Spontaneous and Induced Animal Models for Cancer Research

 ,
,  , ,
, ,  ,
,  , ,
, , 
Abstract
1. Introduction
2. Animal Behavior Research in Oncology
3. Mouse Models Data Bases
4. Genetically Induced Cancer Models
4.1. Methods for Generation of Transgenic Mice
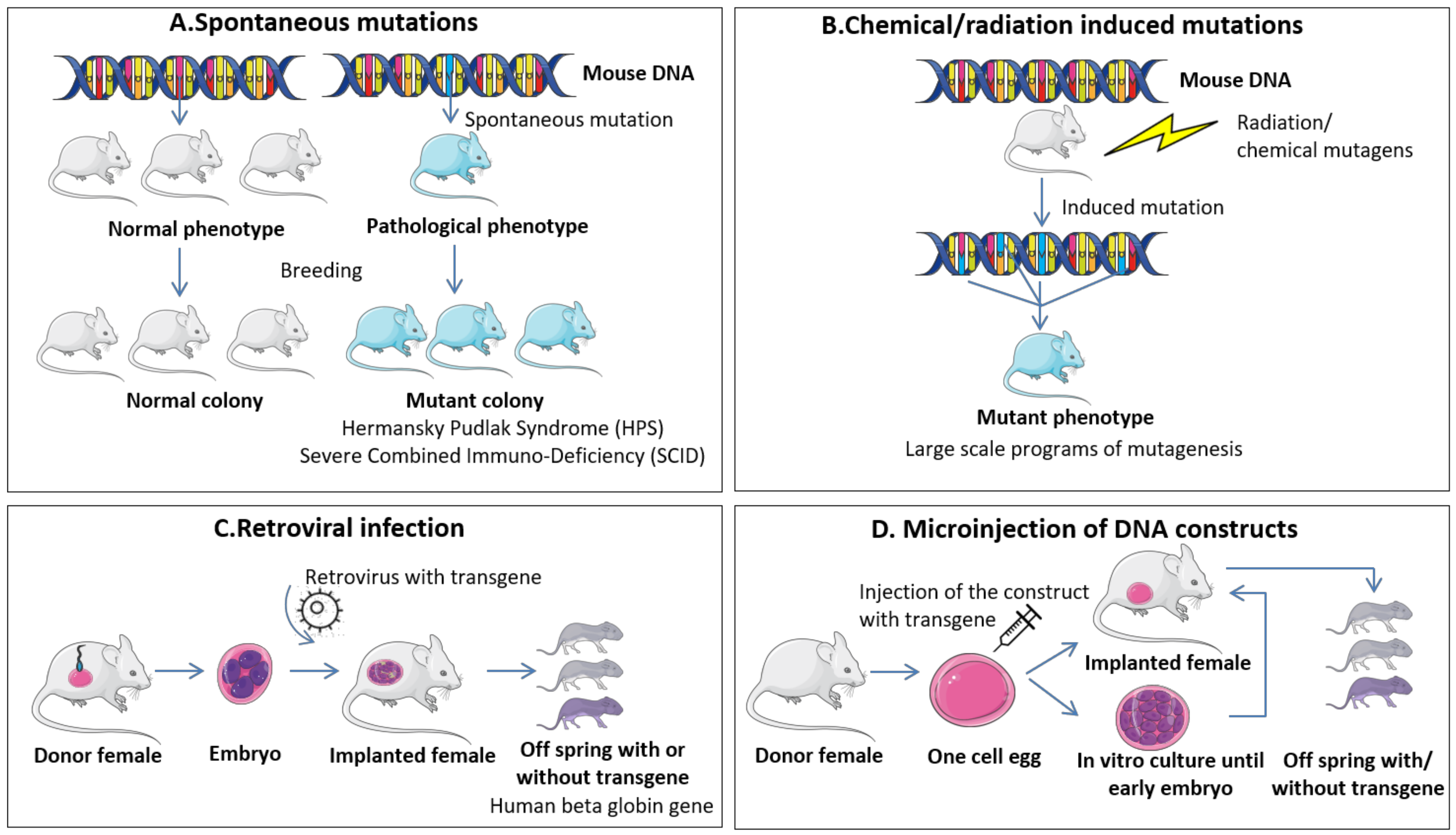
4.1.1. Spontaneous Mutations and Chemical/Radiation Induced Mutations
4.1.2. Retroviral Infection
4.1.3. Microinjection of DNA Constructs
4.1.4. “Gene-Targeted Transgene” Method
4.2. Models of Transgenic Mice in Concordance with the Type of Gene Modification
4.3. Next Generation Mouse Models for Cancer Research
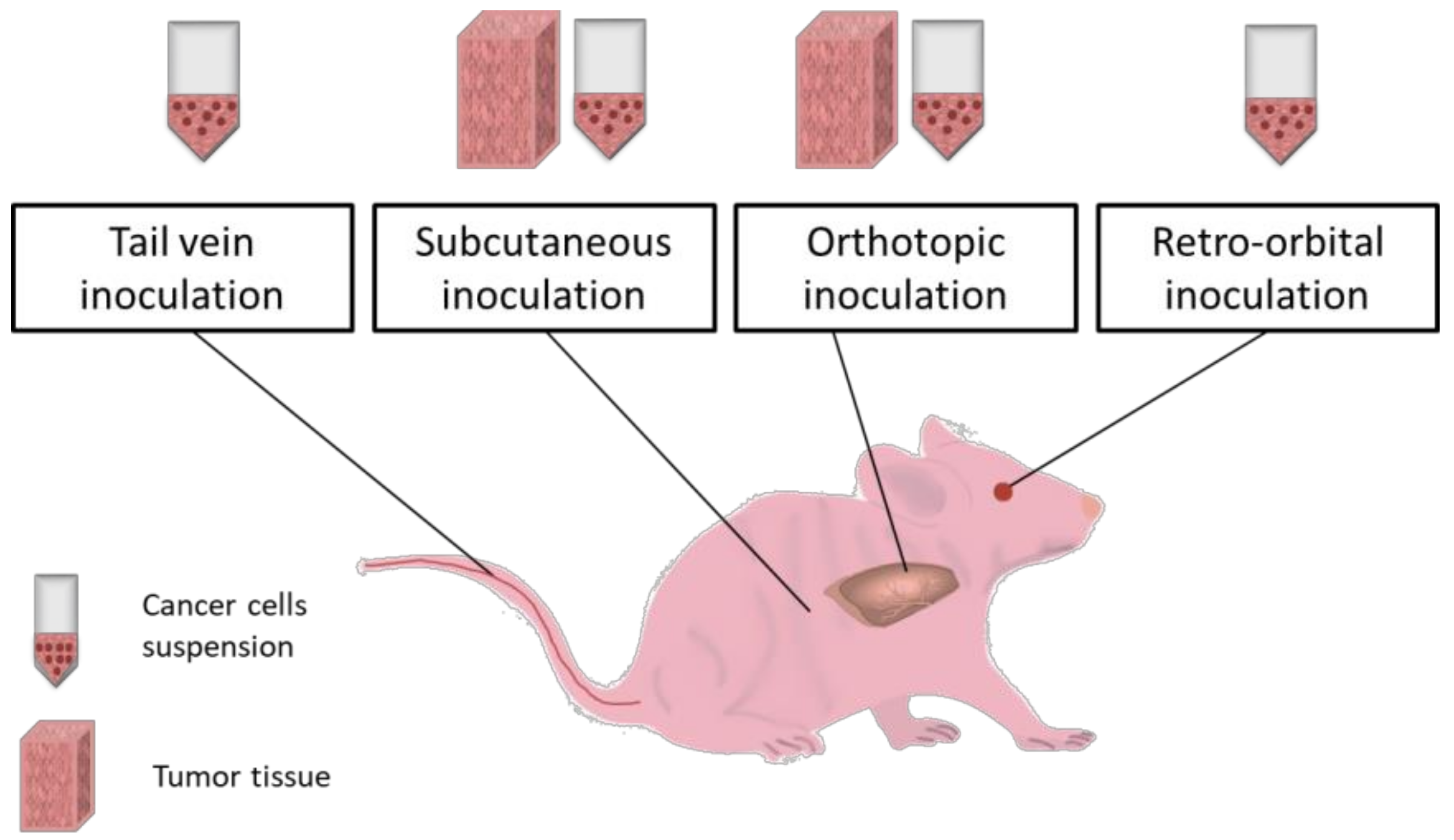
5. Microsurgical Induced Cancer Models
5.1. Subcutaneous Inoculation
5.2. Orthotopic Implantation
5.3. Intraperitoneal Inoculation
5.4. Intravenous Inoculation
5.5. Retro-Orbital Inoculation
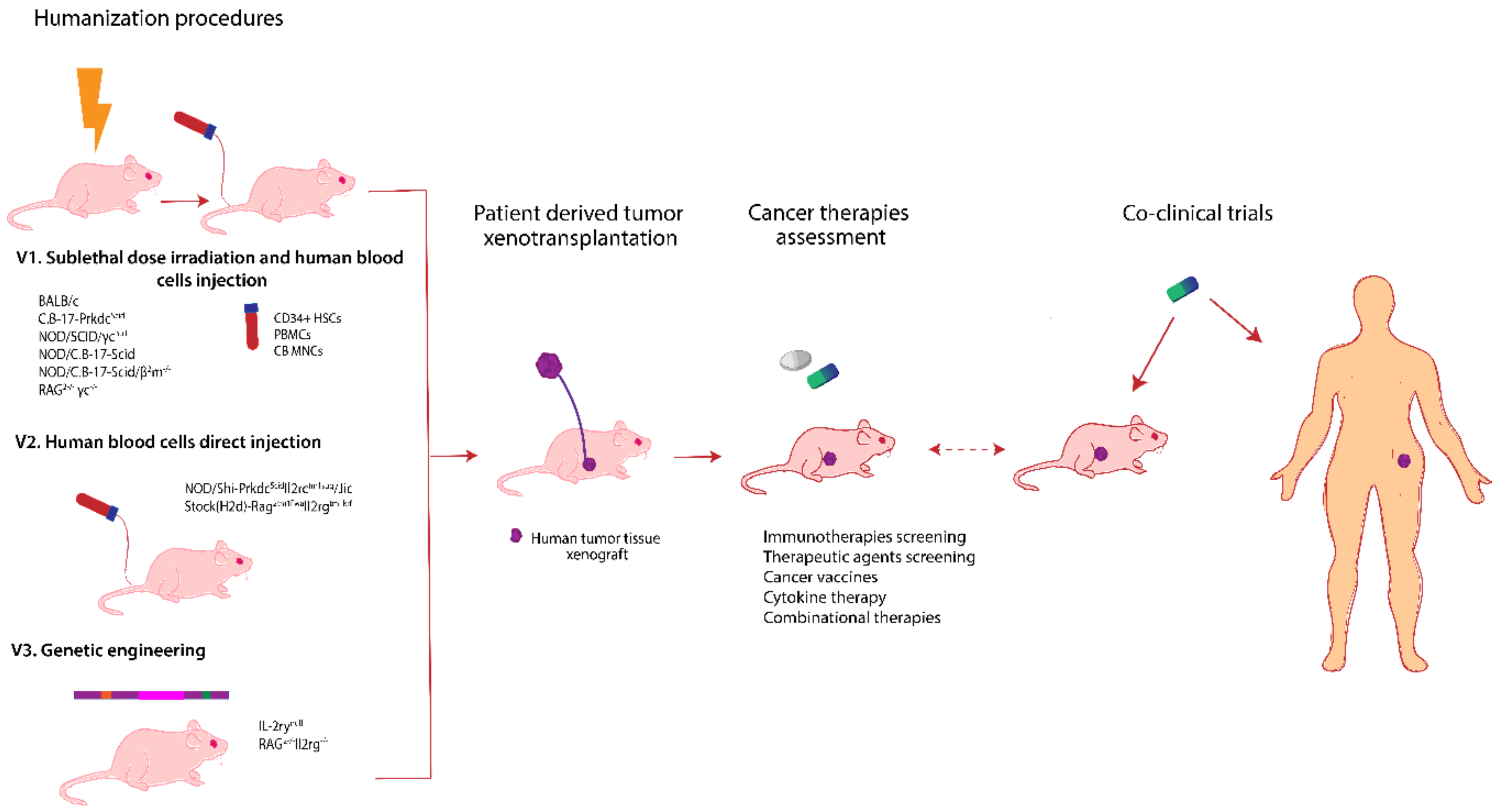
6. Avatar Mouse Models for Personalized Cancer Therapy
6.1. Patient Derived Xenograft Models
6.2. Humanized Mice Models
7. Cancer Metabolism and Animal Models
8. Spontaneous Large Animal Models for Cancer Research
9. Conclusions
Funding
Conflicts of Interest
References
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Berindan-Neagoe, I.; Calin, G.A. Molecular pathways: microRNAs, cancer cells, and microenvironment. Clin. Cancer Res. 2014, 20, 6247–6253. [Google Scholar] [CrossRef] [PubMed]
- Tratar, U.L.; Horvat, S.; Cemazar, M. Transgenic Mouse Models in Cancer Research. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef]
- Workman, P.; An Ad Hoc Committee of the National Cancer Research Institute; Aboagye, E.O.; Balkwill, F.; Balmain, A.; Bruder, G.; Chaplin, D.J.; Double, J.A.; Everitt, J.; Farningham, D.A.H.; et al. Guidelines for the welfare and use of animals in cancer research. Br. J. Cancer 2010, 102, 1555–1577. [Google Scholar] [CrossRef]
- Cheon, D.-J.; Orsulic, S. Mouse Models of Cancer. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 95–119. [Google Scholar] [CrossRef]
- Rudolph, A.; Chang-Claude, J.; Schmidt, M.K. Gene–environment interaction and risk of breast cancer. Br. J. Cancer. 2016, 114, 125–133. [Google Scholar] [CrossRef]
- Samuel, W.; Lovell Jones, C.C.; Hanna, K. Cancer and the Environment: Gene-Environment Interactions; National Academies Press: Washington, DC, USA, 2002. [Google Scholar]
- King, W.D.; Friedenreich, C.M.; Brenner, D.R.; De, P.; Demers, P.A.; Hystad, P.; Nutall, R.; Villeneuve, P.J.; Walter, S.D. The contribution of lifestyle, environment, genetics and chance to cancer risk in individuals and populations. Prev. Med. 2015, 76, 132–134. [Google Scholar] [CrossRef]
- Zimta, A.-A.; Schitcu, V.; Gurzau, E.; Stavaru, C.; Manda, G.; Szedlacsek, S.; Berindan-Neagoe, I. Biological and molecular modifications induced by cadmium and arsenic during breast and prostate cancer development. Environ. Res. 2019, 178, 108700. [Google Scholar] [CrossRef]
- Yee, N.S.; Ignatenko, N.; Finnberg, N.; Lee, N.; Stairs, D. Animal Models of Cancer Biology. Cancer Growth Metastasis 2015, 8, 115–118. [Google Scholar] [CrossRef][Green Version]
- Olson, B.; Li, Y.; Lin, Y.; Liu, E.T.; Patnaik, A. Mouse Models for Cancer Immunotherapy Research. Cancer Discov. 2018, 8, 1358–1365. [Google Scholar] [CrossRef] [PubMed]
- Cekanova, M.; Rathore, K. Animal models and therapeutic molecular targets of cancer: Utility and limitations. Drug Des. Dev. Ther. 2014, 8, 1911–1922. [Google Scholar] [CrossRef] [PubMed]
- Klein, W.M.P.; Bloch, M.; Hesse, B.; McDonald, P.G.; Nebeling, L.; O’Connell, M.E.; Riley, W.T.; Taplin, S.H.; Tesauro, G. Behavioral research in cancer prevention and control: A look to the future. Am. J. Prev. Med. 2014, 46, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Van Meer, P.; Raber, J. Mouse behavioural analysis in systems biology. Biochem. J. 2005, 389, 593–610. [Google Scholar] [CrossRef] [PubMed]
- Chesler, E.J.; Wilson, S.G.; Lariviere, W.R.; Rodriguez-Zas, S.L.; Mogil, J.S. Influences of laboratory environment on behavior. Nat. Neurosci. 2002, 5, 1101–1102. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Smith, M.; Lutgendorf, S.K.; Sood, A.K. Impact of stress on cancer metastasis. Future Oncol. 2010, 6, 1863–1881. [Google Scholar] [CrossRef]
- Nukina, H.; Sudo, N.; Aiba, Y.; Oyama, N.; Koga, Y.; Kubo, C. Restraint stress elevates the plasma interleukin-6 levels in germ-free mice. J. Neuroimmunol. 2001, 115, 46–52. [Google Scholar] [CrossRef]
- Steplewski, Z.; Goldman, P.R.; Vogel, W.H. Effect of housing stress on the formation and development of tumors in rats. Cancer Lett. 1987, 34, 257–261. [Google Scholar] [CrossRef]
- Lelekakis, M.; Moseley, J.M.; Martin, T.J.; Hards, D.; Williams, E.D.; Ho, P.; Lowen, D.; Javni, J.; Miller, F.R.; Slavin, J.; et al. A novel orthotopic model of breast cancer metastasis to bone. Clin. Exp. Metastasis 1999, 17, 163–170. [Google Scholar] [CrossRef]
- Marsland, A.L.; Prather, A.A.; Petersen, K.L.; Cohen, S.; Manuck, S.B. Antagonistic characteristics are positively associated with inflammatory markers independently of trait negative emotionality. Brain Behav. Immun. 2008, 22, 753–761. [Google Scholar] [CrossRef]
- Nelson, R.J. Biology of Aggression; Oxford University Press: New York, NY, USA, 2005. [Google Scholar]
- Takahashi, A.; Miczek, K.A. Neurogenetics of Aggressive Behavior: Studies in Rodents. Curr. Top. Behav. Neurosci. 2013, 17, 3–44. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Huysentruyt, L.C. On the origin of cancer metastasis. Crit. Rev. Oncog. 2013, 18, 43–73. [Google Scholar] [CrossRef] [PubMed]
- Lutgendorf, S.K.; Johnsen, E.L.; Cooper, B.; Anderson, B.; Sorosky, J.I.; E Buller, R.; Sood, A.K. Vascular endothelial growth factor and social support in patients with ovarian carcinoma. Cancer 2002, 95, 808–815. [Google Scholar] [CrossRef] [PubMed]
- Turner-Cobb, J.M.; Sephton, S.E.; Koopman, C.; Blake-Mortimer, J.; Spiegel, D. Social support and salivary cortisol in women with metastatic breast cancer. Psychosom. Med. 2000, 62, 337–345. [Google Scholar] [CrossRef]
- Seeman, T.E.; McEwen, B.S. Impact of social environment characteristics on neuroendocrine regulation. Psychosom. Med. 1996, 58, 459–471. [Google Scholar] [CrossRef]
- Fredriksson, J.M.; Lindquist, J.M.; Bronnikov, G.E.; Nedergaard, J. Norepinephrine Induces Vascular Endothelial Growth Factor Gene Expression in Brown Adipocytes through a β-Adrenoreceptor/cAMP/Protein Kinase A Pathway Involving Src but Independently of Erk1/2. J. Biol. Chem. 2000, 275, 13802–13811. [Google Scholar] [CrossRef]
- Thaker, P.H.; Han, L.Y.; A Kamat, A.; Arevalo, J.M.; Takahashi, R.; Lu, C.; Jennings, N.B.; Armaiz-Pena, G.N.; Bankson, J.A.; Ravoori, M.; et al. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat. Med. 2006, 12, 939–944. [Google Scholar] [CrossRef]
- Pyter, L.M.; Pineros, V.; Galang, J.A.; McClintock, M.K.; Prendergast, B.J. Peripheral tumors induce depressive-like behaviors and cytokine production and alter hypothalamic-pituitary-adrenal axis regulation. Proc. Natl. Acad. Sci. USA 2009, 106, 9069–9074. [Google Scholar] [CrossRef]
- Sharma, A.; Sethi, G.; Tambuwala, M.M.; Aljabali, A.A.A.; Chellappan, D.K.; Dua, K.; Goyal, R. Circadian rhythm disruption and Alzheimer’s disease: The dynamics of a vicious cycle. Curr. Neuropharmacol. 2020, 18, 1–19. [Google Scholar] [CrossRef]
- Sephton, S.E.; Spiegel, D. Circadian disruption in cancer: A neuroendocrine-immune pathway from stress to disease? Brain, Behav. Immun. 2003, 17, 321–328. [Google Scholar] [CrossRef]
- Liu, S.; Madu, C.O.; Lu, Y. The Role of Melatonin in Cancer Development. Oncomedicine 2018, 3, 37–47. [Google Scholar] [CrossRef]
- Blask, D.E.; Dauchy, R.T.; Sauer, L.A. Putting Cancer to Sleep at Night: The Neuroendocrine/Circadian Melatonin Signal. Endocrine 2005, 27, 179–188. [Google Scholar] [CrossRef]
- Blask, D.E. Melatonin, sleep disturbance and cancer risk. Sleep Med. Rev. 2009, 13, 257–264. [Google Scholar] [CrossRef] [PubMed]
- McWhir, J.; Schnieke, A.; Ansell, R.; Wallace, H.; Colman, A.; Scott, A.R.; Kind, A.J. Selective ablation of differentiated cells permits isolation of embryonic stem cell lines from murine embryos with a non–permissive genetic background. Nat. Genet. 1996, 14, 223–226. [Google Scholar] [CrossRef]
- Koehl, M.; Battle, S.E.; Turek, F.W. Sleep in female mice: A strain comparison across the estrous cycle. Sleep 2003, 26, 267–272. [Google Scholar] [CrossRef]
- Gao, J.; Cheon, K.; Nusinowitz, S.; Liu, Q.; Bei, D.; Atkins, K.; Azimi, A.; Daiger, S.P.; Farber, D.B.; Heckenlively, J.R.; et al. Progressive photoreceptor degeneration, outer segment dysplasia, and rhodopsin mislocalization in mice with targeted disruption of the retinitis pigmentosa-1 (Rp1) gene. Proc. Natl. Acad. Sci. USA 2002, 99, 5698–5703. [Google Scholar] [CrossRef] [PubMed]
- Reeb-Whitaker, C.K.; Paigen, B.; Beamer, W.G.; Bronson, R.T.; Churchill, G.A.; Schweitzer, I.B.; Myers, D.D. The impact of reduced frequency of cage changes on the health of mice housed in ventilated cages. Lab. Anim. 2001, 35, 58–73. [Google Scholar] [CrossRef]
