
Molecular Profiling of Merkel Cell Polyomavirus-Associated Merkel Cell Carcinoma and Cutaneous Melanoma

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients’ Samples
2.2. Histology and Immunohistochemistry
2.3. DNA Isolation
2.4. Merkel Cell Polyomavirus Molecular Detection
2.5. BRAF StripAssay
2.6. Next-Generation Sequencing
3. Results
3.1. Clinical Presentations
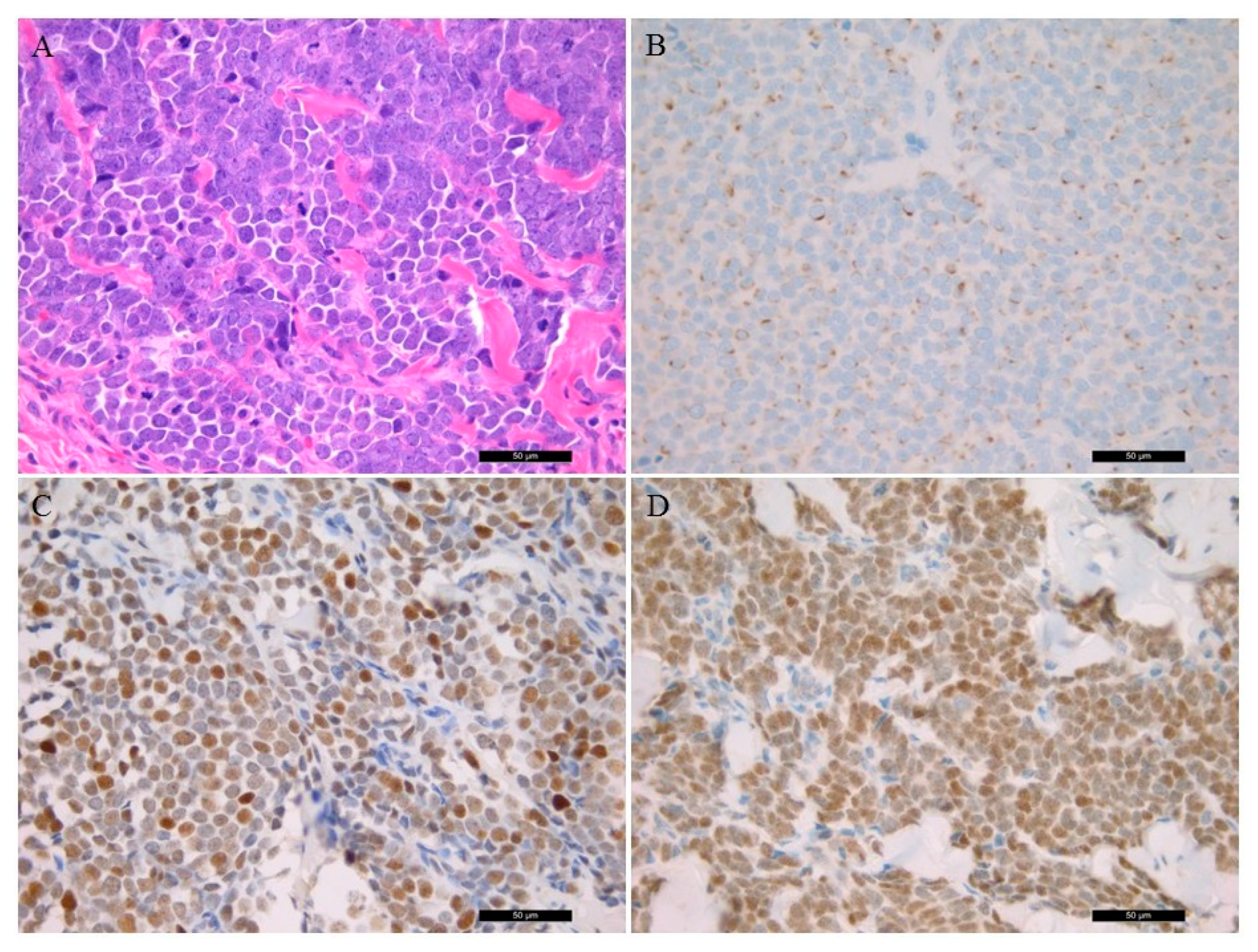
3.2. Histological Features Including Immunohistochemistry
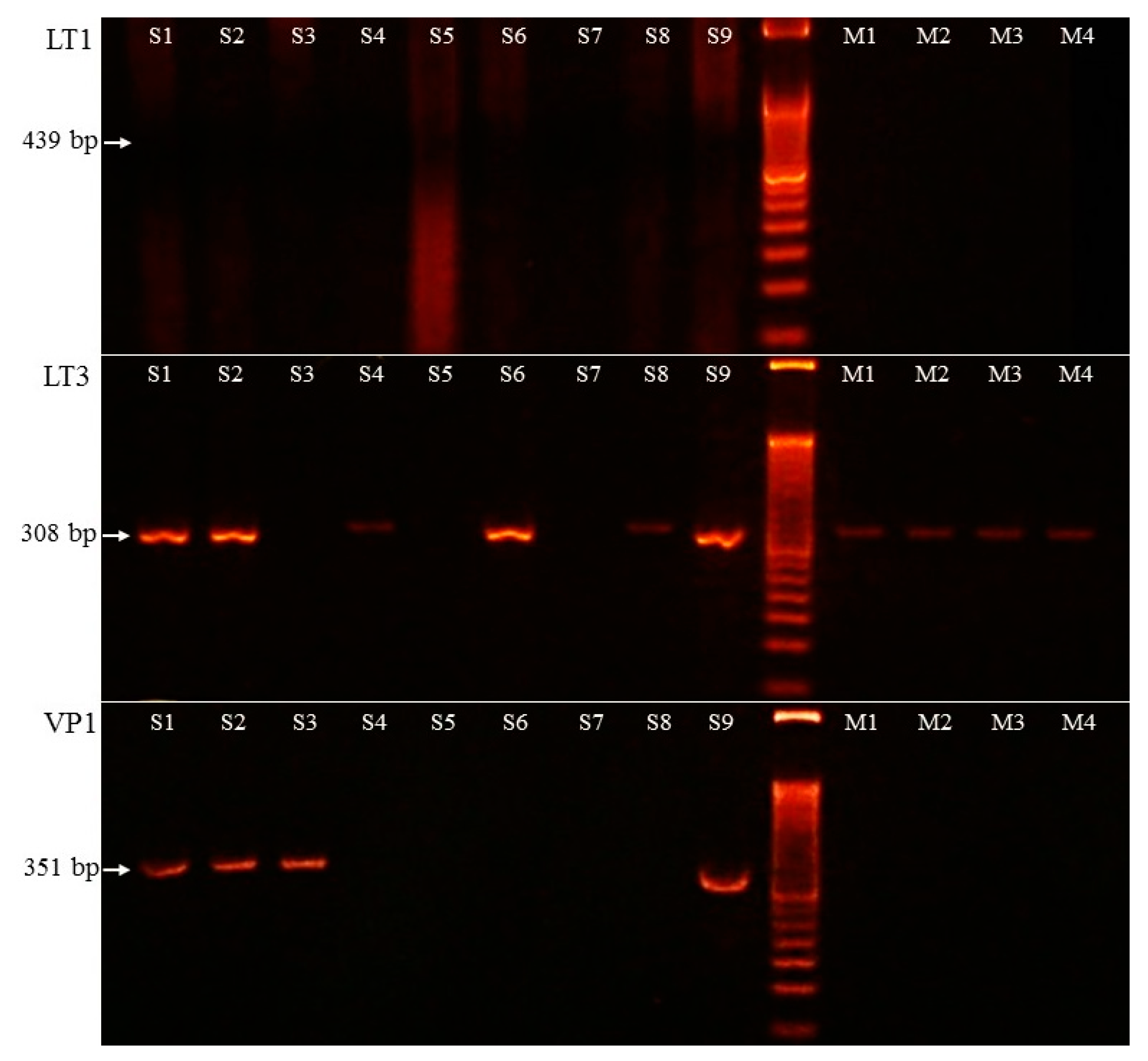
3.3. Merkel Cell Polyomavirus Detection
3.4. Merkel Cell Polyomavirus Amplification in Melanoma Samples
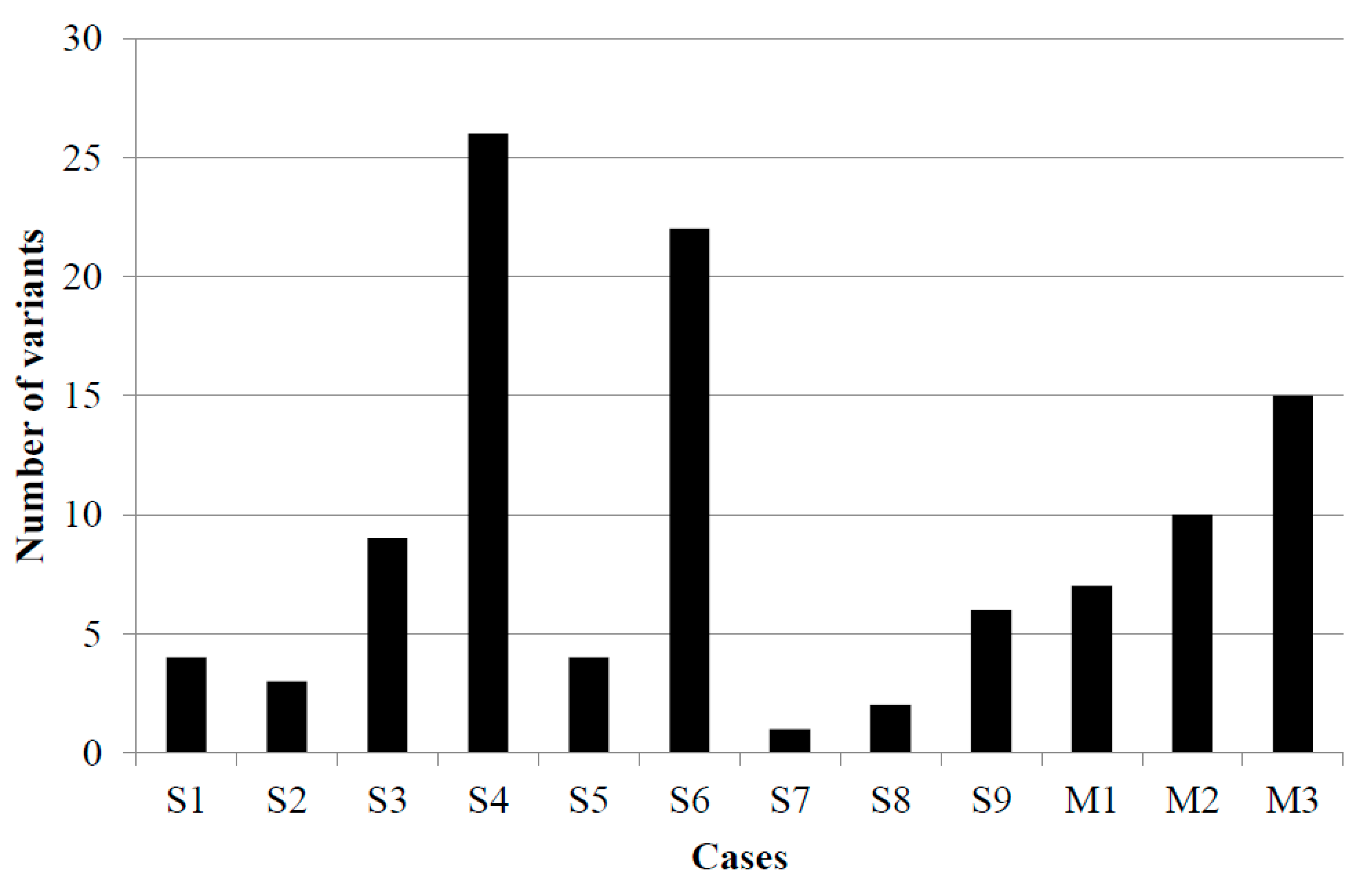
3.5. NGS-Based Mutation Profiling
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Harms, K.L.; Healy, M.A.; Nghiem, P.; Sober, A.J.; Johnson, T.M.; Bichakjian, C.K.; Wong, S.L. Analysis of Prognostic Factors from 9387 Merkel Cell Carcinoma Cases Forms the Basis for the New 8th Edition AJCC Staging System. Ann. Surg Oncol. 2016, 23, 3564–3571. [Google Scholar] [CrossRef] [PubMed]
- Harms, P.W. Update on Merkel Cell Carcinoma. Clin. Lab. Med. 2017, 37, 485–501. [Google Scholar] [CrossRef] [PubMed]
- Coggshall, K.; Tello, T.L.; North, J.P.; Yu, S.S. Merkel Cell Carcinoma: An Update and Review: Pathogenesis, Diagnosis, and Staging. J. Am. Acad Derm. 2018, 78, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Agelli, M.; Clegg, L.X.; Becker, J.C.; Rollison, D.E. The Etiology and Epidemiology of Merkel Cell Carcinoma. Curr. Probl. Cancer 2010, 34, 14–37. [Google Scholar] [CrossRef]
- Fitzgerald, T.L.; Dennis, S.; Kachare, S.D.; Vohra, N.A.; Wong, J.H.; Zervos, E.E. Dramatic Increase in the Incidence and Mortality from Merkel Cell Carcinoma in the United States. Am. Surg. 2015, 81, 802–806. [Google Scholar] [CrossRef] [PubMed]
