
High-Resolution Computed Tomography: Lights and Shadows in Improving Care for SSc-ILD Patients

 ,
,  , , ,
, , ,  and
and
Abstract
:1. Introduction
2. The Role of HRTC in SSc-ILD
3. HRTC vs. Pulmonary Function Tests
4. HRCT Acquisition
5. HRCT Pattern in SSc-ILD
6. Nonpulmonary HRCT Findings in SSc-ILD
7. CT and Radiation Risk
8. Semiquantitative Visual Scoring System
9. The Role of HRTC and Other Imaging Techniques
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cappelli, S.; Bellando Randone, S.; Camiciottoli, G.; De Paulis, A.; Guiducci, S.; Matucci-Cerinic, M. Interstitial lung disease in systemic sclerosis: Where do we stand? Eur. Respir. Rev. 2015, 24, 411–419. [Google Scholar] [CrossRef]
- Fischer, A.; Patel, N.M.; Volkmann, E.R. Interstitial Lung Disease in Systemic Sclerosis: Focus on Early Detection and Intervention. Open Access Rheumatol. 2019, 11, 283–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clukers, J.; Lukers, J.; Lanclus, M.; Belmans, D.; van Holsbeke, C.; de Backer, W.; Vummidi, D.; Cronin, P.; Lavon, B.R.; de Backer, J.; et al. Interstitial lung disease in systemic sclerosis quantification of disease classification and progression with high-resolution computed tomography: An observational study. J. Scleroderma Relat. Disord. 2021, 6, 154–164. [Google Scholar] [CrossRef]
- Khanna, D.; Tashkin, D.P.; Denton, C.P.; Renzoni, E.A.; Desai, S.R.; Varga, J. Etiology, Risk Factors, and Biomarkers in Systemic Sclerosis with Interstitial Lung Disease. Am. J. Respir. Crit. Care Med. 2020, 201, 650–660. [Google Scholar] [CrossRef] [PubMed]
- Tyndall, A.J.; Bannert, B.; Vonk, M.; Airò, P.; Cozzi, F.; Carreira, P.E.; Bancel, D.F.; Allanore, Y.; Müller-Ladner, U.; Distler, O.; et al. Causes and risk factors for death in systemic sclerosis: A study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann. Rheum. Dis. 2010, 69, 1809–1815. [Google Scholar] [CrossRef] [Green Version]
- Winstone, T.A.; Assayag, D.; Wilcox, P.G.; Dunne, J.V.; Hague, C.J.; Leipsic, J.; Collard, H.R.; Ryerson, C.J. Predictors of Mortality and Progression in Scleroderma-Associated Interstitial Lung Disease. Chest 2014, 146, 422–436. [Google Scholar] [CrossRef]
- Elhai, M.; Meune, C.; Avouac, J.; Kahan, A.; Allanore, Y. Trends in mortality in patients with systemic sclerosis over 40 years: A systematic review and meta-analysis of cohort studies. Rheumatology 2012, 51, 1017–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Distler, O.; Assassi, S.; Cottin, V.; Cutolo, M.; Danoff, S.K.; Denton, C.P.; Distler, J.H.; Hoffmann-Vold, A.-M.; Johnson, S.R.; Ladner, U.M.; et al. Predictors of progression in systemic sclerosis patients with interstitial lung disease. Eur. Respir. J. 2020, 55, 1902026. [Google Scholar] [CrossRef] [Green Version]
- Demizio, D.J.; Bernstein, E. Detection and classification of systemic sclerosis-related interstitial lung disease: A review. Curr. Opin. Rheumatol. 2019, 31, 553–560. [Google Scholar] [CrossRef]
- Wells, A.U.; Hansell, D.M.; Rubens, M.B.; Cullinan, P.; Haslam, P.L.; Black, C.M.; Du Bois, R.M. Fibrosing alveolitis in systemic sclerosis. Bronchoalveolar lavage findings in relation to computed tomographic appearance. Am. J. Respir. Crit. Care Med. 1994, 150, 462–468. [Google Scholar] [CrossRef]
- Walker, U.A.; Tyndall, A.; Czirjak, L.; Denton, C.; Farge-Bancel, D.; Kowal-Bielecka, O.; Muller-Ladner, U.; Bocelli-Tyndall, C.; Matucci-Cerinic, M. Clinical risk assessment of organ manifestations in systemic sclerosis: A report from the EULAR Scleroderma Trials and Research group database. Ann. Rheum. Dis. 2007, 66, 754–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaeger, V.; Wirz, E.G.; Allanore, Y.; Rossbach, P.; Riemekasten, G.; Hachulla, E.; Distler, O.; Airò, P.; Carreira, P.E.; Gurman, A.B.; et al. Incidences and Risk Factors of Organ Manifestations in the Early Course of Systemic Sclerosis: A Longitudinal EUSTAR Study. PLoS ONE 2016, 11, e0163894. [Google Scholar] [CrossRef] [PubMed]