- Shimamura, M.; Kuratani, K.; Kinoshita, M. A new automated and high-throughput system for analysis of the forced swim test in mice based on magnetic field changes. J. Pharmacol. Toxicol. Methods 2007, 55, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Krupke, D.M.; Bult, C.J.; Näf, D.; Sundberg, J.P.; Eppig, J.T. Electronic access to data from mouse cancer models: The Mouse Tumor Biology database. Nat. Genet. 2001, 27, 65–66. [Google Scholar] [CrossRef][Green Version]
- Betty Tarnowski, S.; Guruswami, U.; Wagner, D.; George, S.; Pandya, M.; Piparo, N.; Schroedl, J.; Hadfield, C.; Marks, M.H. Improvements and additions to caMOD: Cancer Models Database. Cancer Res. 2007, 67, 3864. [Google Scholar]
- Schofield, P.N.; Gruenberger, M.; Sundberg, J.P.; Gruenberger, M. Pathbase and the MPATH Ontology. Vet. Pathol. 2010, 47, 1016–1020. [Google Scholar] [CrossRef] [PubMed]
- Grouse, L.H.; Greenhut, S.F.; Jia, L.; Gerhard, D.S.; Cgap Research Team. The cancer genome anatomy project: Web-based analysis tools for deciphering the molecular expression profiles of cancer. Cancer Res. 2004, 64, 1110–1111. [Google Scholar]
- Eppig, J.T.; Motenko, H.; Richardson, J.E.; Richards-Smith, B.; Smith, C. The International Mouse Strain Resource (IMSR): Cataloging worldwide mouse and ES cell line resources. Mamm. Genome 2015, 26, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Fuentes, V.; The IMPC Consortium; Cacheiro, P.; Meehan, T.F.; Aguilar-Pimentel, J.A.; Brown, S.D.M.; Flenniken, A.M.; Flicek, P.; Galli, A.; Mashhadi, H.H.; et al. The International Mouse Phenotyping Consortium (IMPC): A functional catalogue of the mammalian genome that informs conservation. Conserv. Genet. 2018, 19, 995–1005. [Google Scholar] [CrossRef]
- LAMHDI. The search for animal models. Lab Anim. 2014, 43, 236. [Google Scholar] [CrossRef]
- Eppig, J.T. Mouse Genome Informatics (MGI) Resource: Genetic, Genomic, and Biological Knowledgebase for the Laboratory Mouse. ILAR J. 2017, 58, 17–41. [Google Scholar] [CrossRef]
- Aidinis, V.; Chandras, C.; Manoloukos, M.; Thanassopoulou, A.; Kranidioti, K.; Armaka, M.; Douni, E.; Kontoyiannis, D.L.; Zouberakis, M.; Kollias, G.; et al. MUGEN mouse database; Animal models of human immunological diseases. Nucleic Acids Res. 2007, 36, D1048–D1054. [Google Scholar] [CrossRef]
- Cacheiro, P.; The International Mouse Phenotyping Consortium and the Monarch Initiative; Haendel, M.; Smedley, D.; Meehan, T. New models for human disease from the International Mouse Phenotyping Consortium. Mamm. Genome 2019, 30, 143–150. [Google Scholar] [CrossRef]
- Kinoshita, J.; Clark, T. Alzforum. Breast Cancer 2007, 401, 365–381. [Google Scholar]
- Banerjee-Basu, S.; Packer, A. SFARI Gene: An evolving database for the autism research community. Dis. Model. Mech. 2010, 3, 133–135. [Google Scholar] [CrossRef]
- House, C.D.; Hernandez, L.F.; Annunziata, C.M. Recent Technological Advances in Using Mouse Models to Study Ovarian Cancer. Front. Oncol. 2014, 4, 26. [Google Scholar] [CrossRef]
- Smith, H.W.; Muller, W.J. Transgenic Mouse Models--A Seminal Breakthrough in Oncogene Research. Cold Spring Harb. Protoc. 2013, 2013, 17–26. [Google Scholar] [CrossRef]
- Hanahan, D.; Wagner, E.F.; Palmiter, R.D. The origins of oncomice: A history of the first transgenic mice genetically engineered to develop cancer. Genes Dev. 2007, 21, 2258–2270. [Google Scholar] [CrossRef] [PubMed]
- Rülicke, T.; Montagutelli, X.; Pintado, B.; Thon, R.; Hedrich, H.J. FELASA guidelines for the production and nomenclature of transgenic rodents. Lab. Anim. 2007, 41, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Swank, R.T.; Novak, E.K.; McGarry, M.P.; Rusiniak, M.E.; Feng, L. Mouse Models of Hermansky Pudlak Syndrome: A Review. Pigment. Cell Res. 1998, 11, 60–80. [Google Scholar] [CrossRef] [PubMed]
- Bosma, G.C.; Custer, R.P.; Bosma, M.J. A severe combined immunodeficiency mutation in the mouse. Nature 1983, 301, 527–530. [Google Scholar] [CrossRef]
- Stanford, W.L.; Cohn, J.B.; Cordes, S.P. Gene-trap mutagenesis: Past, present and beyond. Nat. Rev. Genet. 2001, 2, 756–768. [Google Scholar] [CrossRef] [PubMed]
- Doyle, A.; McGarry, M.P.; Lee, N.A.; Lee, J.J. The construction of transgenic and gene knockout/knockin mouse models of human disease. Transgenic Res. 2011, 21, 327–349. [Google Scholar] [CrossRef]
- Russell, W.L.; Kelly, E.M.; Hunsicker, P.R.; Bangham, J.W.; Maddux, S.C.; Phipps, E.L. Specific-locus test shows ethylnitrosourea to be the most potent mutagen in the mouse. Proc. Natl. Acad. Sci. USA 1979, 76, 5818–5819. [Google Scholar] [CrossRef]
- Soriano, P.; Cone, R.; Mulligan, R.; Jaenisch, R. Tissue-specific and ectopic expression of genes introduced into transgenic mice by retroviruses. Science 1986, 234, 1409–1413. [Google Scholar] [CrossRef]
- Lois, C.; Hong, E.J.; Pease, S.; Brown, E.J.; Baltimore, D. Germline Transmission and Tissue-Specific Expression of Transgenes Delivered by Lentiviral Vectors. Science 2002, 295, 868–872. [Google Scholar] [CrossRef] [PubMed]
- Costantini, F.; Lacy, E. Introduction of a rabbit β-globin gene into the mouse germ line. Nature 1981, 294, 92–94. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.W.; Ruddle, F.H. Integration and stable germ line transmission of genes injected into mouse pronuclei. Science 1981, 214, 1244–1246. [Google Scholar] [CrossRef] [PubMed]
- Harbers, K.; Jähner, D.; Jaenisch, R. Microinjection of cloned retroviral genomes into mouse zygotes: Integration and expression in the animal. Nature 1981, 293, 540–542. [Google Scholar] [CrossRef]
- Wagner, E.F.; Stewart, T.A.; Mintz, B. The human beta-globin gene and a functional viral thymidine kinase gene in developing mice. Proc. Natl. Acad. Sci. USA 1981, 78, 5016–5020. [Google Scholar] [CrossRef]
- Wagner, T.E.; Hoppe, P.C.; Jollick, J.D.; Scholl, D.R.; Hodinka, R.L.; Gault, J.B. Microinjection of a rabbit beta-globin gene into zygotes and its subsequent expression in adult mice and their offspring. Proc. Natl. Acad. Sci. USA 1981, 78, 6376–6380. [Google Scholar] [CrossRef]
- DeMayo, J.L.; Wang, J.; Liang, D.; Zhang, R.; DeMayo, F.J. Genetically Engineered Mice by Pronuclear DNA microinjection. Curr. Protoc. Mouse Biol. 2012, 2, 245–262. [Google Scholar] [CrossRef]
- Van Der Weyden, L.; Bradley, A. Mouse Chromosome Engineering for Modeling Human Disease. Annu. Rev. Genom. Hum. Genet. 2006, 7, 247–276. [Google Scholar] [CrossRef]
- Koller, B.H.; Smithies, O. Altering Genes in Animals by Gene Targeting. Annu. Rev. Immunol. 1992, 10, 705–730. [Google Scholar] [CrossRef]
- Kumar, T.R.; Larson, M.; Wang, H.; McDermott, J.; Bronshteyn, I. Transgenic Mouse Technology: Principles and Methods. Advanced Structural Safety Studies 2009, 590, 335–362. [Google Scholar]
- Le, Y.; Sauer, B. Conditional Gene Knockout Using Cre Recombinase. Mol. Biotechnol. 2001, 17, 269–276. [Google Scholar] [CrossRef]
- Loussouarn, G.; Barã, I.; Escande, D.; Baró, I. Tissue-Specific Transgenic and Knockout Mice; Springer: Berlin/Heidelberg, Germany, 2006; Volume 337, pp. 185–205. [Google Scholar]
- Perl, A.-K.; Wilgenbus, P.; Dahl, U.; Semb, H.; Christofori, G. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature 1998, 392, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.-C. Generation of General and Tissue-Specific Gene Knockout Mouse Models. Adv. Struct. Saf. Stud. 2013, 1027, 253–271. [Google Scholar]
- Polato, F.; Rusconi, P.; Zangrossi, S.; Morelli, F.; Boeri, M.; Musi, A.; Marchini, S.; Castiglioni, V.; Scanziani, E.; Torri, V.; et al. DRAGO (KIAA0247), a New DNA Damage–Responsive, p53-Inducible Gene That Cooperates With p53 as Oncosupprossor. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Walrath, J.C.; Hawes, J.J.; Van Dyke, T.; Reilly, K.M. Genetically Engineered Mouse Models in Cancer Research. Adv. Cancer Res. 2010, 106, 113–164. [Google Scholar]
- Branda, C.S.; Dymecki, S.M. Talking about a Revolution. Dev. Cell 2004, 6, 7–28. [Google Scholar] [CrossRef]
- Metzger, D.; Chambon, P. Site- and Time-Specific Gene Targeting in the Mouse. Methods 2001, 24, 71–80. [Google Scholar] [CrossRef]
- Iwakuma, T.; Lozano, G. Crippling p53 activities via knock-in mutations in mouse models. Oncogene 2007, 26, 2177–2184. [Google Scholar] [CrossRef]
- Casola, S. Mouse Models for miRNA Expression: The ROSA26 Locus. Adv. Struct. Saf. Stud. 2010, 667, 145–163. [Google Scholar]
- Hohenstein, P.; Slight, J.; Ozdemir, D.D.; Burn, S.; Berry, R.; Hastie, N.D. High-efficiency Rosa26 knock-in vector construction for Cre-regulated overexpression and RNAi. Pathogenetics 2008, 1, 3. [Google Scholar] [CrossRef]