- Tetzlaff, M.T.; Harms, P.W. Danger Is Only Skin Deep: Aggressive Epidermal Carcinomas. An Overview of the Diagnosis, Demographics, Molecular-Genetics, Staging, Prognostic Biomarkers, and Therapeutic Advances in Merkel Cell Carcinoma. Mod. Pathol. 2020, 33, 42–55. [Google Scholar] [CrossRef]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal Integration of a Polyomavirus in Human Merkel Cell Carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef]
- Moshiri, A.S.; Doumani, R.; Yelistratova, L.; Blom, A.; Lachance, K.; Shinohara, M.M.; Delaney, M.; Chang, O.; McArdle, S.; Thomas, H.; et al. Polyomavirus-Negative Merkel Cell Carcinoma: A More Aggressive Subtype Based on Analysis of 282 Cases Using Multimodal Tumor Virus Detection. J. Investig. Derm. 2017, 137, 819–827. [Google Scholar] [CrossRef]
- Andea, A.A.; Patel, R.; Ponnazhagan, S.; Kumar, S.; DeVilliers, P.; Jhala, D.; Eltoum, I.E.; Siegal, G.P. Merkel Cell Carcinoma: Correlation of KIT Expression with Survival and Evaluation of KIT Gene Mutational Status. Hum. Pathol. 2010, 41, 1405–1412. [Google Scholar] [CrossRef]
- Nardi, V.; Song, Y.; Santamaria-Barria, J.A.; Cosper, A.K.; Lam, Q.; Faber, A.C.; Boland, G.M.; Yeap, B.Y.; Bergethon, K.; Scialabba, V.L.; et al. Activation of PI3K Signaling in Merkel Cell Carcinoma. Clin. Cancer Res. 2012, 18, 1227–1236. [Google Scholar] [CrossRef]
- Kuromi, T.; Matsushita, M.; Iwasaki, T.; Nonaka, D.; Kuwamoto, S.; Nagata, K.; Kato, M.; Akizuki, G.; Kitamura, Y.; Hayashi, K. Association of Expression of the Hedgehog Signal with Merkel Cell Polyomavirus Infection and Prognosis of Merkel Cell Carcinoma. Hum. Pathol. 2017, 69, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.Q.; Waldeck, K.; Vergara, I.A.; Schröder, J.; Madore, J.; Wilmott, J.S.; Colebatch, A.J.; De Paoli-Iseppi, R.; Li, J.; Lupat, R.; et al. UV-Associated Mutations Underlie the Etiology of MCV-Negative Merkel Cell Carcinomas. Cancer Res. 2015, 75, 5228–5234. [Google Scholar] [CrossRef] [PubMed]
- Goh, G.; Walradt, T.; Markarov, V.; Blom, A.; Riaz, N.; Doumani, R.; Stafstrom, K.; Moshiri, A.; Yelistratova, L.; Levinsohn, J.; et al. Mutational Landscape of MCPyV-Positive and MCPyV-Negative Merkel Cell Carcinomas with Implications for Immunotherapy. Oncotarget 2016, 7, 3403–3415. [Google Scholar] [CrossRef] [PubMed]