- Smith, V.; Scirè, C.A.; Talarico, R.; Airo, P.; Alexander, T.; Allanore, Y.; Bruni, C.; Codullo, V.; Dalm, V.; De Vries-Bouwstra, J.; et al. Systemic sclerosis: State of the art on clinical practice guidelines. RMD Open 2018, 4, e000782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, K.T.; Reveille, J.D. The clinical relevance of autoantibodies in scleroderma. Arthritis Res. Ther. 2003, 5, 80–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulli, A.; Ruaro, B.; Smith, V.; Pizzorni, C.; Zampogna, G.; Gallo, M.; Cutolo, M. Progression of Nailfold Microvascular Damage and Antinuclear Antibody Pattern in Systemic Sclerosis. J. Rheumatol. 2013, 40, 634–639. [Google Scholar] [CrossRef] [PubMed]
- Launay, D.; Remy-Jardin, M.; Michon-Pasturel, U.; Mastora, I.; Hachulla, E.; Lambert, M.; Delannoy, V.; Queyrel, V.; Duhamel, A.; Matran, R.; et al. High resolution computed tomography in fibrosing alveolitis associated with systemic sclerosis. J. Rheumatol. 2006, 33, 1789–1801. [Google Scholar] [PubMed]
- Wells, A.U.; Hansell, D.M.; Corrin, B.; Harrison, N.K.; Goldstraw, P.; Black, C.M.; Du Bois, R.M. High resolution computed tomography as a predictor of lung histology in systemic sclerosis. Thorax 1992, 47, 738–742. [Google Scholar] [CrossRef] [Green Version]
- Bouros, D.; Wells, A.U.; Nicholson, A.G.; Colby, T.V.; Polychronopoulos, V.; Pantelidis, P.; Haslam, P.L.; Vassilakis, D.A.; Black, C.M.; Du Bois, R.M. Histopathologic Subsets of Fibrosing Alveolitis in Patients with Systemic Sclerosis and Their Relationship to Outcome. Am. J. Respir. Crit. Care Med. 2002, 165, 1581–1586. [Google Scholar] [CrossRef]
- Desai, S.R.; Veeraraghavan, S.; Hansell, D.M.; Nikolakopolou, A.; Goh, N.S.L.; Nicholson, A.G.; Colby, T.V.; Denton, C.P.; Black, C.M.; Du Bois, R.M.; et al. CT Features of Lung Disease in Patients with Systemic Sclerosis: Comparison with Idiopathic Pulmonary Fibrosis and Nonspecific Interstitial Pneumonia. Radiology 2004, 232, 560–567. [Google Scholar] [CrossRef]
- Akiyama, M.; Kaneko, Y.; Takeuchi, T. Ground Glass Opacity with Mixed Consolidation on Chest Computed Tomography Reflects the Severe Condition of Pneumocystis Pneumonia in Association with a Poor Prognosis in Patients with Connective Tissue Diseases. Intern Med. 2019, 58, 3379–3383. [Google Scholar] [CrossRef] [Green Version]
- Volkmann, E.R.; Tashkin, D.P.; Sim, M.; Li, N.; Goldmuntz, E.; Keyes-Elstein, L.; Pinckney, A.; Furst, D.E.; Clements, P.J.; Khanna, D.; et al. Short-term progression of interstitial lung disease in systemic sclerosis predicts long-term survival in two independent clinical trial cohorts. Ann. Rheum. Dis. 2018, 78, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Ruaro, B.; Confalonieri, M.; Salton, F.; Wade, B.; Baratella, E.; Geri, P.; Confalonieri, P.; Kodric, M.; Biolo, M.; Bruni, C. The Relationship between Pulmonary Damage and Peripheral Vascular Manifestations in Systemic Sclerosis Patients. Pharmaceuticals 2021, 14, 403. [Google Scholar] [CrossRef]
- Wells, A.U. High-resolution computed tomography and scleroderma lung disease. Rheumatology 2008, 47, 59–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanna, D.; Clements, P.J.; Furst, D.E.; Chon, Y.; Elashoff, R.; Roth, M.D.; Sterz, M.G.; Chung, J.; FitzGerald, J.; Seibold, J.R.; et al. Correlation of the degree of dyspnea with health-related quality of life, functional abilities, and diffusing capacity for carbon monoxide in patients with systemic sclerosis and active alveolitis: Results from the scleroderma lung study. Arthritis Rheum. 2005, 52, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Distler, O.; Volkmann, E.R.; Hoffmann-Vold, A.M.; Maher, T. Current and future perspectives on management of systemic sclerosis-associated interstitial lung disease. Expert Rev. Clin. Immunol. 2019, 15, 1009–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann-Vold, A.-M.; Allanore, Y.; Bendstrup, E.; Bruni, C.; Distler, O.; Maher, T.M.; Wijsenbeek, M.; Kreuter, M. The need for a holistic approach for SSc-ILD—Achievements and ambiguity in a devastating disease. Respir. Res. 2020, 21, 197. [Google Scholar] [CrossRef]