- Sportoletti, P.; Varasano, E.; Rossi, R.; Bereshchenko, O.; Cecchini, D.; Gionfriddo, I.; Bolli, N.; Tiacci, E.; Intermesoli, T.; Zanghì, P.; et al. The human NPM1 mutation A perturbs megakaryopoiesis in a conditional mouse model. Blood 2013, 121, 3447–3458. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.W.; Goudswaard, C.S.; Van Putten, W.; Bijl, M.A.; Sanders, M.A.; Hugens, W.; Uitterlinden, A.G.; Erpelinck, C.A.J.; Delwel, R.; Löwenberg, B.; et al. Mutations in nucleophosmin (NPM1) in acute myeloid leukemia (AML): Association with other gene abnormalities and previously established gene expression signatures and their favorable prognostic significance. Blood 2005, 106, 3747–3754. [Google Scholar] [CrossRef] [PubMed]
- Lakso, M.; Sauer, B.; Mosinger, B.; Lee, E.J.; Manning, R.W.; Yu, S.H.; Mulder, K.L.; Westphal, H. Targeted oncogene activation by site-specific recombination in transgenic mice. Proc. Natl. Acad. Sci. USA 1992, 89, 6232–6236. [Google Scholar] [CrossRef] [PubMed]
- Jackson, E.L.; Willis, N.; Mercer, K.; Bronson, R.T.; Crowley, D.; Montoya, R.; Jacks, T.; Tuveson, D.A. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001, 15, 3243–3248. [Google Scholar] [CrossRef]
- Abe, T.; Fujimori, T. Reporter Mouse Lines for Fluorescence Imaging. Dev. Growth Differ. 2013, 55, 390–405. [Google Scholar] [CrossRef]
- Momota, H.; Holland, E.C. Bioluminescence technology for imaging cell proliferation. Curr. Opin. Biotechnol. 2005, 16, 681–686. [Google Scholar] [CrossRef]
- Sattarzadeh, A.; Saberianfar, R.; Zipfel, W.R.; Menassa, R.; Hanson, M.R. Green to red photoconversion of GFP for protein tracking in vivo. Sci. Rep. 2015, 5, 11771. [Google Scholar] [CrossRef]
- Bauer, C.A.; Kim, E.Y.; Marangoni, F.; Carrizosa, E.; Claudio, N.M.; Mempel, T.R. Dynamic Treg interactions with intratumoral APCs promote local CTL dysfunction. J. Clin. Investig. 2014, 124, 2425–2440. [Google Scholar] [CrossRef]
- Kersten, K.; de Visser, K.E.; van Miltenburg, M.H.; Jonkers, J. Genetically engineered mouse models in oncology research and cancer medicine. EMBO Mol. Med. 2016, 9, 137–153. [Google Scholar] [CrossRef]
- Zitvogel, L.; Pitt, J.M.; Daillere, R.; Smyth, M.J.; Kroemer, G. Mouse models in oncoimmunology. Nat. Rev. Cancer 2016, 16, 759–773. [Google Scholar] [CrossRef]
- Heyer, J.; Kwong, L.N.; Lowe, S.W.; Chin, L. Non-germline genetically engineered mouse models for translational cancer research. Nat. Rev. Cancer 2010, 10, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Rajendra, Y.; Baldi, L.; Hacker, D.L.; Wurm, F.M. Comparison of three transposons for the generation of highly productive recombinant CHO cell pools and cell lines. Biotechnol. Bioeng. 2015, 113, 1234–1243. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Guo, H.; Tammana, S.; Jung, Y.-C.; Mellgren, E.; Bassi, P.; Cao, Q.; Tu, Z.J.; Kim, Y.C.; Ekker, S.C.; et al. Gene Transfer Efficiency and Genome-Wide Integration Profiling of Sleeping Beauty, Tol2, and PiggyBac Transposons in Human Primary T Cells. Mol. Ther. 2010, 18, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, H.; Jaenisch, R. Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nat. Protoc. 2014, 9, 1956–1968. [Google Scholar] [CrossRef]
- Gulei, D.; Berindan-Neagoe, I. CRISPR/Cas9: A Potential Life-Saving Tool. What’s next? Mol. Ther. Nucleic Acids 2017, 9, 333–336. [Google Scholar] [CrossRef]
- Zarei, A.; Razban, V.; Hosseini, S.E.; Tabei, S.M.B. Creating cell and animal models of human disease by genome editing using CRISPR/Cas9. J. Gene Med. 2019, 21, e3082. [Google Scholar] [CrossRef]
- Platt, R.J.; Chen, S.; Zhou, Y.; Yim, M.J.; Swiech, L.; Kempton, H.R.; Dahlman, J.E.; Parnas, O.; Eisenhaure, T.M.; Jovanovic, M.; et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 2014, 159, 440–455. [Google Scholar] [CrossRef]
- Chiou, S.-H.; Winters, I.; Wang, J.; Naranjo, S.; Dudgeon, C.; Tamburini, F.B.; Brady, J.J.; Yang, D.; Grüner, B.M.; Chuang, C.-H.; et al. Pancreatic cancer modeling using retrograde viral vector delivery and in vivo CRISPR/Cas9-mediated somatic genome editing. Genes Dev. 2015, 29, 1576–1585. [Google Scholar] [CrossRef]
- Wang, G.; Chow, R.D.; Ye, L.; Guzman, C.D.; Dai, X.; Dong, M.B.; Zhang, F.; Sharp, P.A.; Platt, R.J.; Chen, S. Mapping a functional cancer genome atlas of tumor suppressors in mouse liver using AAV-CRISPR–mediated direct in vivo screening. Sci. Adv. 2018, 4, eaao5508. [Google Scholar] [CrossRef]
- Gaj, T.; Gersbach, C.A.; Barbas, I.C.F. ZFN, TALEN and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef]
- Steel, C.D.; Stephens, A.L.; Hahto, S.M.; Singletary, S.J.; Ciavarra, R.P. Comparison of the lateral tail vein and the retro-orbital venous sinus as routes of intravenous drug delivery in a transgenic mouse model. Lab Anim. 2008, 37, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Yardeni, T.; Eckhaus, M.; Morris, H.D.; Huizing, M.; Hoogstraten-Miller, S. Retro-orbital injections in mice. Lab Anim. 2011, 40, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Cuadrado, L.; Tracey, N.; Ma, R.; Qian, B.; Brunton, V.G. Mouse models of metastasis: Progress and prospects. Dis. Model. Mech. 2017, 10, 1061–1074. [Google Scholar] [CrossRef] [PubMed]
- Lwin, T.M.; Hoffman, R.M.; Bouvet, M. Advantages of patient-derived orthotopic mouse models and genetic reporters for developing fluorescence-guided surgery. J. Surg. Oncol. 2018, 118, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, T.; Fu, X.; Kubota, T.; Watanabe, M.; Kitajima, M.; Hoffman, R.M. Nude mouse metastatic models of human stomach cancer constructed using orthotopic implantation of histologically intact tissue. Cancer Res. 1993, 53, 1204–1208. [Google Scholar]
- Rao, Q.; You, A.; Guo, Z.; Zuo, B.; Gao, X.; Zhang, T.; Du, Z.; Wu, C.; Yin, H. Intrahepatic Tissue Implantation Represents a Favorable Approach for Establishing Orthotopic Transplantation Hepatocellular Carcinoma Mouse Models. PLoS ONE 2016, 11, e0148263. [Google Scholar] [CrossRef]
- Mayer, P.; Sivakumar, N.; Pritz, M.; Varga, M.; Mehmann, A.; Lee, S.; Salvatore, A.; Magno, M.; Pharr, M.; Johannssen, H.C.; et al. Flexible and Lightweight Devices for Wireless Multi-Color Optogenetic Experiments Controllable via Commercial Cell Phones. Front. Mol. Neurosci. 2019, 13. [Google Scholar] [CrossRef]
- Zhu, Y.; Hu, Y.; Cheng, M.; Zeng, C.-Y.; Yang, Z.; Zhou, X.-D.; Chen, J.; Lu, N. Establishment and Characterization of a Nude Mouse Model of Subcutaneously Implanted Tumors and Abdominal Metastasis in Gastric Cancer. Gastroenterol. Res. Pract. 2017, 2017, 1–6. [Google Scholar] [CrossRef]
- Kyriazis, A.A.; Kyriazis, A.P. Preferential sites of growth of human tumors in nude mice following subcutaneous transplantation. Cancer Res. 1980, 40, 4509–4511. [Google Scholar]
- A McDonald, T.; Zepeda, M.L.; Tomlinson, M.J.; Bee, W.H.; Ivens, I.A. Subcutaneous administration of biotherapeutics: Current experience in animal models. Curr. Opin. Mol. Ther. 2010, 12, 461–470. [Google Scholar]
- Turner, P.V.; Brabb, T.; Pekow, C.; Vasbinder, M.A. Administration of Substances to Laboratory Animals: Routes of Administration and Factors to Consider. J. Am. Assoc. Lab. Anim. Sci. 2011, 50, 600–613. [Google Scholar]
- Mertens, B.; Nogueira, T.C.D.A.; Topalis, D.; Stránská, R.; Snoeck, R.; Andrei, G. Investigation of tumor-tumor interactions in a double human cervical carcinoma xenograft model in nude mice. Oncotarget 2018, 9, 21978–22000. [Google Scholar] [CrossRef] [PubMed]
- Tomayko, M.M.; Reynolds, C.P. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother. Pharmacol. 1989, 24, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Rashid, O.M.; Nagahashi, M.; Ramachandran, S.; Dumur, C.I.; Schaum, J.C.; Yamada, A.; Aoyagi, T.; Milstien, S.; Spiegel, S.; Takabe, K. Is tail vein injection a relevant breast cancer lung metastasis model? J. Thorac. Dis. 2013, 5, 385–392. [Google Scholar] [PubMed]
- Smith, L.P.; Thomas, G.R. Animal models for the study of squamous cell carcinoma of the upper aerodigestive tract: A historical perspective with review of their utility and limitations. Part A. Chemically-inducedde novo cancer, syngeneic animal models of HNSCC, animal models of transplanted xenogeneic human tumors. Int. J. Cancer 2006, 118, 2111–2122. [Google Scholar] [CrossRef]
- Woodfield, S.E.; Shi, Y.; Patel, R.H.; Jin, J.; Major, A.; Sarabia, S.F.; Starosolski, Z.; Zorman, B.; Gupta, S.S.; Chen, Z.; et al. A Novel Cell Line Based Orthotopic Xenograft Mouse Model That Recapitulates Human Hepatoblastoma. Sci. Rep. 2017, 7, 17751. [Google Scholar] [CrossRef] [PubMed]
- Bibby, M. Orthotopic models of cancer for preclinical drug evaluation. Eur. J. Cancer 2004, 40, 852–857. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, G.; Sun, X.; Cao, K.; Ma, C.; Nan, N.; Yang, G.; Yu, M.; Wang, X. Establishment of a murine breast tumor model by subcutaneous or orthotopic implantation. Oncol. Lett. 2018, 15, 6233–6240. [Google Scholar] [CrossRef]
- Murakami, T.; Zhang, Y.; Wang, X.; Hiroshima, Y.; Kasashima, H.; Yashiro, M.; Hirakawa, K.; Miwa, A.; Kiyuna, T.; Matsuyama, R.; et al. Orthotopic Implantation of Intact Tumor Tissue Leads to Metastasis of OCUM-2MD3 Human Gastric Cancer in Nude Mice Visualized in Real Time by Intravital Fluorescence Imaging. Anticancer Res. 2016, 36, 2125–2130. [Google Scholar]