- Harms, P.W.; Harms, K.L.; Moore, P.S.; DeCaprio, J.A.; Nghiem, P.; Wong, M.K.K.; Brownell, I.; International Workshop on Merkel Cell Carcinoma Research (IWMCC) Working Group. The Biology and Treatment of Merkel Cell Carcinoma: Current Understanding and Research Priorities. Nat. Rev. Clin. Oncol. 2018, 15, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Varga, E.; Kiss, M.; Szabó, K.; Kemény, L. Detection of Merkel Cell Polyomavirus DNA in Merkel Cell Carcinomas. Br. J. Derm. 2009, 161, 930–932. [Google Scholar] [CrossRef]
- Sirikanjanapong, S.; Melamed, J.; Patel, R.R. Intraepidermal and Dermal Merkel Cell Carcinoma with Squamous Cell Carcinoma in Situ: A Case Report with Review of Literature. J. Cutan. Pathol. 2010, 37, 881–885. [Google Scholar] [CrossRef]
- Shuda, M.; Arora, R.; Kwun, H.J.; Feng, H.; Sarid, R.; Fernández-Figueras, M.-T.; Tolstov, Y.; Gjoerup, O.; Mansukhani, M.M.; Swerdlow, S.H.; et al. Human Merkel Cell Polyomavirus Infection I. MCV T Antigen Expression in Merkel Cell Carcinoma, Lymphoid Tissues and Lymphoid Tumors. Int. J. Cancer 2009, 125, 1243–1249. [Google Scholar] [CrossRef]
- Jung, H.S.; Choi, Y.-L.; Choi, J.-S.; Roh, J.H.; Pyon, J.K.; Woo, K.-J.; Lee, E.H.; Jang, K.-T.; Han, J.; Park, C.-S.; et al. Detection of Merkel Cell Polyomavirus in Merkel Cell Carcinomas and Small Cell Carcinomas by PCR and Immunohistochemistry. Histol. Histopathol. 2011, 26, 1231–1241. [Google Scholar] [CrossRef]
- Leitz, M.; Stieler, K.; Grundhoff, A.; Moll, I.; Brandner, J.M.; Fischer, N. Merkel Cell Polyomavirus Detection in Merkel Cell Cancer Tumors in Northern Germany Using PCR and Protein Expression. J. Med. Virol. 2014, 86, 1813–1819. [Google Scholar] [CrossRef]