- Ruaro, B.; Confalonieri, M.; Matucci-Cerinic, M.; Salton, F.; Confalonieri, P.; Santagiuliana, M.; Citton, G.; Baratella, E.; Bruni, C. The Treatment of Lung Involvement in Systemic Sclerosis. Pharmaceuticals 2021, 14, 154. [Google Scholar] [CrossRef]
- Le Gouellec, N.; Duhamel, A.; Perez, T.; Hachulla, A.L.; Sobanski, V.; Faivre, J.B.; Morell-Dubois, S.; Lambert, M.; Hatron, P.Y.; Hachulla, E.; et al. Predictors of lung function test severity and outcome in systemic sclerosis-associated interstitial lung disease. PLoS ONE 2017, 12, e0181692. [Google Scholar] [CrossRef]
- Hoang-Duc, H.; Pham-Huy, Q.; Vu-Minh, T.; Duong-Quy, S. Study of the Correlation between HRCT Semi-quantitative Scoring, Concentration of Alveolar Nitric Oxide, and Clinical-functional Parameters of Systemic Sclerosis-induced Interstitial Lung Disease. Yale J. Biol. Med. 2020, 93, 657–667. [Google Scholar]
- Khanna, D.; Nagaraja, V.; Tseng, C.-H.; Abtin, F.; Suh, R.; Kim, G.; Wells, A.; Furst, D.E.; Clements, P.J.; Roth, M.D.; et al. Predictors of lung function decline in scleroderma-related interstitial lung disease based on high-resolution computed tomography: Implications for cohort enrichment in systemic sclerosis–associated interstitial lung disease trials. Arthritis Res. 2015, 17, 372. [Google Scholar] [CrossRef] [Green Version]
- Çetinçakmak, M.G.; Göya, C.; Hamidi, C.; Tekbaş, G.; Abakay, Ö.; Batmaz, I.; Hattapoğlu, S.; Yavuz, A.; Bilici, A. Quantitative volumetric assessment of pulmonary involvement in patients with systemic sclerosis. Quant. Imaging Med. Surg. 2016, 6, 50–56. [Google Scholar] [CrossRef]
- Ooi, G.C.; Mok, M.Y.; Tsang, K.W.T.; Wong, Y.; Khong, P.L.; Fung, P.C.W.; Chan, S.; Tse, H.F.; Wong, R.W.S.; Lam, W.K.; et al. Interstitial Lung disease in Systemic Sclerosis. An HRCT-clinical correlative study. Acta Radiol. 2003, 44, 258–264. [Google Scholar] [CrossRef]
- Yoon, R.G.; Seo, J.B.; Kim, N.; Lee, H.J.; Lee, S.M.; Lee, Y.K.; Song, J.W.; Song, J.W.; Kim, D.S. Quantitative assessment of change in regional disease patterns on serial HRCT of fibrotic interstitial pneumonia with texture-based automated quantification system. Eur. Radiol. 2013, 23, 692–701. [Google Scholar] [CrossRef] [PubMed]
- Margaritopoulos, G.A.; Antoniou, K.M.; Denton, C.; Wells, A.U. Interstitial Lung Disease in Systemic Sclerosis. Semin. Respir. Crit. Care Med. 2014, 35, 213–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goh, N.S.; Desai, S.R.; Veeraraghavan, S.; Hansell, D.M.; Copley, S.J.; Maher, T.M.; Corte, T.J.; Sander, C.R.; Ratoff, J.; Devaraj, A.; et al. Interstitial lung disease in systemic sclerosis: A simple staging system. Am. J. Respir. Crit. Care Med. 2008, 177, 1248–1254. [Google Scholar] [CrossRef]
- Schurawitzki, H.; Stiglbauer, R.; Graninger, W.; Herold, C.; Pölzleitner, D.; Burghuber, O.; Tscholakoff, D. Interstitial lung disease in progressive systemic sclerosis: High-resolution CT versus radiography. Radiology 1990, 176, 755–759. [Google Scholar] [CrossRef]
- Hoffmann-Vold, A.-M.; Aaløkken, T.M.; Lund, M.B.; Garen, T.; Midtvedt, Ø.; Brunborg, C.; Gran, J.T.; Molberg, Ø. Predictive Value of Serial High-Resolution Computed Tomography Analyses and Concurrent Lung Function Tests in Systemic Sclerosis. Arthritis Rheumatol. 2015, 67, 2205–2212. [Google Scholar] [CrossRef]
- Showalter, K.; Hoffmann, A.; Rouleau, G.; Aaby, D.; Lee, J.; Richardson, C.; Dematte, J.; Agrawal, R.; Chang, R.W.; Hinchcliff, M. Performance of Forced Vital Capacity and Lung Diffusion Cutpoints for Associated Radiographic Interstitial Lung Disease in Systemic Sclerosis. J. Rheumatol. 2018, 45, 1572–1576. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann-Vold, A.-M.; Fretheim, H.; Halse, A.-K.; Seip, M.; Bitter, H.; Wallenius, M.; Garen, T.; Salberg, A.; Brunborg, C.; Midtvedt, Ø.; et al. Tracking Impact of Interstitial Lung Disease in Systemic Sclerosis in a Complete Nationwide Cohort. Am. J. Respir. Crit. Care Med. 2019, 200, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Martinović Kaliterna, D.; Petrić, M. Biomarkers of skin and lung fibrosis in systemic sclerosis. Expert Rev. Clin. Immunol. 2019, 15, 1215–1223. [Google Scholar] [CrossRef]