- Soares, K.C.; Foley, K.; Olino, K.; Leubner, A.; Mayo, S.C.; Jain, A.; Jaffee, E.; Schulick, R.D.; Yoshimura, K.; Edil, B.H.; et al. A preclinical murine model of hepatic metastases. J. Vis. Exp. 2014, 91, e51677. [Google Scholar] [CrossRef]
- Lauber, D.T.; Fülöp, A.; Kovács, T.; Szigeti, K.; Máthé, D.; Szijártó, A. State of the art in vivo imaging techniques for laboratory animals. Lab. Anim. 2017, 51, 465–478. [Google Scholar] [CrossRef]
- Smith, B.R.; Gambhir, S.S. Nanomaterials for In Vivo Imaging. Chem. Rev. 2017, 117, 901–986. [Google Scholar] [CrossRef]
- Busato, A.; Feruglio, P.F.; Parnigotto, P.; Marzola, P.; Sbarbati, A. In vivo imaging techniques: A new era for histochemical analysis. Eur. J. Histochem. 2016, 60. [Google Scholar] [CrossRef] [PubMed]
- Busuttil, R.A.; Liu, D.S.; Di Costanzo, N.; Schröder, J.; Mitchell, C.; Boussioutas, A. An orthotopic mouse model of gastric cancer invasion and metastasis. Sci. Rep. 2018, 8, 825. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Cai, J.; Zhang, Y.; Zhu, Y.; Yang, P.; Wang, Z. Establishment of two ovarian cancer orthotopic xenograft mouse models for in vivo imaging: A comparative study. Int. J. Oncol. 2017, 51, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Zinn, K.R.; Chaudhuri, T.R.; Szafran, A.A.; O’Quinn, D.; Weaver, C.T.; Dugger, K.; Lamar, D.; Kesterson, R.A.; Wang, X.; Frank, S.J. Noninvasive Bioluminescence Imaging in Small Animals. ILAR J. 2008, 49, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, T.; Anayama, T.; Matsuda, Y.; Hwang, D.M.; McVeigh, P.Z.; Wilson, B.C.; Zheng, G.; Keshavjee, S.; Yasufuku, K. Orthotopic Lung Cancer Murine Model by Nonoperative Transbronchial Approach. Ann. Thorac. Surg. 2014, 97, 1771–1775. [Google Scholar] [CrossRef]
- Saar, M.; Körbel, C.; Linxweiler, J.; Jung, V.; Kamradt, J.; Hasenfus, A.; Stöckle, M.; Unteregger, G.; Menger, M.D. Orthotopic tumorgrafts in nude mice: A new method to study human prostate cancer. Prostate 2015, 75, 1526–1537. [Google Scholar] [CrossRef]
- Melsens, E.; De Vlieghere, E.; Descamps, B.; Vanhove, C.; De Wever, O.; Ceelen, W.; Pattyn, P. Improved xenograft efficiency of esophageal adenocarcinoma cell lines through in vivo selection. Oncol. Rep. 2017, 38, 71–81. [Google Scholar] [CrossRef]
- Coccolini, F.; Gheza, F.; Lotti, M.; Virzì, S.; Iusco, D.; Ghermandi, C.; Melotti, R.M.; Baiocchi, G.L.; Giulini, S.M.; Ansaloni, L.; et al. Peritoneal carcinomatosis. World J. Gastroenterol. 2013, 19, 6979–6994. [Google Scholar] [CrossRef]
- Weiss, L. Metastatic inefficiency: Intravascular and intraperitoneal implantation of cancer cells. Infect. Complicat. Cancer Patients 1996, 82, 1–11. [Google Scholar] [CrossRef]
- Hornung, R.; Major, A.L.; McHale, M.; Liaw, L.-H.L.; Sabiniano, L.A.; Tromberg, B.J.; Berns, M.W.; Tadir, Y. In vivo detection of metastatic ovarian cancer by means of 5-aminolevulinic acid-induced fluorescence in a rat model. J. Am. Assoc. Gynecol. Laparosc. 1998, 5, 141–148. [Google Scholar] [CrossRef]
- Yao, Y.; Zhou, Y.; Su, X.; Dai, L.; Yu, L.; Deng, H.; Gou, L.-T.; Yang, J. Establishment and characterization of intraperitoneal xenograft models by co-injection of human tumor cells and extracellular matrix gel. Oncol. Lett. 2015, 10, 3450–3456. [Google Scholar] [CrossRef] [PubMed]
- Frisk, T.W.; Rydholm, S.; Andersson, H.; Stemme, G.; Brismar, H. A concept for miniaturized 3-D cell culture using an extracellular matrix gel. Electrophoresis 2005, 26, 4751–4758. [Google Scholar] [CrossRef] [PubMed]
- A Engbring, J.; Kleinman, H.K. The basement membrane matrix in malignancy. J. Pathol. 2003, 200, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Kuwada, K.; Kagawa, S.; Yoshida, R.; Sakamoto, S.; Ito, A.; Watanabe, M.; Ieda, T.; Kuroda, S.; Kikuchi, S.; Tazawa, H.; et al. The epithelial-to-mesenchymal transition induced by tumor-associated macrophages confers chemoresistance in peritoneally disseminated pancreatic cancer. J. Exp. Clin. Cancer Res. 2018, 37, 307. [Google Scholar] [CrossRef] [PubMed]
- Lau, T.-S.; Chan, L.K.-Y.; Wong, C.-H.; Hui, C.W.-C.; Sneddon, K.; Cheung, T.-H.; Yim, S.-F.; Lee, J.H.-S.; Yeung, C.S.-Y.; Chung, T.K.H.; et al. A loop of cancer-stroma-cancer interaction promotes peritoneal metastasis of ovarian cancer via TNFα-TGFα-EGFR. Oncogene 2017, 36, 3576–3587. [Google Scholar] [CrossRef]
- Mohanty, S.; Xu, L. Experimental Metastasis Assay. J. Vis. Exp. 2010, 42. [Google Scholar] [CrossRef]
- Warren, S.; Gates, O. The Fate of Intravenously Injected Tumor Cells. Am. J. Cancer 1936, 27, 485–492. [Google Scholar] [CrossRef]
- Masuda, J.; Takayama, E.; Strober, W.; Satoh, A.; Morimoto, Y.; Honjo, Y.; Ichinohe, T.; Tokuno, S.; Ishizuka, T.; Nakata, T.; et al. Tumor growth limited to subcutaneous site vs. tumor growth in pulmonary site exhibit differential effects on systemic immunities. Oncol. Rep. 2017, 38, 449–455. [Google Scholar] [CrossRef]
- Kuchimaru, T.; Kataoka, N.; Nakagawa, K.; Isozaki, T.; Miyabara, H.; Minegishi, M.; Kadonosono, T.; Kizaka-Kondoh, S. A reliable murine model of bone metastasis by injecting cancer cells through caudal arteries. Nat. Commun. 2018, 9, 2981. [Google Scholar] [CrossRef] [PubMed]
- Kähkönen, T.E.; Bernoulli, J.; Halleen, J.M.; Suominen, M.I. Novel and Conventional Preclinical Models to Investigate Bone Metastasis. Curr. Mol. Biol. Rep. 2019, 5, 48–54. [Google Scholar] [CrossRef]
- Zhao, H.-Y.; Gong, Y.; Ye, F.-G.; Ling, H.; Hu, X. Incidence and prognostic factors of patients with synchronous liver metastases upon initial diagnosis of breast cancer: A population-based study. Cancer Manag. Res. 2018, 10, 5937–5950. [Google Scholar] [CrossRef] [PubMed]
- Engstrand, J.; Nilsson, H.; Strömberg, C.; Jonas, E.; Freedman, J. Colorectal cancer liver metastases—A population-based study on incidence, management and survival. BMC Cancer 2018, 18, 78. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.C.; McCarthy, L.P.; Mehdi, S. Isolated Hepatic Metastasis from Prostate Carcinoma. Urol. Case Rep. 2016, 10, 51–53. [Google Scholar] [CrossRef]
- Goddard, E.T.; Fischer, J.; Schedin, P.J. A Portal Vein Injection Model to Study Liver Metastasis of Breast Cancer. J. Vis. Exp. 2016, 118, e54903. [Google Scholar] [CrossRef]
- Chen, J.-J.; Zhou, W.; Cai, N.; Chang, G. In Vivo Murine Model of Leukemia Cell-Induced Spinal Bone Destruction. BioMed Res. Int. 2017, 2017, 1–6. [Google Scholar] [CrossRef]
- Saland, E.; Boutzen, H.; Castellano, R.; Pouyet, L.; Griessinger, E.; Larrue, C.; De Toni, F.; Scotland, S.; David, M.; Danet-Desnoyers, G.; et al. A robust and rapid xenograft model to assess efficacy of chemotherapeutic agents for human acute myeloid leukemia. Blood Cancer J. 2015, 5, e297. [Google Scholar] [CrossRef]
- Okuda, H.; Yokoyama, A. In vivo Leukemogenesis Model Using Retrovirus Transduction. BIO-PROTOCOL 2017, 7. [Google Scholar] [CrossRef]
- McGill, C.M.; Brown, T.J.; Cheng, Y.-Y.; Fisher, L.N.; Shanmugavelandy, S.S.; Gustafson, S.J.; Dunlap, K.L.; Lila, M.A.; Kester, M.; Toran, P.T.; et al. Therapeutic Effect of Blueberry Extracts for Acute Myeloid Leukemia. Int. J. Biopharm. Sci. 2018, 1, 102. [Google Scholar]
- Somervaille, T.C.P.; Cleary, M.L. Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia. Cancer Cell 2006, 10, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.; Zha, J.; Jiang, Z.; Jia, X.; Shi, Y.; Li, P.; Chen, X.L.; Fang, Z.-H.; Du, Z.; Xu, B. Apatinib exhibits anti-leukemia activity in preclinical models of acute lymphoblastic leukemia. J. Transl. Med. 2018, 16, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Veys, P.; Wynn, R.F.; Ahn, K.W.; Samarasinghe, S.; He, W.; Bonney, D.; Craddock, J.; Cornish, J.; Davies, S.M.; Dvorak, C.C.; et al. Impact of immune modulation with in vivo T-cell depletion and myleoablative total body irradiation conditioning on outcomes after unrelated donor transplantation for childhood acute lymphoblastic leukemia. Blood 2012, 119, 6155–6161. [Google Scholar] [CrossRef] [PubMed]
- Andre, N.D.; Silva, V.A.O.; Ariza, C.B.; Watanabe, M.A.E.; De Lucca, F.L. In vivo knockdown of CXCR4 using jetPEI/CXCR4 shRNA nanoparticles inhibits the pulmonary metastatic potential of B16-F10 melanoma cells. Mol. Med. Rep. 2012, 12, 8320–8326. [Google Scholar] [CrossRef] [PubMed]
- Galaup, A.; Cazes, A.; Le Jan, S.; Philippe, J.; Connault, E.; Le Coz, E.; Mekid, H.; Mir, L.M.; Opolon, P.; Corvol, P.; et al. Angiopoietin-like 4 prevents metastasis through inhibition of vascular permeability and tumor cell motility and invasiveness. Proc. Natl. Acad. Sci. USA 2006, 103, 18721–18726. [Google Scholar] [CrossRef]
- Benjamin, D.C.; Hynes, R.O. Intravital imaging of metastasis in adult Zebrafish. BMC Cancer 2017, 17, 660. [Google Scholar] [CrossRef]