- Leroux-Kozal, V.; Lévêque, N.; Brodard, V.; Lesage, C.; Dudez, O.; Makeieff, M.; Kanagaratnam, L.; Diebold, M.-D. Merkel Cell Carcinoma: Histopathologic and Prognostic Features According to the Immunohistochemical Expression of Merkel Cell Polyomavirus Large T Antigen Correlated with Viral Load. Hum. Pathol. 2015, 46, 443–453. [Google Scholar] [CrossRef]
- Swick, B.L.; Srikantha, R.; Messingham, K.N. Specific Analysis of KIT and PDGFR-Alpha Expression and Mutational Status in Merkel Cell Carcinoma. J. Cutan. Pathol. 2013, 40, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Figueras, M.T.; Puig, L.; Musulen, E.; Gilaberte, M.; Ferrándiz, C.; Lerma, E.; Ariza, A. Prognostic Significance of P27Kip1, P45Skp2 and Ki67 Expression Profiles in Merkel Cell Carcinoma, Extracutaneous Small Cell Carcinoma, and Cutaneous Squamous Cell Carcinoma. Histopathology 2005, 46, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Henderson, S.A.; Tetzlaff, M.T.; Pattanaprichakul, P.; Fox, P.; Torres-Cabala, C.A.; Bassett, R.L.; Prieto, V.G.; Richards, H.W.; Curry, J.L. Detection of Mitotic Figures and G2+ Tumor Nuclei with Histone Markers Correlates with Worse Overall Survival in Patients with Merkel Cell Carcinoma. J. Cutan. Pathol. 2014, 41, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.L.; Frieling, G.W. Merkel Cell Carcinoma: A Review and Update on Current Concepts. Diagn. Histopathol. 2016, 22, 127–133. [Google Scholar] [CrossRef]
- Kwun, H.J.; Shuda, M.; Feng, H.; Camacho, C.J.; Moore, P.S.; Chang, Y. Merkel Cell Polyomavirus Small T Antigen Controls Viral Replication and Oncoprotein Expression by Targeting the Cellular Ubiquitin Ligase SCFFbw7. Cell Host Microbe 2013, 14, 125–135. [Google Scholar] [CrossRef]
- Harms, K.L.; Lazo de la Vega, L.; Hovelson, D.H.; Rahrig, S.; Cani, A.K.; Liu, C.-J.; Fullen, D.R.; Wang, M.; Andea, A.A.; Bichakjian, C.K.; et al. Molecular Profiling of Multiple Primary Merkel Cell Carcinoma to Distinguish Genetically Distinct Tumors From Clonally Related Metastases. JAMA Derm. 2017, 153, 505–512. [Google Scholar] [CrossRef]