- Kim, H.J.; Tashkin, D.P.; Gjertson, D.W.; Brown, M.S.; Kleerup, E.; Chong, S.; Belperio, J.A.; Roth, M.D.; Abtin, F.; Elashoff, R.; et al. Transitions to different patterns of interstitial lung disease in scleroderma with and without treatment. Ann. Rheum. Dis. 2016, 75, 1367–1371. [Google Scholar] [CrossRef]
- Hoffmann-Vold, A.-M.; Allanore, Y.; Alves, M.; Brunborg, C.; Airó, P.; Ananieva, L.P.; Czirják, L.; Guiducci, S.; Hachulla, E.; Li, M.; et al. Progressive interstitial lung disease in patients with systemic sclerosis-associated interstitial lung disease in the EUSTAR database. Ann. Rheum. Dis. 2020, 80, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Hax, V.; Bredemeier, M.; Moro, A.L.D.; Pavan, T.R.; Vieira, M.V.; Pitrez, E.H.; Chakr, R.; Xavier, R.M. Clinical algorithms for the diagnosis and prognosis of interstitial lung disease in systemic sclerosis. Semin. Arthritis Rheum. 2017, 47, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Behr, J.; Furst, D.E. Pulmonary function tests. Rheumatology 2008, 47, v65–v67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caron, M.; Hoa, S.; Hudson, M.; Schwartzman, K.; Steele, R. Pulmonary function tests as outcomes for systemic sclerosis interstitial lung disease. Eur. Respir. Rev. 2018, 27, 148. [Google Scholar] [CrossRef] [Green Version]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef]
- Steen, V.D.; Conte, C.; Owens, G.R.; Medsger, T.A., Jr. Severe restrictive lung disease in systemic sclerosis. Arthritis Rheum. 1994, 37, 1283–1289. [Google Scholar] [CrossRef]
- Khanna, D.; Tseng, C.-H.; Farmani, N.; Steen, V.; Furst, D.E.; Clements, P.J.; Roth, M.D.; Goldin, J.; Elashoff, R.; Seibold, J.R.; et al. Clinical course of lung physiology in patients with scleroderma and interstitial lung disease: Analysis of the Scleroderma Lung Study Placebo Group. Arthritis Rheum. 2011, 63, 3078–3085. [Google Scholar] [CrossRef] [Green Version]
- Suliman, Y.A.; Dobrota, R.; Huscher, D.; Nguyen-Kim, T.D.L.; Maurer, B.; Jordan, S.; Speich, R.; Frauenfelder, T.; Distler, O. Brief Report: Pulmonary Function Tests: High Rate of False-Negative Results in the Early Detection and Screening of Scleroderma-Related Interstitial Lung Disease. Arthritis Rheumatol. 2015, 67, 3256–3261. [Google Scholar] [CrossRef]
- Goh, N.S.; Hoyles, R.K.; Denton, C.P.; Hansell, D.M.; Renzoni, E.; Maher, T.M.; Nicholson, A.G.; Wells, A.U. Short-Term Pulmonary Function Trends Are Predictive of Mortality in Interstitial Lung Disease Associated with Systemic Sclerosis. Arthritis Rheumatol. 2017, 69, 1670–1678. [Google Scholar] [CrossRef]
- Man, A.; Davidyock, T.; Ferguson, L.T.; Ieong, M.; Zhang, Y.; Simms, R.W. Changes in forced vital capacity over time in systemic sclerosis: Application of group-based trajectory modelling. Rheumatology 2015, 54, 1464–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tashkin, D.P.; Volkmann, E.R.; Tseng, C.-H.; Kim, H.J.; Goldin, J.; Clements, P.; Furst, D.; Khanna, D.; Kleerup, E.; Roth, M.D.; et al. Relationship between quantitative radiographic assessments of interstitial lung disease and physiological and clinical features of systemic sclerosis. Ann. Rheum. Dis. 2014, 75, 374–381. [Google Scholar] [CrossRef]
- Stock, C.J.W.; Hoyles, R.K.; Daccord, C.; Kokosi, M.; Visca, D.; De Lauretis, A.; Alfieri, V.; Kouranos, V.; Margaritopoulos, G.; George, P.M.; et al. Serum markers of pulmonary epithelial damage in systemic sclerosis-associated interstitial lung disease and disease progression. Respirology 2021, 26, 461–468. [Google Scholar] [CrossRef]
- Baratella, E.; Ruaro, B.; Giudici, F.; Wade, B.; Santagiuliana, M.; Salton, F.; Confalonieri, P.; Simbolo, M.; Scarpa, A.; Tollot, S.; et al. Evaluation of Correlations between Genetic Variants and High-Resolution Computed Tomography Patterns in Idiopathic Pulmonary Fibrosis. Diagnostics 2021, 11, 762. [Google Scholar] [CrossRef]
- Orlandi, M.; Landini, N.; Sambataro, G.; Nardi, C.; Tofani, L.; Bruni, C.; Randone, S.B.; Blagojevic, J.; Melchiorre, D.; Hughes, M.; et al. The role of chest CT in deciphering interstitial lung involvement: Systemic sclerosis versus COVID-19. Rheumatology 2021, 28, 1–10. [Google Scholar] [CrossRef]