- Maddipati, R.; Stanger, B.Z. Pancreatic Cancer Metastases Harbor Evidence of Polyclonality. Cancer Discov. 2015, 5, 1086–1097. [Google Scholar] [CrossRef]
- Kim, C.; Kim, I.H.; Kim, S.-I.; Kim, Y.S.; Kang, S.H.; Moon, S.H.; Kim, T.-S.; Kim, S.-K. Comparison of the Intraperitoneal, Retroorbital and per Oral Routes for F-18 FDG Administration as Effective Alternatives to Intravenous Administration in Mouse Tumor Models Using Small Animal PET/CT Studies. Nucl. Med. Mol. Imaging 2011, 45, 169–176. [Google Scholar] [CrossRef][Green Version]
- Shin, S.-S.; Jeong, B.-S.; Wall, B.A.; Li, J.; Shan, N.L.; Wen, Y.; Goydos, J.S.; Chen, S. Participation of xCT in melanoma cell proliferation in vitro and tumorigenesis in vivo. Oncogenesis 2018, 7, 86. [Google Scholar] [CrossRef]
- Barrett, D.M.; Zhao, Y.; Liu, X.; Jiang, S.; Carpenito, C.; Kalos, M.; Carroll, R.G.; June, C.H.; Grupp, S.A. Treatment of Advanced Leukemia in Mice with mRNA Engineered T Cells. Hum. Gene Ther. 2011, 22, 1575–1586. [Google Scholar] [CrossRef]
- Smirnova, T.; Adomako, A.; Locker, J.; Van Rooijen, N.; Prystowsky, M.B.; Segall, J.E. In Vivo Invasion of Head and Neck Squamous Cell Carcinoma Cells Does Not Require Macrophages. Am. J. Pathol. 2011, 178, 2857–2865. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kijewska, M.; Viski, C.; Turrell, F.; Fitzpatrick, A.; Van Weverwijk, A.; Gao, Q.; Iravani, M.; Isacke, C.M. Using an in-vivo syngeneic spontaneous metastasis model identifies ID2 as a promoter of breast cancer colonisation in the brain. Breast Cancer Res. 2019, 21, 4. [Google Scholar] [CrossRef] [PubMed]
- Janker, F.; Weder, W.; Jang, J.-H.; Jungraithmayr, W. Preclinical, non-genetic models of lung adenocarcinoma: A comparative survey. Oncotarget 2018, 9, 30527–30538. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, T.; Yokobori, T.; Naito, T.; Kakinuma, C.; Hagiwara, S.; Nishiyama, M.; Asao, T. Investigation into metastatic processes and the therapeutic effects of gemcitabine on human pancreatic cancer using an orthotopic SUIT-2 pancreatic cancer mouse model. Oncol. Lett. 2017, 15, 3091–3099. [Google Scholar] [CrossRef]
- Havens, A.M.; Pedersen, E.A.; Shiozawa, Y.; Ying, C.; Jung, Y.; Sun, Y.; Neeley, C.; Wang, J.; Mehra, R.; Keller, E.T.; et al. An In Vivo Mouse Model for Human Prostate Cancer Metastasis. Neoplasia 2008, 10, 371–379. [Google Scholar] [CrossRef]
- Heo, E.J.; Cho, Y.J.; Cho, W.C.; Hong, J.E.; Jeon, H.-K.; Oh, O.-Y.; Choi, Y.-L.; Song, S.Y.; Choi, J.-J.; Bae, D.-S.; et al. Patient-Derived Xenograft Models of Epithelial Ovarian Cancer for Preclinical Studies. Cancer Res. Treat. 2017, 49, 915–926. [Google Scholar] [CrossRef]
- Boonstra, M.C.; Van Driel, P.B.; Van Willigen, D.M.; Stammes, M.A.; Prevoo, H.A.; Tummers, Q.R.; Mazar, A.P.; Beekman, F.J.; Kuppen, P.; Van De Velde, C.J.; et al. uPAR-targeted multimodal tracer for pre- and intraoperative imaging in cancer surgery. Oncotarget 2015, 6, 14260–14273. [Google Scholar] [CrossRef]
- Soto, F.; Chrostowski, R. Frontiers of Medical Micro/Nanorobotics: In vivo Applications and Commercialization Perspectives Toward Clinical Uses. Front. Bioeng. Biotechnol. 2018, 6. [Google Scholar] [CrossRef]
- Li, S.; Jiang, Q.; Liu, S.; Zhang, Y.; Tian, Y.; Song, C.; Wang, J.; Zou, Y.; Anderson, G.J.; Han, J.-Y.; et al. A DNA nanorobot functions as a cancer therapeutic in response to a molecular trigger in vivo. Nat. Biotechnol. 2018, 36, 258–264. [Google Scholar] [CrossRef]
- Cook, N.; Jodrell, D.I.; Tuveson, D.A. Predictive in vivo animal models and translation to clinical trials. Drug Discov. Today 2012, 17, 253–260. [Google Scholar] [CrossRef]
- Willyard, C. The mice with human tumours: Growing pains for a popular cancer model. Nature 2018, 560, 156–157. [Google Scholar] [CrossRef] [PubMed]
- Tentler, J.J.; Tan, A.C.; Weekes, C.D.; Jimeno, A.; Leong, S.; Pitts, T.M.; Arcaroli, J.J.; Messersmith, W.A.; Eckhardt, S.G. Patient-derived tumour xenografts as models for oncology drug development. Nat. Rev. Clin. Oncol. 2012, 9, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.V.; Faversani, A.; Gatti, S.; Ricca, D.; Del Gobbo, A.; Ferrero, S.; Palleschi, A.; Vaira, V.; Bosari, S. A New Mouse Avatar Model of Non-Small Cell Lung Cancer. Front. Oncol. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Suárez, C.; Martinez, M.; Trilla, E.; Jimenez-Valerio, G.; De Torres, I.; Morales-Herrera, R.; Jimenez, J.; Lorente, D.; Vivancos, A.; Nuciforo, P.; et al. Patient-derived AVATAR mouse models to predict prognosis in advanced renal cell carcinoma. J. Clin. Oncol. 2016, 34, 551. [Google Scholar] [CrossRef]
- Garralda, E.; Paz, K.; López-Casas, P.P.; Jones, S.; Katz, A.; Kann, L.M.; López-Rios, F.; Sarno, F.; Al-Shahrour, F.; Vasquez, D.; et al. Integrated next-generation sequencing and avatar mouse models for personalized cancer treatment. Clin. Cancer Res. 2014, 20, 2476–2484. [Google Scholar] [CrossRef] [PubMed]
- Malaney, P.; Nicosia, S.V.; Davé, V. One mouse, one patient paradigm: New avatars of personalized cancer therapy. Cancer Lett. 2014, 344, 1–12. [Google Scholar] [CrossRef]
- Moro, M.; Casanova, M.; Roz, L. Patient-derived xenografts, a multi-faceted in vivo model enlightening research on rare liver cancer biology. Hepatobiliary Surg. Nutr. 2017, 6, 344–346. [Google Scholar] [CrossRef]
- Tiao, G.; Geller, J.; Timchenko, N. Generation of pediatric liver cancer patient-derived xenograft platforms for pediatric liver cancer: A critical stage in the development of anticancer treatments. Hepatology 2016, 64, 1017–1019. [Google Scholar] [CrossRef]
- Nicolle, D.; Fabre, M.; Simon-Coma, M.; Gorse, A.; Kappler, R.; Nonell, L.; Mallo, M.; Haidar, H.; Déas, O.; Mussini, C.; et al. Patient-derived xenografts from pediatric liver cancer predict tumor recurrence and advise clinical management. Hepatology 2016, 64, 1121–1135. [Google Scholar] [CrossRef]
- Wang, J.; Xing, B.; Liu, W.; Li, J.; Wang, X.; Li, J.; Yang, J.; Ji, C.; Li, Z.; Dong, B.; et al. Molecularly annotation of mouse avatar models derived from patients with colorectal cancer liver metastasis. Theranostics 2019, 9, 3485–3500. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, Z.; Wang, J.; Zhang, M.; Li, Z.; Wang, S.; Dong, B.; Zhang, C.; Gao, J.; Shen, L. Mouse avatar models of esophageal squamous cell carcinoma proved the potential for EGFR-TKI afatinib and uncovered Src family kinases involved in acquired resistance. J. Hematol. Oncol. 2018, 11, 109. [Google Scholar] [CrossRef]
- Zayed, A.A.; Mandrekar, S.J.; Haluska, P. Molecular and clinical implementations of ovarian cancer mouse avatar models. Chin. Clin. Oncol. 2015, 4, 30. [Google Scholar] [CrossRef] [PubMed]
- Valencia, A.; Hidalgo, M. Getting personalized cancer genome analysis into the clinic: The challenges in bioinformatics. Genome Med. 2012, 4, 61. [Google Scholar] [CrossRef]
- Ledford, H. Cancer-genome study challenges mouse ‘avatars’. Nature 2017. [Google Scholar] [CrossRef]
- Janssen, L.M.; Ramsay, E.E.; Logsdon, C.D.; Overwijk, W.W. The immune system in cancer metastasis: Friend or foe? J. Immunother. Cancer 2017, 5, 79. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.-C.; Liang, Q.; Ali, H.; Bayliss, L.; Beasley, A.; Bloomfield-Gerdes, T.; Bonoli, L.; Brown, R.; Campbell, J.; Carpenter, A.; et al. Complete humanization of the mouse immunoglobulin loci enables efficient therapeutic antibody discovery. Nat. Biotechnol. 2014, 32, 356–363. [Google Scholar] [CrossRef]
- Murphy, A.; Macdonald, L.E.; Stevens, S.; Karow, M.; Dore, A.T.; Pobursky, K.; Huang, T.T.; Poueymirou, W.T.; Esau, L.; Meola, M.; et al. Mice with megabase humanization of their immunoglobulin genes generate antibodies as efficiently as normal mice. Proc. Natl. Acad. Sci. USA 2014, 111, 5153–5158. [Google Scholar] [CrossRef]
- Macdonald, L.E.; Karow, M.; Stevens, S.; Auerbach, W.; Poueymirou, W.T.; Yasenchak, J.; Frendewey, D.; Valenzuela, D.M.; Giallourakis, C.C.; Alt, F.W.; et al. Precise and in situ genetic humanization of 6 Mb of mouse immunoglobulin genes. Proc. Natl. Acad. Sci. USA 2014, 111, 5147–5152. [Google Scholar] [CrossRef]
- Rongvaux, A.; Willinger, T.; Martínek, J.; Strowig, T.; Gearty, S.V.; Teichmann, L.L.; Saito, Y.; Marches, F.; Halene, S.; Palucka, A.K.; et al. Development and function of human innate immune cells in a humanized mouse model. Nat. Biotechnol. 2014, 32, 364–372. [Google Scholar] [CrossRef]
- Kalscheuer, H.; Danzl, N.; Onoe, T.; Faust, T.; Winchester, R.; Goland, R.; Greenberg, E.; Spitzer, T.R.; Savage, D.G.; Tahara, H.; et al. A Model for Personalized in Vivo Analysis of Human Immune Responsiveness. Sci. Transl. Med. 2012, 4, 125ra30. [Google Scholar] [CrossRef]
- Brainard, D.M.; Seung, E.; Frahm, N.; Cariappa, A.; Bailey, C.C.; Hart, W.K.; Shin, H.-S.; Brooks, S.F.; Knight, H.L.; Eichbaum, Q.; et al. Induction of Robust Cellular and Humoral Virus-Specific Adaptive Immune Responses in Human Immunodeficiency Virus-Infected Humanized BLT Mice. J. Virol. 2009, 83, 7305–7321. [Google Scholar] [CrossRef] [PubMed]
