I.P.; Carducci, C.; Miele, E.; Carai, A.; Di Loreto, S.; Marrazzo, A.; Cacchione, A.; Cecinati, V.; Tornesello, A.; et al. MRI features as a helpful tool to predict the molecular subgroups of medulloblastoma: State of the art. Ther. Adv. Neurol. Disord. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.T.; Asthana, S.; Gao, S.P.; Lee, B.H.; Chapman, J.S.; Kandoth, C.; Gao, J.; Socci, N.D.; Solit, D.B.; Olshen, A.B.; et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat. Biotechnol. 2016, 34, 155–163. [Google Scholar] [CrossRef]
- Fortuno, C.; Pesaran, T.; Dolinsky, J.; Yussuf, A.; McGoldrick, K.; Kho, P.F.; James, P.A.; Spurdle, A. p53 major hotspot variants are associated with poorer prognostic features in hereditary cancer patients. Cancer Genet. 2019, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Baugh, E.H.; Ke, H.; Levine, A.J.; A Bonneau, R.; Chan, C.S. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 2018, 25, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Tabori, U.; Sung, L.; Hukin, J.; Laperriere, N.; Crooks, B.; Carret, A.-S.; Silva, M.; Odame, I.; Mpofu, C.; Strother, D.; et al. Medulloblastoma in the second decade of life: A specific group with respect to toxicity and management. Cancer 2005, 103, 1874–1880. [Google Scholar] [CrossRef] [PubMed]
- Satge, D.; Rickert, C. A Medical Enigma: Persons with Down Syndrome Do Not Develop Medulloblastoma. Neuroepidemiology 2008, 32, 164. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GENE | RefSeq | HGVS | DBSNP | ACMG | Location |
|---|---|---|---|---|---|
| TP53 | NM_000546.5 | c.743G>A p.Arg248Gln | rs11540652 | Pathogenic | 17:7577538 |
| CTNNB1 | NM_003072.3 | c.3727C>T p.Arg1243Trp | rs121913400 | Likely pathogenic | 3:41266101 |
| SMARCA4 | NM_001024847.2 | c.118G>A p.Asp40Asn | Likely pathogenic | 19:11144146 | |
| FBXW7 | NM_018315.4 | c.1154G>A p.Arg385His | rs1057519895 | Likely pathogenic | 4:153249384 |
| Reference (Number of Patients) | Presence of Another CPS | Age | Histological Classification | Molecular Finding | Outcome |
|---|---|---|---|---|---|
| Benesch M. et al., Pediatr Blood Cancer, 2009 (9) [1] | No | 4 years | Medulloblastoma | Group 3 (personal communication: Capper et al.: 2015 [6]) | Alive at 60 months in complete remission |
| Mangum R. et al., Childs Nerv Syst, 2016 (10) [1] | Yes (Gorlin Syndrome) | 21 months | Medulloblastoma desmoplastic/nodular Synaptofisin Neu-N: positive GFAP (Glial fibrillary acidic protein), Neurofilament proteins: negative β-catenin: negative N-myc/C-myc amplification: negative | SHH subgroup Heterozygous PTCH1 variant (c.834G>A) predicted to result in premature protein termination (p.Trp278 *) | Not available |
| Our case (1) | No | 10 years | Classic Medulloblastoma with focal anaplasia grade IV (according to WHO 2016) Synaptofisina, Neu-N: positive YAP1: diffusely expressed GAB1: negative β-catenin negative (but a pathogenic CTNNB1 variant in exon 3) p53: positive N-myc/C-myc amplification: negative | WNT subgroup | Died at 11 months from diagnosis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boni, A.; Ranalli, M.; Del Baldo, G.; Carta, R.; Lodi, M.; Agolini, E.; Rinelli, M.; Valentini, D.; Rossi, S.; Alesi, V.; et al. Medulloblastoma Associated with Down Syndrome: From a Rare Event Leading to a Pathogenic Hypothesis. Diagnostics 2021, 11, 254. https://doi.org/10.3390/diagnostics11020254
Boni A, Ranalli M, Del Baldo G, Carta R, Lodi M, Agolini E, Rinelli M, Valentini D, Rossi S, Alesi V, et al. Medulloblastoma Associated with Down Syndrome: From a Rare Event Leading to a Pathogenic Hypothesis. Diagnostics. 2021; 11(2):254. https://doi.org/10.3390/diagnostics11020254
Chicago/Turabian StyleBoni, Alessandra, Marco Ranalli, Giada Del Baldo, Roberto Carta, Mariachiara Lodi, Emanuele Agolini, Martina Rinelli, Diletta Valentini, Sabrina Rossi, Viola Alesi, and et al. 2021. "Medulloblastoma Associated with Down Syndrome: From a Rare Event Leading to a Pathogenic Hypothesis" Diagnostics 11, no. 2: 254. https://doi.org/10.3390/diagnostics11020254
APA StyleBoni, A., Ranalli, M., Del Baldo, G., Carta, R., Lodi, M., Agolini, E., Rinelli, M., Valentini, D., Rossi, S., Alesi, V., Cacchione, A., Miele, E., Alessi, I., Caroleo, A. M., Colafati, G. S., De Ioris, M. A., Boccuto, L., Balducci, M., Carai, A., & Mastronuzzi, A. (2021). Medulloblastoma Associated with Down Syndrome: From a Rare Event Leading to a Pathogenic Hypothesis. Diagnostics, 11(2), 254. https://doi.org/10.3390/diagnostics11020254