- Ferrazza, A.M.; Gigante, A.; Gasperini, M.L.; Ammendola, R.M.; Paone, G.; Carbone, I.; Rosato, E. Assessment of interstitial lung disease in systemic sclerosis using the quantitative CT algorithm CALIPER. Clin. Rheumatol. 2020, 39, 1537–1542. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.A.; Sverzellati, N.; Travis, W.D.; Brown, K.K.; Colby, T.V.; Galvin, J.R.; Goldin, J.G.; Hansell, D.M.; Inoue, Y.; Johkoh, T.; et al. Diagnostic criteria for idiopathic pulmonary fibrosis: A Fleischner Society White Paper. Lancet Respir. Med. 2018, 6, 138–153. [Google Scholar] [CrossRef]
- Hansell, D.M.; Bankier, A.A.; MacMahon, H.; McLoud, T.C.; Müller, N.L.; Remy, J. Fleischner Society: Glossary of Terms for Thoracic Imaging. Radiology 2008, 246, 697–722. [Google Scholar] [CrossRef] [Green Version]
- Wells, A.U. Pulmonary Function Tests in Connective Tissue Disease. Semin. Respir. Crit. Care Med. 2007, 28, 379–388. [Google Scholar] [CrossRef]
- Yoo, H.; Hino, T.; Han, J.; Franks, T.J.; Im, Y.; Hatabu, H.; Chung, M.P.; Lee, K.S. Connective tissue disease-related interstitial lung disease (CTD-ILD) and interstitial lung abnormality (ILA): Evolving concept of CT findings, pathology and management. Eur. J. Radiol. Open 2020, 8, 100311. [Google Scholar] [CrossRef] [PubMed]
- Remy-Jardin, M.; Giraud, F.; Remy, J.; Copin, M.C.; Gosselin, B.; Duhamel, A. Importance of ground-glass attenuation in chronic diffuse infiltrative lung disease: Pathologic-CT correlation. Radiology 1993, 189, 693–698. [Google Scholar] [CrossRef]
- Shah, R.M.; Jimenez, S.; Wechsler, R. Significance of Ground-glass Opacity on HRCT in Long-term Follow-up of Patients With Systemic Sclerosis. J. Thorac. Imaging 2007, 22, 120–124. [Google Scholar] [CrossRef]
- Ruaro, B.; Nallino, M.G.; Casabella, A.; Salton, F.; Confalonieri, P.; De Tanti, A.; Bruni, C. Monitoring the microcirculation in the diagnosis and follow-up of systemic sclerosis patients: Focus on pulmonary and peripheral vascular manifestations. Microcirculation 2020, 27, e12647. [Google Scholar] [CrossRef] [PubMed]
- Winklehner, A.; Berger, N.; Maurer, B.; Distler, O.; Alkadhi, H.; Frauenfelder, T. Screening for interstitial lung disease in systemic sclerosis: The diagnostic accuracy of HRCT image series with high increment and reduced number of slices. Ann. Rheum. Dis. 2012, 71, 549–552. [Google Scholar] [CrossRef] [PubMed]
- Frauenfelder, T.; Winklehner, A.; Nguyen-Kim, T.D.L.; Dobrota, R.; Baumueller, S.; Maurer, B.; Distler, O. Screening for interstitial lung disease in systemic sclerosis: Performance of high-resolution CT with limited number of slices: A prospective study. Ann. Rheum. Dis. 2014, 73, 2069–2073. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Kim, T.D.L.; Maurer, B.; Suliman, Y.A.; Morsbach, F.; Distler, O.; Frauenfelder, T. The impact of slice-reduced computed tomography on histogram-based densitometry assessment of lung fibrosis in patients with systemic sclerosis. J. Thorac. Dis. 2018, 10, 2142–2152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, J.J.; Olson, A.; Fischer, A.; Bull, T.; Brown, K.K.; Raghu, G. Scleroderma lung disease. Eur. Respir. Rev. 2013, 22, 6–19. [Google Scholar] [CrossRef]
- Chaisson, N.F.; Hassoun, P.M. Systemic Sclerosis-Associated Pulmonary Arterial Hypertension. Chest 2013, 144, 1346–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strollo, D.; Goldin, J. Imaging Lung Disease in Systemic Sclerosis. Curr. Rheumatol. Rep. 2010, 12, 156–161. [Google Scholar] [CrossRef] [Green Version]
- Thompson, A.E. A study of the frequency of pericardial and pleural effusions in scleroderma. Rheumatology 1998, 37, 1320–1323. [Google Scholar] [CrossRef] [Green Version]