- Shultz, L.D.; Goodwin, N.; Ishikawa, F.; Hosur, V.; Lyons, B.L.; Greiner, D.L. Human Cancer Growth and Therapy in Immunodeficient Mouse Models. Cold Spring Harb. Protoc. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Shultz, L.D.; Ishikawa, F.; Greiner, D.L. Humanized mice in translational biomedical research. Nat. Rev. Immunol. 2007, 7, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Shultz, L.D.; Lyons, B.L.; Burzenski, L.M.; Gott, B.; Chen, X.; Chaleff, S.; Kotb, M.; Gillies, S.D.; King, M.; Mangada, J.; et al. Human Lymphoid and Myeloid Cell Development in NOD/LtSz-scid IL2Rγnull Mice Engrafted with Mobilized Human Hemopoietic Stem Cells. J. Immunol. 2005, 174, 6477–6489. [Google Scholar] [CrossRef] [PubMed]
- Escobar, G.; Moi, D.; Ranghetti, A.; Ozkal-Baydin, P.; Squadrito, M.L.; Kajaste-Rudnitski, A.; Bondanza, A.; Gentner, B.; De Palma, M.; Mazzieri, R.; et al. Genetic Engineering of Hematopoiesis for Targeted IFN- Delivery Inhibits Breast Cancer Progression. Sci. Transl. Med. 2014, 6, 217ra3. [Google Scholar] [CrossRef] [PubMed]
- Maletzki, C.; Bock, S.; Fruh, P.; Macius, K.; Witt, A.; Prall, F.; Linnebacher, M. NSG mice as hosts for oncological precision medicine. Lab. Investig. 2019, 100, 27–37. [Google Scholar] [CrossRef]
- Lepus, C.M.; Gibson, T.F.; Gerber, S.A.; Kawikova, I.; Szczepanik, M.; Hossain, J.; Ablamunits, V.; Kirkiles-Smith, N.; Herold, K.C.; Donis, R.O.; et al. Comparison of human fetal liver, umbilical cord blood, and adult blood hematopoietic stem cell engraftment in NOD-scid/γc−/−, Balb/c-Rag1−/−γc−/−, and C.B-17-scid/bg immunodeficient mice. Hum. Immunol. 2009, 70, 790–802. [Google Scholar] [CrossRef]
- Kennedy, M.; Awong, G.; Sturgeon, C.M.; Ditadi, A.; LaMotte-Mohs, R.; Zúñiga-Pflücker, J.C.; Keller, G. T Lymphocyte Potential Marks the Emergence of Definitive Hematopoietic Progenitors in Human Pluripotent Stem Cell Differentiation Cultures. Cell Rep. 2012, 2, 1722–1735. [Google Scholar] [CrossRef]
- Aspord, C.; Pedroza-Gonzalez, A.; Gallegos, M.; Tindle, S.; Burton, E.C.; Su, D.; Marches, F.; Banchereau, J.; Palucka, A.K. Breast cancer instructs dendritic cells to prime interleukin 13–secreting CD4+ T cells that facilitate tumor development. J. Exp. Med. 2007, 204, 1037–1047. [Google Scholar] [CrossRef]
- Wulf-Goldenberg, A.; Eckert, K.; Fichtner, I. Intrahepatically transplanted human cord blood cells reduce SW480 tumor growth in the presence of bispecific EpCAM/CD3 antibody. Cytotherapy 2011, 13, 108–113. [Google Scholar] [CrossRef]
- Guichelaar, T.; Mutis, T. Bone marrow provides an environment that prevents suppression of therapeutic graft-vs.-tumor immunity by regulatory T cells. OncoImmunology 2013, 2, e24659. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pino, S.; Brehm, M.A.; Covassin-Barberis, L.; King, M.; Gott, B.; Chase, T.H.; Wagner, J.; Burzenski, L.; Foreman, O.; Greiner, D.L.; et al. Development of novel major histocompatibility complex class I and class II-deficient NOD-SCID IL2R gamma chain knockout mice for modeling human xenogeneic graft-versus-host disease. Methods Mol. Biol. 2010, 602, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Sanmamed, M.F.; Chester, C.; Melero, I.; Kohrt, H.E. Defining the optimal murine models to investigate immune checkpoint blockers and their combination with other immunotherapies. Ann. Oncol. 2016, 27, 1190–1198. [Google Scholar] [CrossRef]
- Fisher, T.S.; Kamperschroer, C.; Oliphant, T.; Love, V.A.; Lira, P.D.; Doyonnas, R.; Bergqvist, S.; Baxi, S.; Rohner, A.; Shen, A.C.; et al. Targeting of 4-1BB by monoclonal antibody PF-05082566 enhances T-cell function and promotes anti-tumor activity. Cancer Immunol. Immunother. 2012, 61, 1721–1733. [Google Scholar] [CrossRef]
- Sanmamed, M.F.; Rodriguez, I.; Schalper, K.A.; Oñate, C.; Azpilikueta, A.; E Rodriguez-Ruiz, M.; Morales-Kastresana, A.; Labiano, S.; Perez-Gracia, J.L.; Martín-Algarra, S.; et al. Nivolumab and Urelumab Enhance Antitumor Activity of Human T Lymphocytes Engrafted in Rag2-/-IL2R?null Immunodeficient Mice. Cancer Res. 2015, 75, 3466–3478. [Google Scholar] [CrossRef]
- Pyo, K.-H.; Kim, J.H.; Lee, J.-M.; Kim, S.E.; Cho, J.S.; Lim, S.M.; Cho, B.C. Promising preclinical platform for evaluation of immuno-oncology drugs using Hu-PBL-NSG lung cancer models. Lung Cancer 2019, 127, 112–121. [Google Scholar] [CrossRef] [PubMed]
- King, M.A.; Covassin, L.; Brehm, M.A.; Racki, W.; Pearson, T.; Leif, J.; Laning, J.; Fodor, W.; Foreman, O.; Burzenski, L.; et al. Human peripheral blood leucocyte non-obese diabetic-severe combined immunodeficiency interleukin-2 receptor gamma chain gene mouse model of xenogeneic graft-versus-host-like disease and the role of host major histocompatibility complex. Clin. Exp. Immunol. 2009, 157, 104–118. [Google Scholar] [CrossRef]
- Nervi, B.; Rettig, M.P.; Ritchey, J.K.; Wang, H.L.; Bauer, G.; Walker, J.; Bonyhadi, M.L.; Berenson, R.J.; Prior, J.L.; Piwnica-Worms, D.; et al. Factors affecting human T cell engraftment, trafficking, and associated xenogeneic graft-vs-host disease in NOD/SCID β2mnull mice. Exp. Hematol. 2007, 35, 1823–1838. [Google Scholar] [CrossRef]
- Choi, Y.; Lee, S.; Kim, K.; Kim, S.-H.; Chung, Y.-J.; Lee, C. Studying cancer immunotherapy using patient-derived xenografts (PDXs) in humanized mice. Exp. Mol. Med. 2018, 50, 99. [Google Scholar] [CrossRef]
- Xia, A.; Zhang, Y.; Xu, J.; Yin, T.; Lu, X.-J. T Cell Dysfunction in Cancer Immunity and Immunotherapy. Front. Immunol. 2019, 10, 1719. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Molon, B.; Calì, B.; Viola, A. T Cells and Cancer: How Metabolism Shapes Immunity. Front. Immunol. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Katz, S.G.; Rabinovich, P.M. T Cell Reprogramming Against Cancer. Methods Mol. Biol. 2019, 3–44. [Google Scholar] [CrossRef]
- Galon, J.; Costes, A.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Zinzindohoué, F.; Bruneval, P.; Cugnenc, P.-H.; et al. Type, Density, and Location of Immune Cells Within Human Colorectal Tumors Predict Clinical Outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed]
- Yee, C.; Thompson, J.A.; Byrd, D.; Riddell, S.R.; Roche, P.; Celis, E.; Greenberg, P.D. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: In vivo persistence, migration, and antitumor effect of transferred T cells. Proc. Natl. Acad. Sci. USA 2002, 99, 16168–16173. [Google Scholar] [CrossRef]
- Jespersen, H.; Lindberg, M.F.; Donia, M.; Söderberg, E.M.V.; Andersen, R.; Keller, U.; Ny, L.; Svane, I.M.; Nilsson, L.M.; Nilsson, J.A. Clinical responses to adoptive T-cell transfer can be modeled in an autologous immune-humanized mouse model. Nat. Commun. 2017, 8, 707. [Google Scholar] [CrossRef]
- Johanna, I.; Straetemans, T.; Heijhuurs, S.; Aarts-Riemens, T.; Norell, H.; Bongiovanni, L.; De Bruin, A.; Sebestyen, Z.; Kuball, J. Evaluating in vivo efficacy – toxicity profile of TEG001 in humanized mice xenografts against primary human AML disease and healthy hematopoietic cells. J. Immunother. Cancer 2019, 7, 69. [Google Scholar] [CrossRef]
- Yip, H.; Haupt, C.; Maresh, G.; Zhang, X.; Li, L. Humanized mice for immune checkpoint blockade in human solid tumors. Am. J. Clin. Exp. Urol. 2019, 7, 313–320. [Google Scholar]
- Chen, Q.; Wang, J.; Liu, W.N.; Zhao, Y. Cancer Immunotherapies and Humanized Mouse Drug Testing Platforms. Transl. Oncol. 2019, 12, 987–995. [Google Scholar] [CrossRef]
- Piruat, J.I.; Millán-Uclés, Á. Genetically Modeled Mice with Mutations in Mitochondrial Metabolic Enzymes for the Study of Cancer. Front. Oncol. 2014, 4. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Flores, R.E.; Poff, A.M.; D’Agostino, M.P. Cancer as a metabolic disease: Implications for novel therapeutics. Carcinogenesis 2013, 35, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Chevrollier, A.; Loiseau, D.; Reynier, P.; Stepien, G. Adenine nucleotide translocase 2 is a key mitochondrial protein in cancer metabolism. Biochim. Biophys. Acta (BBA) Gen. Subj. 2011, 1807, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Poljšak, B.; Kovac, V.; Dahmane, R.; Levec, T.; Starc, A. Cancer Etiology: A Metabolic Disease Originating from Life’s Major Evolutionary Transition? Oxidative Med. Cell. Longev. 2019, 2019, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Millán-Uclés, Á.; García-Flores, P.; Báez, A.; Perez-Simón, J.A.; López-Barneo, J.; Piruat, J.A. A Conditional Mouse Mutant in the Tumor Suppressor SdhD Gene Unveils a Link between p21WAF1/Cip1 Induction and Mitochondrial Dysfunction. PLoS ONE 2014, 9, e85528. [Google Scholar] [CrossRef]
- Ashrafian, H.; O’Flaherty, L.; Adam, J.; Steeples, V.; Chung, Y.-L.; East, P.; Vanharanta, S.; Lehtonen, H.; Nye, E.; Hatipoglu, E.; et al. Expression Profiling in Progressive Stages of Fumarate-Hydratase Deficiency: The Contribution of Metabolic Changes to Tumorigenesis. Cancer Res. 2010, 70, 9153–9165. [Google Scholar] [CrossRef]