- Schrama, D.; Peitsch, W.K.; Zapatka, M.; Kneitz, H.; Houben, R.; Eib, S.; Haferkamp, S.; Moore, P.S.; Shuda, M.; Thompson, J.F.; et al. Merkel Cell Polyomavirus Status Is Not Associated with Clinical Course of Merkel Cell Carcinoma. J. Investig. Derm. 2011, 131, 1631–1638. [Google Scholar] [CrossRef]
- Knepper, T.C.; Montesion, M.; Russell, J.S.; Sokol, E.S.; Frampton, G.M.; Miller, V.A.; Albacker, L.A.; McLeod, H.L.; Eroglu, Z.; Khushalani, N.I.; et al. The Genomic Landscape of Merkel Cell Carcinoma and Clinicogenomic Biomarkers of Response to Immune Checkpoint Inhibitor Therapy. Clin. Cancer Res. 2019, 25, 5961–5971. [Google Scholar] [CrossRef]
- Koburger, I.; Meckbach, D.; Metzler, G.; Fauser, U.; Garbe, C.; Bauer, J. Absence of Merkel Cell Polyoma Virus in Cutaneous Melanoma. Exp. Derm. 2011, 20, 78–79. [Google Scholar] [CrossRef]
- Helmbold, P.; Lahtz, C.; Herpel, E.; Schnabel, P.A.; Dammann, R.H. Frequent Hypermethylation of RASSF1A Tumour Suppressor Gene Promoter and Presence of Merkel Cell Polyomavirus in Small Cell Lung Cancer. Eur. J. Cancer 2009, 45, 2207–2211. [Google Scholar] [CrossRef]
- Katano, H.; Ito, H.; Suzuki, Y.; Nakamura, T.; Sato, Y.; Tsuji, T.; Matsuo, K.; Nakagawa, H.; Sata, T. Detection of Merkel Cell Polyomavirus in Merkel Cell Carcinoma and Kaposi’s Sarcoma. J. Med. Virol. 2009, 81, 1951–1958. [Google Scholar] [CrossRef] [PubMed]
- Dworkin, A.M.; Tseng, S.Y.; Allain, D.C.; Iwenofu, O.H.; Peters, S.B.; Toland, A.E. Merkel Cell Polyomavirus in Cutaneous Squamous Cell Carcinoma of Immunocompetent Individuals. J. Investig. Derm. 2009, 129, 2868–2874. [Google Scholar] [CrossRef] [PubMed]
- Kassem, A.; Technau, K.; Kurz, A.K.; Pantulu, D.; Löning, M.; Kayser, G.; Stickeler, E.; Weyers, W.; Diaz, C.; Werner, M.; et al. Merkel Cell Polyomavirus Sequences Are Frequently Detected in Nonmelanoma Skin Cancer of Immunosuppressed Patients. Int. J. Cancer 2009, 125, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Csoboz, B.; Rasheed, K.; Sveinbjørnsson, B.; Moens, U. Merkel Cell Polyomavirus and Non-Merkel Cell Carcinomas: Guilty or Circumstantial Evidence? APMIS 2020, 128, 104–120. [Google Scholar] [CrossRef] [PubMed]