- Bastos, A.D.L.; Corrêa, R.D.A.; Ferreira, G.A. Tomography patterns of lung disease in systemic sclerosis. Radiol. Bras. 2016, 49, 316–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Brown, M.S.; Elashoff, R.; Li, G.; Gjertson, D.W.; Lynch, D.A.; Strollo, D.C.; Kleerup, E.C.; Chong, D.; Shah, S.K.; et al. Quantitative texture-based assessment of one-year changes in fibrotic reticular patterns on HRCT in scleroderma lung disease treated with oral cyclophosphamide. Eur. Radiol. 2011, 21, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Salaffi, F.; Carotti, M.; Bosello, S.; Ciapetti, A.; Gutierrez, M.; Bichisecchi, E.; Giuseppetti, G.; Ferraccioli, G. Computer-Aided Quantification of Interstitial Lung Disease from High Resolution Computed Tomography Images in Systemic Sclerosis: Correlation with Visual Reader-Based Score and Physiologic Tests. BioMed Res. Int. 2015, 2015, 834262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picano, E.; Matucci-Cerinic, M. Unnecessary radiation exposure from medical imaging in the rheumatology patient. Rheumatology 2011, 50, 1537–1539. [Google Scholar] [CrossRef] [Green Version]
- Faletra, F.F.; D’Angeli, I.; Klersy, C.; Averaimo, M.; Klimusina, J.; Pasotti, E.; Pedrazzini, G.B.; Curti, M.; Carraro, C.; DiLiberto, R.; et al. Estimates of lifetime attributable risk of cancer after a single radiation exposure from 64-slice computed tomographic coronary angiography. Heart 2010, 96, 927–932. [Google Scholar] [CrossRef]
- Haaf, K.T.; Jeon, J.; Tammemagi, M.; Han, S.S.; Kong, C.Y.; Plevritis, S.K.; Feuer, E.J.; De Koning, H.J.; Steyerberg, E.W.; Meza, R. Risk prediction models for selection of lung cancer screening candidates: A retrospective validation study. PLoS Med. 2017, 14, e1002277. [Google Scholar] [CrossRef]
- Brenner, D.J. Radiation risks potentially associated with low-dose CT screening of adult smokers for lung cancer. Radiology 2004, 231, 440–445. [Google Scholar] [CrossRef]
- Bach, P.B.; Mirkin, J.; Oliver, T.K.; Azzoli, C.G.; Berry, D.A.; Brawley, O.W.; Byers, T.; Colditz, G.; Gould, M.K.; Jett, J.R.; et al. Benefits and Harms of CT Screening for Lung Cancer. JAMA 2012, 307, 2418–2429. [Google Scholar] [CrossRef] [Green Version]
- Salaffi, F.; Carotti, M.; Di Donato, E.; Di Carlo, M.; Ceccarelli, L.; Giuseppetti, G. Computer-Aided Tomographic Analysis of Interstitial Lung Disease (ILD) in Patients with Systemic Sclerosis (SSc). Correlation with Pulmonary Physiologic Tests and Patient-Centred Measures of Perceived Dyspnea and Functional Disability. PLoS ONE 2016, 11, e0149240. [Google Scholar] [CrossRef]
- Antoniou, K.M.; Trachalaki, A.; Tzouvelekis, A.; Poletti, V.; Vasarmidi, E.; Sfikakis, P.; Bouros, D. A role of antifibrotics in the treatment of Scleroderma-ILD? Pulmonology 2020, 26, 1–2. [Google Scholar] [CrossRef]
- Moore, O.A.; Goh, N.; Corte, T.; Rouse, H.; Hennessy, O.; Thakkar, V.; Byron, J.; Sahhar, J.; Roddy, J.; Gabbay, E.; et al. Extent of disease on high-resolution computed tomography lung is a predictor of decline and mortality in systemic sclerosis-related interstitial lung disease. Rheumatology 2012, 52, 155–160. [Google Scholar] [CrossRef] [Green Version]
- Lynch, D.A. Quantitative CT of Fibrotic Interstitial Lung Disease. Chest 2007, 131, 643–644. [Google Scholar] [CrossRef]
- Lynch, D.A.; Godwin, J.D.; Safrin, S.; Starko, K.M.; Hormel, P.; Brown, K.K.; Raghu, G.; King, T.E., Jr.; Bradford, W.Z.; Schwartz, D.A.; et al. Idiopathic Pulmonary Fibrosis Study Group. High-resolution computed tomography in idiopathic pulmonary fibrosis: Diagnosis and prognosis. Am. J. Respir. Crit. Care Med. 2005, 172, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Tashkin, D.P.; Elashoff, R.; Clements, P.J.; Goldin, J.; Roth, M.D.; Furst, D.E.; Arriola, E.; Silver, R.; Strange, C.; Bolster, M.; et al. Cyclophosphamide versus Placebo in Scleroderma Lung Disease. N. Engl. J. Med. 2006, 354, 2655–2666. [Google Scholar] [CrossRef] [Green Version]
- Collins, C.; Wells, A.; Hansell, D.; Morgan, R.; MacSweeney, J.; Du Bois, R.; Rubens, M. Observer variation in pattern type and extent of disease in fibrosing alveolitis on thin section computed tomography and chest radiography. Clin. Radiol. 1994, 49, 236–240. [Google Scholar] [CrossRef]
- Johkoh, T.; Müller, N.L.; Colby, T.V.; Ichikado, K.; Taniguchi, H.; Kondoh, Y.; Fujimoto, K.; Kinoshita, M.; Arakawa, H.; Yamada, H.; et al. Nonspecific Interstitial Pneumonia: Correlation between Thin-Section CT Findings and Pathologic Subgroups in 55 Patients. Radiology 2002, 225, 199–204. [Google Scholar] [CrossRef]