- Gardner, H.L.; Fenger, J.M.; London, C. Dogs as a Model for Cancer. Annu. Rev. Anim. Biosci. 2015, 4, 199–222. [Google Scholar] [CrossRef]
- Gunnes, G.; Borge, K.S.; Lingaas, F. A statistical assessment of the biological relationship between simultaneous canine mammary tumours. Vet. Comp. Oncol. 2015, 15, 355–365. [Google Scholar] [CrossRef]
- Salas-Araujo, Y.J.; Márquez, A.; Diaz, D.; Romero, L. Epidemiological Study of Mammary Tumors in Female Dogs Diagnosed during the Period 2002-2012: A Growing Animal Health Problem. PLoS ONE 2015, 10, e0127381. [Google Scholar] [CrossRef]
- Ettlin, J.; Clementi, E.; Amini, P.; Malbon, A.J.; Markkanen, E. Analysis of Gene Expression Signatures in Cancer-Associated Stroma from Canine Mammary Tumours Reveals Molecular Homology to Human Breast Carcinomas. Int. J. Mol. Sci. 2017, 18, 1101. [Google Scholar] [CrossRef]
- Sahabi, K.; Selvarajah, G.T.; Abdullah, R.; Cheah, Y.K.; Tan, G.C. Comparative aspects of microRNA expression in canine and human cancers. J. Vet. Sci. 2018, 19, 162–171. [Google Scholar] [CrossRef]
- Shao, Y.W.; Wood, G.A.; Lu, J.; Tang, Q.-L.; Liu, J.; Molyneux, S.; Chen, Y.; Fang, H.; Adissu, H.; McKee, T.; et al. Cross-species genomics identifies DLG2 as a tumor suppressor in osteosarcoma. Oncogene 2018, 38, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Abadie, J.; Nguyen, F.; Loussouarn, D.; Pena, L.; Gama, A.; Rieder, N.; Belousov, A.; Bemelmans, I.; Jaillardon, L.; Ibisch, C.; et al. Canine invasive mammary carcinomas as models of human breast cancer. Part 2: Immunophenotypes and prognostic significance. Breast Cancer Res. Treat. 2017, 167, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Dobson, J.M. Breed-Predispositions to Cancer in Pedigree Dogs. ISRN Veter- Sci. 2013, 2013, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Knapp, D.W.; Ramos-Vara, J.A.; Moore, G.E.; Dhawan, D.; Bonney, P.L.; Young, K.E. Urinary Bladder Cancer in Dogs, a Naturally Occurring Model for Cancer Biology and Drug Development. ILAR J. 2014, 55, 100–118. [Google Scholar] [CrossRef] [PubMed]
- Marconato, L.; Gelain, M.E.; Comazzi, S. The dog as a possible animal model for human non-Hodgkin lymphoma: A review. Hematol. Oncol. 2012, 31, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Komazawa, S.; Sakai, H.; Itoh, Y.; Kawabe, M.; Murakami, M.; Mori, T.; Maruo, K. Canine tumor development and crude incidence of tumors by breed based on domestic dogs in Gifu prefecture. J. Vet. Med. Sci. 2016, 78, 1269–1275. [Google Scholar] [CrossRef]
- Davis, B.W.; Ostrander, E.A. Domestic Dogs and Cancer Research: A Breed-Based Genomics Approach. ILAR J. 2014, 55, 59–68. [Google Scholar] [CrossRef]
- Hernández, B.; Adissu, H.; Wei, B.-R.; Michael, H.T.; Merlino, G.; Simpson, R.M. Naturally Occurring Canine Melanoma as a Predictive Comparative Oncology Model for Human Mucosal and Other Triple Wild-Type Melanomas. Int. J. Mol. Sci. 2018, 19, 394. [Google Scholar] [CrossRef]
- Van Der Weyden, L.; Patton, E.E.; A Wood, G.; Foote, A.K.; Brenn, T.; Arends, M.J.; Adams, D.J. Cross-species models of human melanoma. J. Pathol. 2015, 238, 152–165. [Google Scholar] [CrossRef]
- Prouteau, A.; Andre, C. Canine Melanomas as Models for Human Melanomas: Clinical, Histological, and Genetic Comparison. Genes 2019, 10, 501. [Google Scholar] [CrossRef]
- Abdelmegeed, S.M.; Mohammed, S. Canine mammary tumors as a model for human disease. Oncol. Lett. 2018, 15, 8195–8205. [Google Scholar] [CrossRef] [PubMed]
- Pinho, S.; Carvalho, S.; Cabral, J.; A Reis, C.; Gärtner, F. Canine tumors: A spontaneous animal model of human carcinogenesis. Transl. Res. 2012, 159, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Schiffman, J.D.; Breen, M. Comparative oncology: What dogs and other species can teach us about humans with cancer. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20140231. [Google Scholar] [CrossRef] [PubMed]
- De Araujo, M.R.; Campos, L.; Damasceno, K.; Gamba, C.; Ferreira, E.; Cassali, G.D. HER-2, EGFR, Cox-2 and Ki67 expression in lymph node metastasis of canine mammary carcinomas: Association with clinical-pathological parameters and overall survival. Res. Vet. Sci. 2016, 106, 121–130. [Google Scholar] [CrossRef]
- Carvalho, M.I.; Pires, I.; Prada, J.; Lobo, L.; Queiroga, F.L. Ki-67 and PCNA Expression in Canine Mammary Tumors and Adjacent Nonneoplastic Mammary Glands. Vet. Pathol. 2016, 53, 1138–1146. [Google Scholar] [CrossRef]
- Raposo-Ferreira, T.M.M.; Bueno, R.C.; Terra, E.M.; Avante, M.L.; Tinucci-Costa, M.; Carvalho, M.; Cassali, G.D.; Drigo, S.A.; Rogatto, S.R.; Laufer-Amorim, R. Downregulation ofATMGene and Protein Expression in Canine Mammary Tumors. Vet. Pathol. 2016, 53, 1154–1159. [Google Scholar] [CrossRef]
- Spoerri, M.; Guscetti, F.; Hartnack, S.; Boos, A.; Oei, C.; Balogh, O.; Nowaczyk, R.M.; Michel, E.; Reichler, I.; Kowalewski, M. Endocrine control of canine mammary neoplasms: Serum reproductive hormone levels and tissue expression of steroid hormone, prolactin and growth hormone receptors. BMC Vet. Res. 2015, 11, 1–10. [Google Scholar] [CrossRef][Green Version]
- Yoshikawa, Y.; Morimatsu, M.; Ochiai, K.; Ishiguro-Oonuma, T.; Wada, S.; Orino, K.; Watanabe, K. Reduced canine BRCA2 expression levels in mammary gland tumors. BMC Vet. Res. 2015, 11, 159. [Google Scholar] [CrossRef]
- Keller, J.M.; Schade, G.R.; Ives, K.; Cheng, X.; Rosol, T.J.; Piert, M.; Siddiqui, J.; Roberts, W.W.; Keller, E.T. A novel canine model for prostate cancer. Prostate 2013, 73, 952–959. [Google Scholar] [CrossRef]
- Sun, F.; Báez-Díaz, C.; Sánchez-Margallo, F.M. Canine prostate models in preclinical studies of minimally invasive interventions: Part I, canine prostate anatomy and prostate cancer models. Transl. Androl. Urol. 2017, 6, 538–546. [Google Scholar] [CrossRef]
- Fulkerson, C.M.; Dhawan, D.; Ratliff, T.L.; Hahn, N.M.; Knapp, D.W. Naturally Occurring Canine Invasive Urinary Bladder Cancer: A Complementary Animal Model to Improve the Success Rate in Human Clinical Trials of New Cancer Drugs. Int. J. Genom. 2017, 2017, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, D.; Hahn, N.M.; Ramos-Vara, J.A.; Knapp, D.W. Naturally-occurring canine invasive urothelial carcinoma harbors luminal and basal transcriptional subtypes found in human muscle invasive bladder cancer. PLoS Genet. 2018, 14, e1007571. [Google Scholar] [CrossRef] [PubMed]
- John, B.A.; Said, N. Insights from animal models of bladder cancer: Recent advances, challenges, and opportunities. Oncotarget 2017, 8, 57766–57781. [Google Scholar] [CrossRef]
- Scott, M.C.; Temiz, N.A.; Sarver, A.E.; LaRue, R.S.; Rathe, S.K.; Varshney, J.; Wolf, N.K.; Moriarity, B.S.; O’Brien, T.D.; Spector, L.G.; et al. Comparative Transcriptome Analysis Quantifies Immune Cell Transcript Levels, Metastatic Progression, and Survival in Osteosarcoma. Cancer Res. 2017, 78, 326–337. [Google Scholar] [CrossRef]
- Brown, D.C.; Boston, R.C.; Coyne, J.C.; Farrar, J.T. A novel approach to the use of animals in studies of pain: Validation of the canine brief pain inventory in canine bone cancer. Pain Med. 2008, 10, 133–142. [Google Scholar] [CrossRef]
- McDonald, J.T.; Kritharis, A.; Beheshti, A.; Pilichowska, M.; Burgess, K.; Ricks-Santi, L.; McNiel, E.; London, C.B.; Ravi, D.; Evens, A.M. Comparative oncology DNA sequencing of canine T cell lymphoma via human hotspot panel. Oncotarget 2018, 9, 22693–22702. [Google Scholar] [CrossRef] [PubMed]
- Ito, D.; Frantz, A.M.; Modiano, J.F. Canine lymphoma as a comparative model for human non-Hodgkin lymphoma: Recent progress and applications. Vet. Immunol. Immunopathol. 2014, 159, 192–201. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Base Name | Provided Content and Resources | Ref. |
|---|---|---|
| Cancer Models (caMOD) | Pathobiology with images Genetics of induced experimental models in mouse | [42] |
| Pathbase | Histopathology photomicrographs and macroscopic images derived from mutant or genetically manipulated mice | [43] |
| Cancer Genome Anatomy Project | Gene expression profiles from normal, precancerous, and cancerous tissues from mice and humans | [44] |
| International Mouse Strain Resource (ISMR) | Mouse strains, stocks, and mutant embryonic stem cell lines available worldwide, including inbred, mutant, and genetically engineered strains | [45] |
| International Mouse Phenotyping Consortium (IMPC) | The function of every protein coding gene in the mouse genome | [46] |
| Link Animal Models to Human Disease (LAMHDI) | The ideal animal models for different human diseases | [47] |
| Mouse Genome Informatics (MGI) | Integrated genetics, genomics and biological data for human health and disease studies | [48] |
| MUGEN mouse data base (MMdb) | Murine models of immune processes and immunological diseases | [49] |
| Phenotype comparisons for Disease Genes and Models (PhenoDigm) | Gene–disease associations by analyzing phenotype information | [50] |