- Reiman, A.; Kikuchi, H.; Scocchia, D.; Smith, P.; Tsang, Y.W.; Snead, D.; Cree, I.A. Validation of an NGS Mutation Detection Panel for Melanoma. BMC Cancer 2017, 17, 150. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.-L.; Zhou, L.; Sadri, N. Comparison of Targeted next Generation Sequencing (NGS) versus Isolated BRAF V600E Analysis in Patients with Metastatic Melanoma. Virchows Arch. 2018, 473, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Mancini, I.; Simi, L.; Salvianti, F.; Castiglione, F.; Sonnati, G.; Pinzani, P. Analytical Evaluation of an NGS Testing Method for Routine Molecular Diagnostics on Melanoma Formalin-Fixed, Paraffin-Embedded Tumor-Derived DNA. Diagnostics 2019, 9, 117. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Patient | Gender | Age (years) | Location | Size (cm) | Depth (cm) | Treatment | Metastasis | Survival (Month) |
|---|---|---|---|---|---|---|---|---|
| 1 | M | 89 | left knee | 4 | 1 | excision and chemotherapy | n.a. | 7 |
| 2 | M | 80 | right face | 1.5 | 0.9 | excision | lost follow-up | |
| 3 | M | 88 | right chest | 2.2 | 0.5 | excision | lost follow-up | |
| 4 | F | 68 | left shoulder | 1.1 | 0.7 | neoadjuvant chemotherapy | axillary lymph node | 24 |
| 5 | M | 83 | right thigh | 2.4 | 2.2 | excision | lost follow-up | |
| 6 | M | 76 | right 3rd finger | 1 | 6.7 | excision | n.a. | 9 |
| 7 | F | 85 | right face | 1.4 | 1.6 | radiotherapy | lost follow-up | |
| 8 | F | 63 | right gluteus | 2 | 0.4 | radiotherapy | n.a. | 6 |
| 9 | M | 68 | right forearm | 6 | 1 | excision | lymp node | 3 |
| Cases | Lymphocyte Infiltration | Ki-67 (%) | Cytokeratin 20 | Synaptophysin | Chromogranin A | Thyroid Transcription Factor 1 | p53 | MCPyV IHC | LT1 PCR | LT3 PCR | VP1 PCR |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | low | 60 | + | + | + | - | diffuse + | + | - | + | + |
| 2 | low | 60 | + | + | + | - | patchy + | + | - | + | + |