- Camiciottoli, G.; Orlandi, I.; Bartolucci, M.; Meoni, E.; Nacci, F.; Diciotti, S.; Barcaroli, C.; Conforti, M.L.; Pistolesi, M.; Matucci-Cerinic, M.; et al. Lung CT densitometry in systemic sclerosis: Correlation with lung function, exercise testing, and quality of life. Chest 2007, 131, 672–681. [Google Scholar] [CrossRef] [PubMed]
- Madani, A.; Zanen, J.; De Maertelaer, V.; Gevenois, P.A. Pulmonary Emphysema: Objective Quantification at Multi–Detector Row CT—Comparison with Macroscopic and Microscopic Morphometry. Radiology 2006, 238, 1036–1043. [Google Scholar] [CrossRef] [PubMed]
- Hartley, P.G.; Galvin, J.R.; Hunninghake, G.W.; Merchant, J.A.; Yagla, S.J.; Speakman, S.B.; Schwartz, D.A. High-resolution CT-derived measures of lung density are valid indexes of interstitial lung disease. J. Appl. Physiol. 1994, 76, 271–277. [Google Scholar] [CrossRef]
- Best, A.C.; Lynch, A.M.; Bozic, C.M.; Miller, D.; Grunwald, G.K.; Lynch, D.A. Quantitative CT Indexes in Idiopathic Pulmonary Fibrosis: Relationship with Physiologic Impairment. Radiology 2003, 228, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Kohz, P.; Stäbler, A.; Beinert, T.; Behr, J.; Egge, T.; Heuck, A.; Reiser, M. Reproducibility of quantitative, spirometrically controlled CT. Radiology 1995, 197, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Beinert, T.; Kohz, P.; Seemann, M.; Egge, T.; Reiser, M.; Behr, J. Spirometrically controlled high resolution computed tomography—Quantitative assessment of density distribution in patients with diffuse fibrosing alveolitis. Eur. J. Med. Res. 1996, 1, 269–272. [Google Scholar] [PubMed]
- Lamers, R.J.S.; Kemerink, G.J.; Drent, M.; Van Engelshoven, J.M.A. Reproducibility of spirometrically controlled CT lung densitometry in a clinical setting. Eur. Respir. J. 1998, 11, 942–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kauczor, H.U.; Heussel, C.P.; Fischer, B.; Klamm, R.; Mildenberger, P.; Thelen, M. Assessment of lung volumes using helical CT at inspiration and expiration: Comparison with pulmonary function tests. Am. J. Roentgenol. 1998, 171, 1091–1095. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; van Beek, E.J.; Hwanjo, Y.; Guo, J.; McLennan, G.; Hoffman, E.A. Computer-aided Classification of Interstitial Lung Diseases Via MDCT: 3D Adaptive Multiple Feature Method (3D AMFM). Acad. Radiol. 2006, 13, 969–978. [Google Scholar] [CrossRef]
- Delle Sedie, A.; Doveri, M.; Frassi, F.; Gargani, L.; D’Errico, G.; Pepe, P.; Bazzichi, L.; Riente, L.; Caramella, D.; Bombardieri, S. Ultrasound lung comets in systemic sclerosis: A useful tool to detect lung interstitial fibrosis. Clin. Exp. Rheumatol. 2010, 28, S54. [Google Scholar] [PubMed]
- Gargani, L.; Doveri, M.; D’Errico, L.; Frassi, F.; Bazzichi, M.L.; Sedie, A.D.; Scali, M.C.; Monti, S.; Mondillo, S.; Bombardieri, S.; et al. Ultrasound lung comets in systemic sclerosis: A chest sonography hallmark of pulmonary interstitial fibrosis. Rheumatology 2009, 48, 1382–1387. [Google Scholar] [CrossRef] [Green Version]
- Gigante, A.; Fanelli, F.R.; Lucci, S.; Barilaro, G.; Quarta, S.; Barbano, B.; Giovannetti, A.; Amoroso, A.; Rosato, E. Lung ultrasound in systemic sclerosis: Correlation with high-resolution computed tomography, pulmonary function tests and clinical variables of disease. Intern. Emerg. Med. 2015, 11, 213–217. [Google Scholar] [CrossRef] [Green Version]
- Tardella, M.; Di Carlo, M.; Carotti, M.; Filippucci, E.; Grassi, W.; Salaffi, F. Ultrasound B-lines in the evaluation of interstitial lung disease in patients with systemic sclerosis. Medicine 2018, 97, e0566. [Google Scholar] [CrossRef]
- Vizioli, L.; Ciccarese, F.; Forti, P.; Chiesa, A.M.; Giovagnoli, M.; Mughetti, M.; Zompatori, M.; Zoli, M. Integrated Use of Lung Ultrasound and Chest X-Ray in the Detection of Interstitial Lung Disease. Respiration 2017, 93, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Pinal-Fernandez, I.; Pallisa-Nuñez, E.; Selva-O’Callaghan, A.; Castella-Fierro, E.; SimeOn-Aznar, C.P.; Fonollosa-Pla, V.; Vilardell-Tarres, M. Pleural irregularity, a new ultrasound sign for the study of interstitial lung disease in systemic sclerosis and antisynthetase syndrome. Clin. Exp. Rheumatol. 2015, 33, S136–S141. [Google Scholar] [PubMed]