| ALZFORUM | Selected rodent models of neurodegenerative disease, including Alzheimer’s, Parkinson’s, and Amyotrophic lateral sclerosis | [51] |
| SFARI Gene | Genes implicated in autism susceptibility | [52] |
| Method of Induction | Advantages | Disadvantages |
|---|---|---|
| A. Spontaneous mutations | - Discovery of novel mutations associated with specific traits/pathologies - No cost in induction of mutations | - Low mutation frequency - Hard to detect if not associated with phenotypic changes - Extensive validation to confirm the unique role of the mutation |
| B. Chemical/radiation induced mutations | - High mutational rate - Minimal cost for induction of mutation | - Random integrative mutations - Hard to associate specific mutations with pathologies - Extensive validation to confirm the unique role of the mutation |
| C. Retroviral infection | - Insertion of specific gene - Low controlled events | - De novo DNA methylation - Vector capacity in carrying large genes - Random integration in the genome |
| D. Microinjection of DNA constructs | - Direct insertion of specific gene - Medium controlled events - High controlled event with CRISPR/Cas9 | - DNA silencing mechanisms - Insertion of multiple copies in tandem - Random integration in the genome |
| Model | Type of Gene Modification | Application | Example (Oncology) | Ref. | |
|---|---|---|---|---|---|
| Loss of function | Constitutive Knockout | The gene inactivation is encountered in every cell and is also permanent | Overall changes in the phenotypical traits; identification of new genes involved in cancer | Analyzed gene: DRAGO Function: p53 connected gene in response to DNA interference drugs Model of study: p53−/− or p53+/− mice with wild-type of deleted Drago (both alleles) End point observation: rapid tumor development and shorter survival in p53−/− or p53+/− mice with Drago deletion. | [76,77] |
| Conditional Knockout | The gene inactivation is inducible and can be time and tissue specific | Mirroring of spontaneous cancer development in a more accurate manner—at specific point during the life of the organisms and also in specific cells/tissue. | Key components: bacterial Cre and yeast FLP enzymes (their expression can be controlled both spatially and temporally) for recombination between specific 34-bp loxP and FRT sites that flank the gene of interest Spatial control: the recombinase is under the control of a tissue specific promoter Temporal control: tetracycline and tamoxifen-inducible systems that control the activity of Cre. | [78,79,80] | |
| Gain of function | Constitutive Random Insertion Model | The transgene is incorporated in random spots of the genome by DNA microinjection in the pronucleus of fertilized oocytes or transfection of embryos with viral vector constructs | Activity of genes (especially oncogenes) in installation and sustenance of carcinogenesis | Analyzed gene: mutant TP53 Function: oncogenic function and ability to sustain carcinogenesis Model of study: knock-in alleles with mutations that mirror the ones found in human cancers End point observation: mutations able to individualize the functions of apoptosis and cell cycle arrest are able to slow down the malignant development (indicating that both tasks are important for tumor suppression); each model of study (different mutated spots) exhibits a distinct phenotype, showing the complex interconnection between the dynamics of TP53 genetics and heterogeneity of cancer. | [4,81] |
| Knock-in Permissive Locus Model | Specific insertion of the gene into the genome via homologous recombination; widely used spot for insertion - Rosa26 locus due to lack of critical genes and stable gene expression in different cellular entities | Activity of genes (especially oncogenes) in installation and sustenance of carcinogenesis | Analyzed gene: mutated Npm1 (altered in AML), type A, hematopoietic compartment Function: Model of study: Npm1-TCTG/WT;Cre(+) mice generated by insertion of transgenic gene in the Rosa26 locus with expression regulated via Cre-recombinase. End point observation: no development of the targeted disease (AML); perturbed megakaryocytic development and upregulation of specific miRNA profile similar to those found in humans with mutated Npm1: miR-10a, miR-10b, and miR-20a | [82,83,84,85] | |
| Conditional Knock-in Model | Adapted Constitutive Random Insertion Model, where the expression of the target gene is regulated through temporal and spatial control | Activity of genes (especially oncogenes) in installation and sustenance of carcinogenesis in a time and spatial specific manner | Key components: bacterial Cre and yeast FLP enzymes (their expression can be controlled both spatially and temporally) for recombination between specific 34-bp loxP and FRT sites that flank the gene of interest Spatial control: use of tissue specific promoters or through insertion of a STOP cassette between the promoter and the sequence of interest that is also flanked by loxP or FRT sites. Under the expression of Cre or FLP recombinase the STOP cassette is removed, and the transcription of the transgene is possible. Temporal control: control of Cre or FLP recombinase activity | [86,87] | |
| Reporter Knock-in Model | The expression of the transgene is followed by incorporation of tracking proteins—fluorescent/bioluminescent | Activity of genes (especially oncogenes) in installation and sustenance of carcinogenesis in a time and spatial specific manner and real time monitoring; tracking of tumor growth by incorporation of genes encoding tracking proteins; interaction between immune cells toward tumor inhibition | Key components: fluorescent proteins—e.g., GFP, RFP, bioluminescent enzymes - e.g., firefly luciferase Exemple of model: mice models containing firefly luciferase under the control of the human promoter E2F1, which exerts its function in proliferating cells crossed with mice models of cancer | [88,89,90,91] |
| Cancer Localization | Cancer Type | Cell Line/Tissue | Animal Strain | Xenograft Method | Cancer Development Evaluation | Ref. |
|---|---|---|---|---|---|---|
| Skin | Melanoma | SK-mel2 and SK-mel187 | Balb/c nude | Subcutaneous injection | Tumor measurements Western immunoblotting | [162] |
| Blood | Acute myeloid leukemia | Nalm6, Reh, Molt4 and Jurkat | NOD-scid-IL2Rg−/− (NSI) | Irradiation followed by retro-orbital vein injection | Flow cytometry for CD45+ cells Bone marrow and spleen staining | [155] |
| Acute lymphoblastic leukemia | Nalm-6 | NOD-SCID-γc–/– (NSG) | Tail vein injection | Flow cytometry Immunostaining Bioluminiscent imaging | [163] | |
| Head and neck | Head and neck squamous cell carcinoma | FaDu, UMSCC47 | Athymic nude NCI-Frederick | Mouth floor injection | Intravital imaging Immunohistochemistry | [164] |
| Thorax | Breast cancer | 4T1-Luc MDA-MB-231 | Balb/c and NSG | Subcutaneous, Intracranial and Lateral tail vein injection | In vivo imaging Immunohistochemistry | [165] |
| Lewis lung carcinoma | LLC | C57BL/6 and BALB/c | Urethane intraperitoneal administration, Inferior vena cava, and subcutaneous injection | Immunohistochemistry | [166] | |
| Abdomen | Gastric cancer | MKN-45, AGS and MKN-28 | Bl6/Rag2/GammaC double knockout | Orthotopic injection | In vivo bioluminescence imaging Immunohistochemistry | [127] |
| Pancreatic cancer | Capan-1 and SUIT-2 | BALBc nu/nu | Orthotopic injection | Immunohistochemistry | [167] | |
| Pelvis | Prostate cancer | PC-3 | CB.17. SCID | Orthotopic and subcutaneous injection | Bioluminiscence Intravital microscopy Immunohistochemistry | [168] |
| Epithelial ovarian cancer | Fresh tissue | BALB/c nude | Subrenal capsule implantation | Immunohistochemistry Short tandem repeat assay Western blot | [169] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Onaciu, A.; Munteanu, R.; Munteanu, V.C.; Gulei, D.; Raduly, L.; Feder, R.-I.; Pirlog, R.; Atanasov, A.G.; Korban, S.S.; Irimie, A.; et al. Spontaneous and Induced Animal Models for Cancer Research. Diagnostics 2020, 10, 660. https://doi.org/10.3390/diagnostics10090660
Onaciu A, Munteanu R, Munteanu VC, Gulei D, Raduly L, Feder R-I, Pirlog R, Atanasov AG, Korban SS, Irimie A, et al. Spontaneous and Induced Animal Models for Cancer Research. Diagnostics. 2020; 10(9):660. https://doi.org/10.3390/diagnostics10090660
Chicago/Turabian StyleOnaciu, Anca, Raluca Munteanu, Vlad Cristian Munteanu, Diana Gulei, Lajos Raduly, Richard-Ionut Feder, Radu Pirlog, Atanas G. Atanasov, Schuyler S. Korban, Alexandru Irimie, and et al. 2020. "Spontaneous and Induced Animal Models for Cancer Research" Diagnostics 10, no. 9: 660. https://doi.org/10.3390/diagnostics10090660
APA StyleOnaciu, A., Munteanu, R., Munteanu, V. C., Gulei, D., Raduly, L., Feder, R.-I., Pirlog, R., Atanasov, A. G., Korban, S. S., Irimie, A., & Berindan-Neagoe, I. (2020). Spontaneous and Induced Animal Models for Cancer Research. Diagnostics, 10(9), 660. https://doi.org/10.3390/diagnostics10090660