| 3 | low | 50 | + | + | - | - | diffuse + | - | - | - | + |
| 4 | low | 60 | dot-like | marked | marked | - | - | diffuse + | - | + | - |
| 5 | low | 80 | dot-like | marked | focal | - | weakly + | - | - | - | - |
| 6 | moderate | 70 | dot-like | marked | marked | - | - | patchy + | - | + | - |
| 7 | low | 60 | dot-like | dot-like | dot-like | - | - | - | - | - | - |
| 8 | moderate | 70 | dot-like | + | patchy + | - | diffuse + | - | - | + | - |
| 9 | low | 60 | dot-like | dot-like | dot like | - | patchy + | diffuse + | - | + | + |
| Scheme | Gene Symbol | Gene Name | Nucleotide Change | Amino Acid Change | Variant Allele Frequency (%) | Clinical Sgnificance | COSMIC ID |
|---|---|---|---|---|---|---|---|
| S1 (+) | RB1 | retinoblastoma 1 | c.2033A>T | p.His678Leu | 48 | no data available | - |
| S2 (+) | STK11 | serine/threonine kinase 11 | c.1062C>G | p.Phe354Leu | 54.55 | neutral | COSM21360 |
| S3 (+) | EGFR | epidermal growth factor receptor | c.2137G>A | p.Glu713Lys | 30.25 | no data available | - |
| FBXW7 | F-box/WD repeat-containing protein 7 | c.1031C>T | p.Ser344Phe | 89.3 | COSM1177864 | ||
| TP53 | tumor protein P53 | c.832C>T | p.Pro278Ser | 92.4 | pathogenic | COSM10939 | |
| S4 (+) | CTNNB1 | catenin beta-1 | c.59C>T | p.Ala20Val | 9 | pathogenic | COSM5702 |
| S5 (-) | CDKN2A | cyclin-dependent kinase inhibitor 2A | c.442G>A | p.Ala148Thr | 50 | neutral | COSM3774362 |
| FGFR1 | fibroblast growth factor receptor 1 | c.2181-6C>T | - | 48.6 | no data available | - | |
| S6 (+) | ABL1 | tyrosine-protein kinase ABL1 | c.740A>G | p.Lys247Arg | 42.1 | benign | - |
| FOXL2 | forkhead box protein L2 | c.536C>G | p.Ala179Gly | 65.3 | benign | COSM4600643 | |
| HNF1A | hepatocyte nuclear factor 1 A | c.864del | p.Pro291GlnfsTer51 | 7.5 | pathogenic | COSM935974 | |
| S7 (-) | no mutation detected | ||||||
| S8 (+) | JAK3 | Janus kinase 3 | c.2164G>A | p.Val722Ile | 42.5 | neutral | COSM34213 |