- Barskova, T.; Gargani, L.; Guiducci, S.; Randone, S.B.; Bruni, C.; Carnesecchi, G.; Conforti, M.L.; Porta, F.; Pignone, A.; Caramella, D.; et al. Lung ultrasound for the screening of interstitial lung disease in very early systemic sclerosis. Ann. Rheum. Dis. 2012, 72, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Montesi, S.B.; Caravan, P. Novel Imaging Approaches in Systemic Sclerosis-Associated Interstitial Lung Disease. Curr. Rheumatol. Rep. 2019, 21, 25. [Google Scholar] [CrossRef] [PubMed]
- Pinal-Fernandez, I.; Pineda-Sanchez, V.; Pallisa-Nuñez, E.; Simeon-Aznar, C.P.; Selva-O’Callaghan, A.; Fonollosa-Pla, V.; Vilardell-Tarres, M. Fast 1.5 T chest MRI for the assessment of interstitial lung disease extent secondary to systemic sclerosis. Clin. Rheumatol. 2016, 35, 2339–2345. [Google Scholar] [CrossRef] [PubMed]
- Montesi, S.B.; Désogère, P.; Fuchs, B.C.; Caravan, P. Molecular imaging of fibrosis: Recent advances and future directions. J. Clin. Investig. 2019, 129, 24–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, S.L.; Devaraj, A.; Enghelmayer, J.I.; Kishi, K.; Silva, R.S.; Patel, N.; Rossman, M.D.; Valenzuela, C.; Vancheri, C. Role of imaging in progressive-fibrosing interstitial lung diseases. Eur. Respir. Rev. 2018, 27, 180073. [Google Scholar] [CrossRef]
- Cottin, V.; Brown, K.K. Interstitial lung disease associated with systemic sclerosis (SSc-ILD). Respir. Res. 2019, 20, 13. [Google Scholar] [CrossRef]
- Ruaro, B.; Bruni, C.; Wade, B.; Baratella, E.; Confalonieri, P.; Antonaglia, C.; Geri, P.; Biolo, M.; Confalonieri, M.; Salton, F. Laser Speckle Contrast Analysis: Functional Evaluation of Microvascular Damage in Connective Tissue Diseases. Is There Evidence of Correlations with Organ Involvement, Such as Pulmonary Damage? Front. Physiol. 2021, 12, 1759. [Google Scholar] [CrossRef]
- Konopka, K.E.; Myers, J.L. Interstitial lung disease pathology in systemic sclerosis. Ther. Adv. Musculoskelet. Dis. 2021, 13. [Google Scholar] [CrossRef]
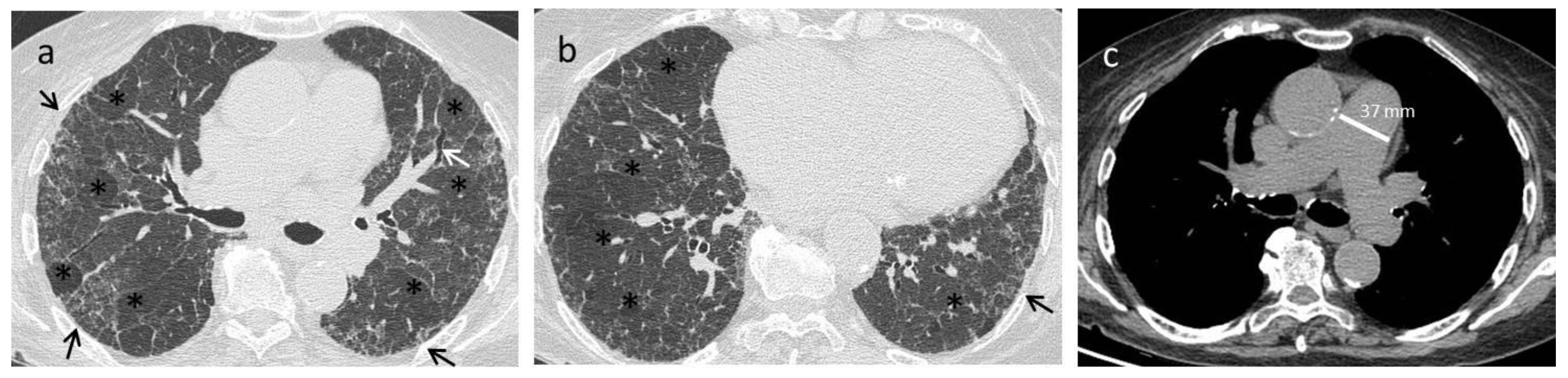


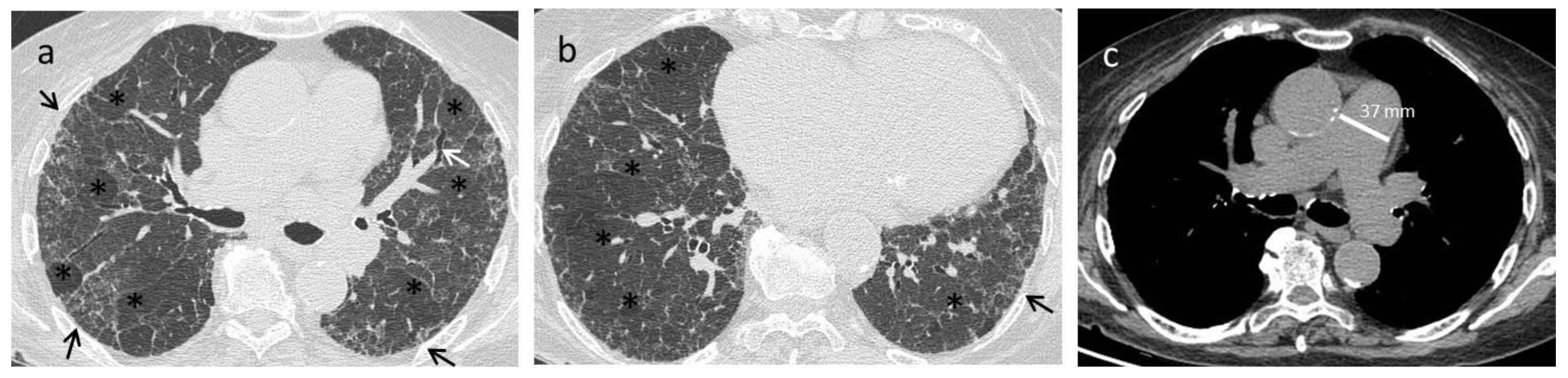
{kind=link}
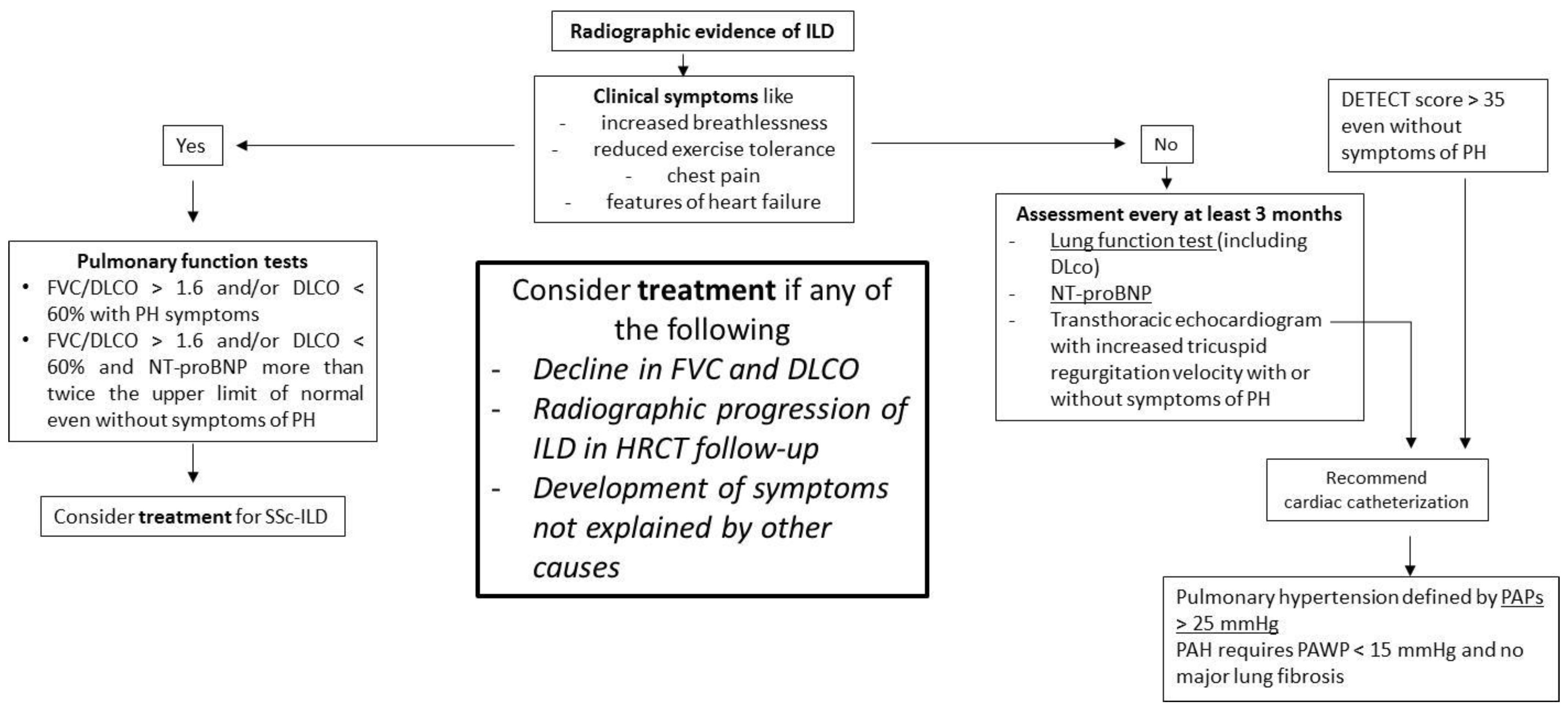
{kind=link}
{kind=link}
{kind=link}
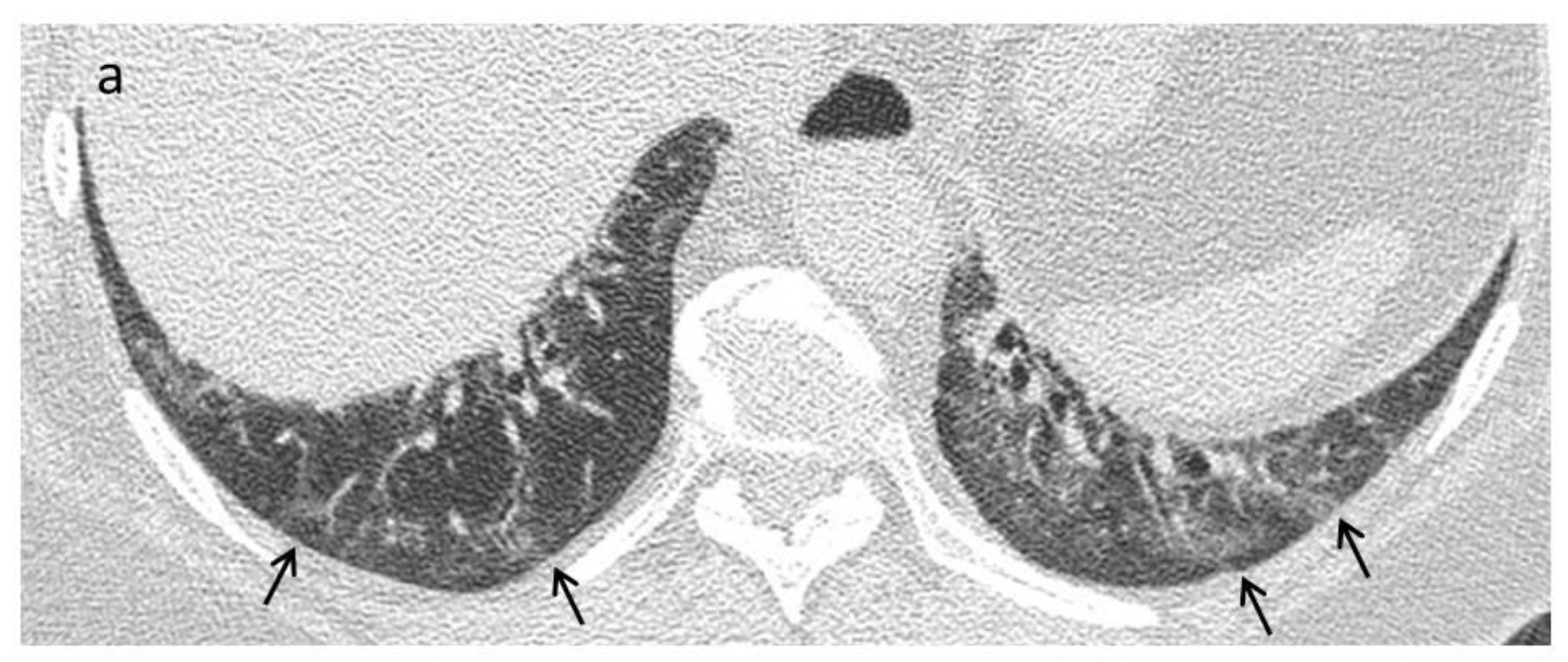
{kind=link}
{kind=link}
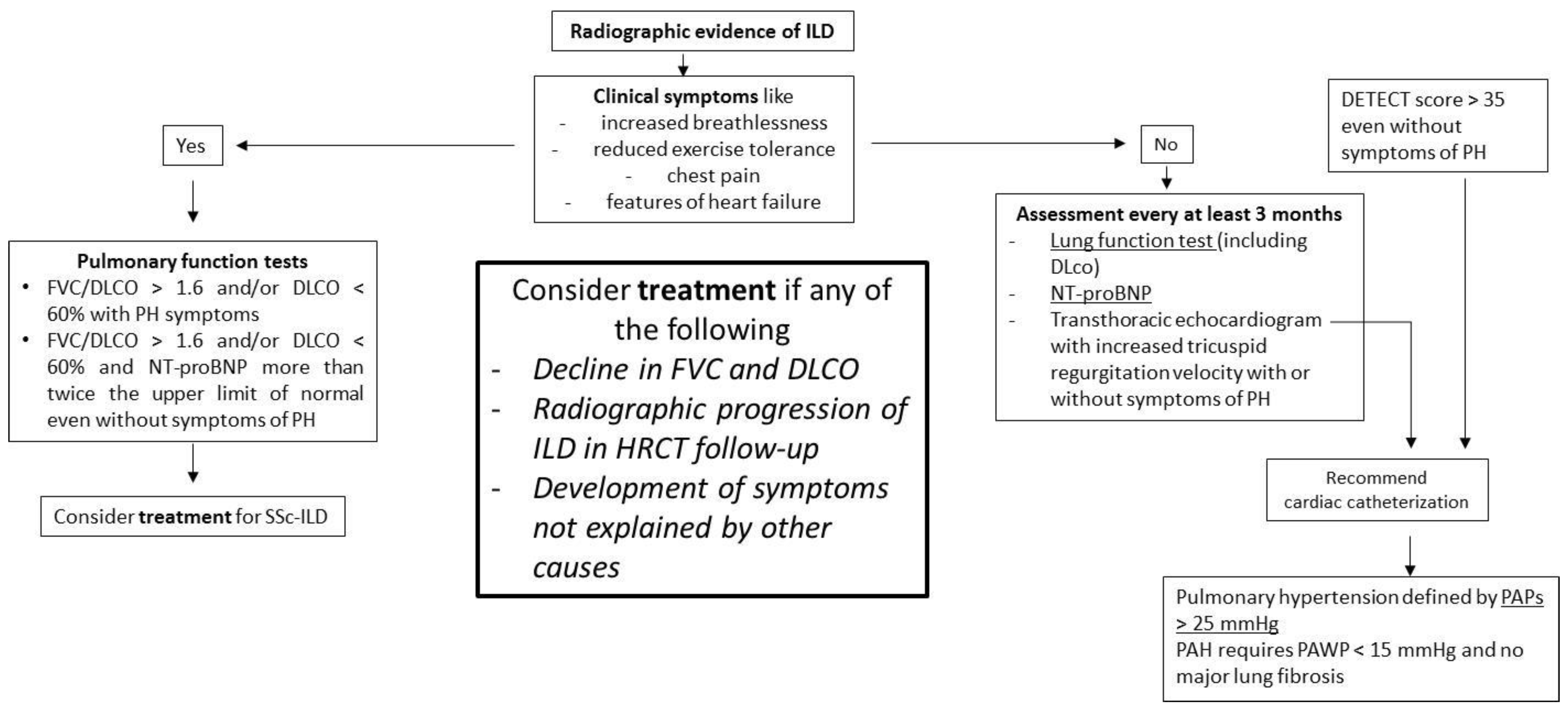
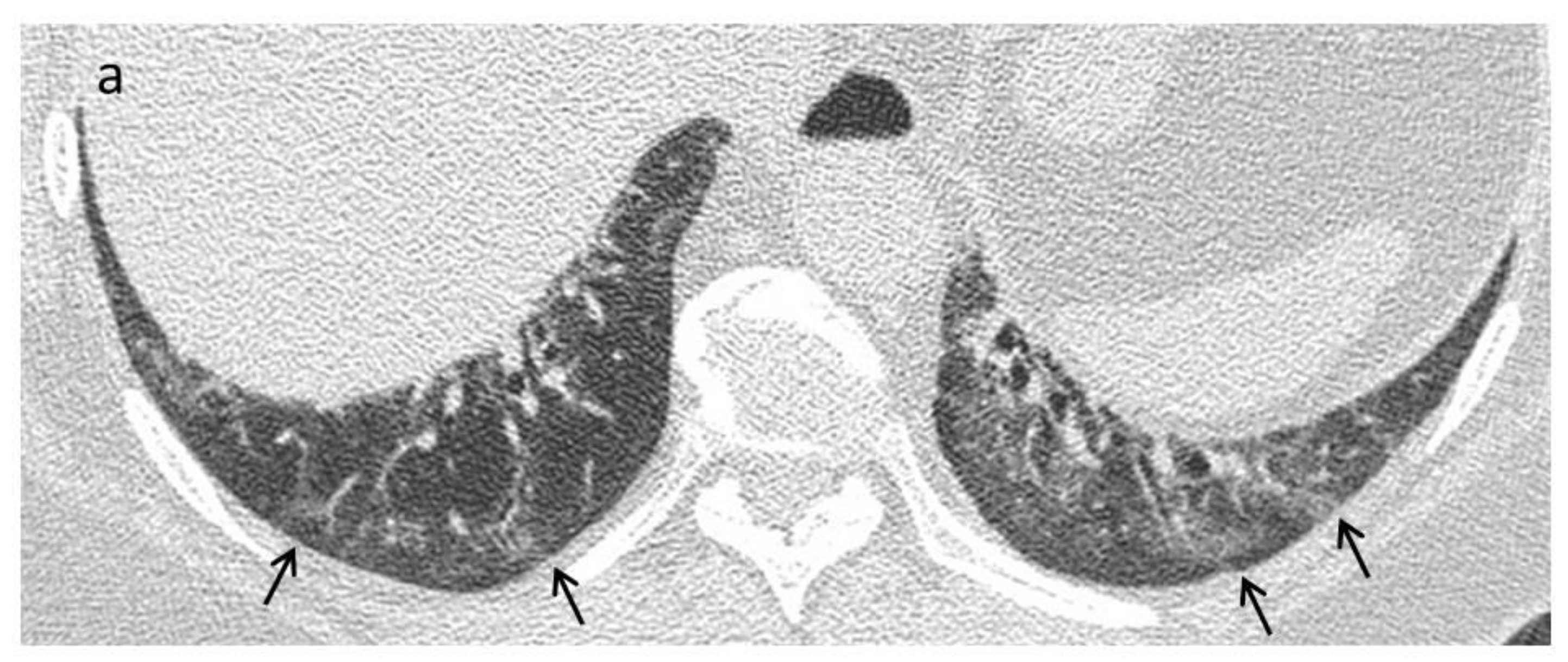
| Early stage | NSIP pattern:
|
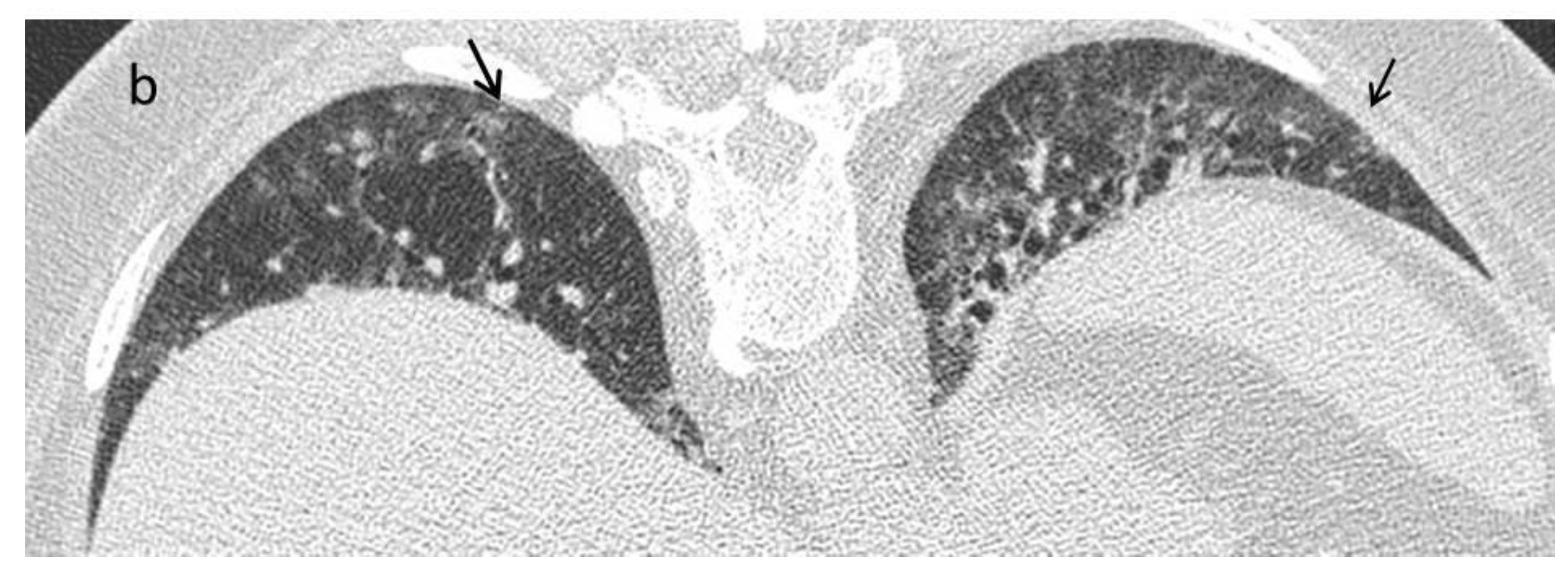
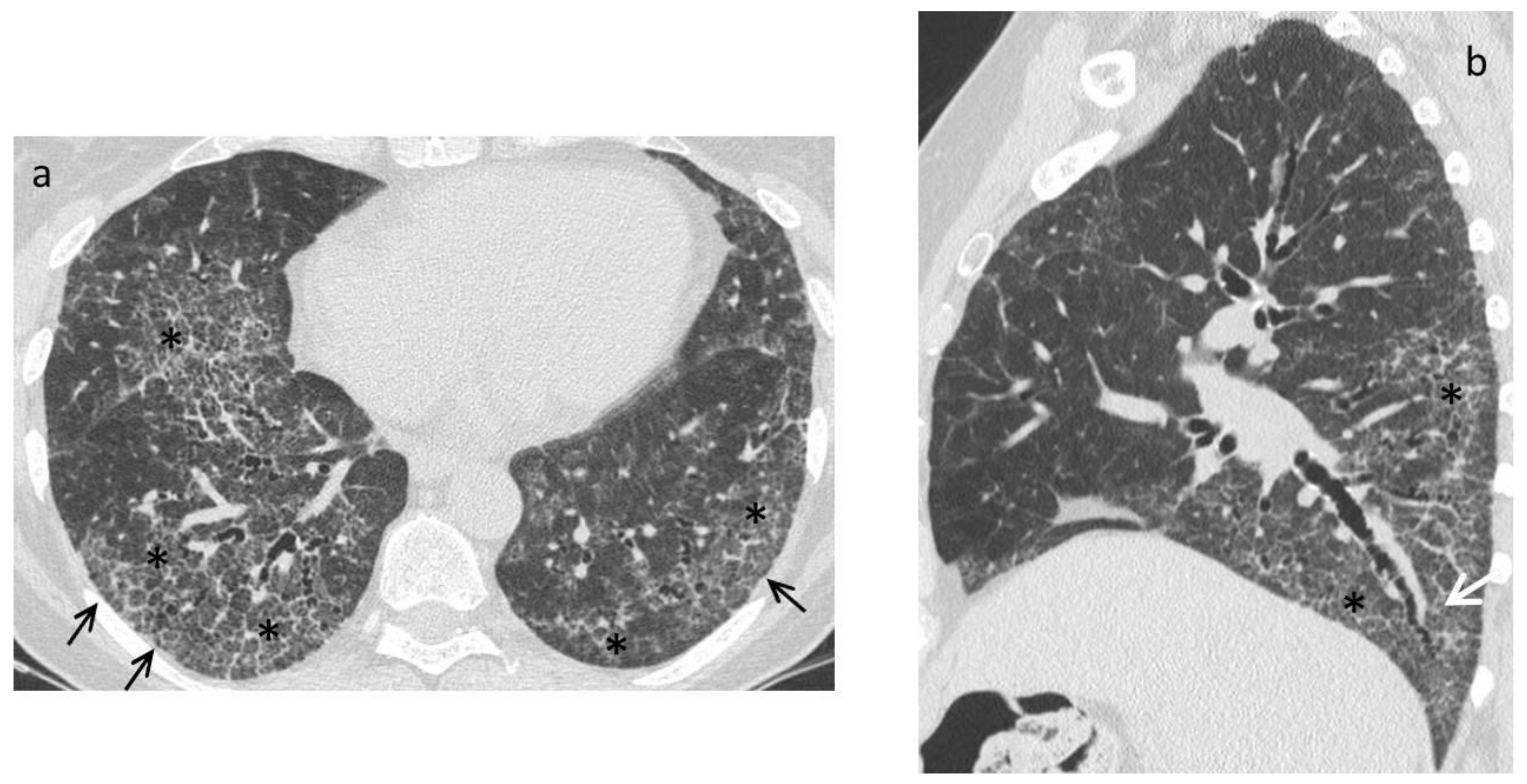
| Late fibrotic stage | Fibrotic NSIP/UIP pattern:
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruaro, B.; Baratella, E.; Confalonieri, P.; Wade, B.; Marrocchio, C.; Geri, P.; Busca, A.; Pozzan, R.; Andrisano, A.G.; Cova, M.A.; et al. High-Resolution Computed Tomography: Lights and Shadows in Improving Care for SSc-ILD Patients. Diagnostics 2021, 11, 1960. https://doi.org/10.3390/diagnostics11111960
Ruaro B, Baratella E, Confalonieri P, Wade B, Marrocchio C, Geri P, Busca A, Pozzan R, Andrisano AG, Cova MA, et al. High-Resolution Computed Tomography: Lights and Shadows in Improving Care for SSc-ILD Patients. Diagnostics. 2021; 11(11):1960. https://doi.org/10.3390/diagnostics11111960
Chicago/Turabian StyleRuaro, Barbara, Elisa Baratella, Paola Confalonieri, Barbara Wade, Cristina Marrocchio, Pietro Geri, Annalisa Busca, Riccardo Pozzan, Alessia Giovanna Andrisano, Maria Assunta Cova, and et al. 2021. "High-Resolution Computed Tomography: Lights and Shadows in Improving Care for SSc-ILD Patients" Diagnostics 11, no. 11: 1960. https://doi.org/10.3390/diagnostics11111960
APA StyleRuaro, B., Baratella, E., Confalonieri, P., Wade, B., Marrocchio, C., Geri, P., Busca, A., Pozzan, R., Andrisano, A. G., Cova, M. A., Confalonieri, M., & Salton, F. (2021). High-Resolution Computed Tomography: Lights and Shadows in Improving Care for SSc-ILD Patients. Diagnostics, 11(11), 1960. https://doi.org/10.3390/diagnostics11111960