| S9 (+) | TP53 | tumor protein P53 | c.-28-4G>A | - | 4.5 | no data available | - |
| M1 (+) | BRAF | serine/threonine kinase BRAF | c.1799_1800delinsAA | p.Val600Glu | 40 | pathogenic | COSM475 |
| PIK3CA | phosphatidylinositol bisphosphate 3-kinase | c.1031T>C | p.Val344Ala | 13.4 | pathogenic | COSM86951 | |
| STK11 | serine/threonine kinase 11 | c.1211C>T | p.Ser404Phe | 35.1 | no data available | - | |
| M2 (+) | BRAF | serine/threonine kinase BRAF | c.1799T>A | p.Val600Glu | 22.2 | pathogenic | COSM476 |
| CDKN2A | cyclin-dependent kinase inhibitor 2A | c.169dup | p.Ala57GlyfsTer63 | 20.6 | pathogenic | COSM110662 | |
| SMAD4 | mothers against decapentaplegic homolog 4 | c.122A>G | p.Glu41Gly | 5.7 | no data available | - | |
| M3 (+) | BRAF | serine/threonine kinase BRAF | c.1799T>A | p.Val600Glu | 15.4 | pathogenic | COSM476 |
| APC | adenomatous polyposis coli | c.3949G>C | p.Glu1317Gln | 50 | pathogenic | COSM19099 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mokánszki, A.; Méhes, G.; Csoma, S.L.; Kollár, S.; Chang Chien, Y.-C. Molecular Profiling of Merkel Cell Polyomavirus-Associated Merkel Cell Carcinoma and Cutaneous Melanoma. Diagnostics 2021, 11, 212. https://doi.org/10.3390/diagnostics11020212
Mokánszki A, Méhes G, Csoma SL, Kollár S, Chang Chien Y-C. Molecular Profiling of Merkel Cell Polyomavirus-Associated Merkel Cell Carcinoma and Cutaneous Melanoma. Diagnostics. 2021; 11(2):212. https://doi.org/10.3390/diagnostics11020212
Chicago/Turabian StyleMokánszki, Attila, Gábor Méhes, Szilvia Lilla Csoma, Sándor Kollár, and Yi-Che Chang Chien. 2021. "Molecular Profiling of Merkel Cell Polyomavirus-Associated Merkel Cell Carcinoma and Cutaneous Melanoma" Diagnostics 11, no. 2: 212. https://doi.org/10.3390/diagnostics11020212
APA StyleMokánszki, A., Méhes, G., Csoma, S. L., Kollár, S., & Chang Chien, Y.-C. (2021). Molecular Profiling of Merkel Cell Polyomavirus-Associated Merkel Cell Carcinoma and Cutaneous Melanoma. Diagnostics, 11(2), 212. https://doi.org/10.3390/diagnostics11020212