Infantile/Congenital High-Grade Gliomas: Molecular Features and Therapeutic Perspectives


 ,
,  , ,
, ,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
3. Clinical Findings and Diagnosis
4. Neuropathology and Molecular Characteristics of iHGGs
5. Treatment Options
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rickert, C.H.; Probst-Cousin, S.; Gullotta, F. Primary Intracranial Neoplasms of Infancy and Early Childhood. Childs Nerv. Syst. 1997, 13, 507–513. Available online: https://www.ncbi.nlm.nih.gov/pubmed/9403197 (accessed on 8 December 2018).
- Duffner, P.K.; Horowitz, M.E.; Krischer, J.P.; Burger, P.C.; Cohen, M.E.; Sanford, R.A.; Friedman, H.S.; Kun, L.E. The treatment of malignant brain tumors in infants and very young children: An update of the Pediatric Oncology Group experience. Neuro-Oncology 1999, 1, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Severino, M.; Schwartz, E.S.; Thurnher, M.M.; Rydland, J.; Nikas, I.; Rossi, A. Congenital tumors of the central nervous system. Neuroradiology 2010, 52, 531–548. [Google Scholar] [CrossRef] [PubMed]
- Milani, H.J.; Araujo Júnior, E.; Cavalheiro, S.; Oliveira, P.S.; Hisaba, W.J.; Barreto, E.Q.S.; Barbosa, M.M.; Nardozza, L.M.; Moron, A.F. Fetal brain tumors: Prenatal diagnosis by ultrasound and magnetic resonance imaging. World J. Radiol. 2015, 7, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Dunham, C.; Pillai, S.; Steinbok, P. Infant brain tumors: A neuropathologic population-based institutional reappraisal. Hum. Pathol. 2012, 43, 1668–1676. [Google Scholar] [CrossRef] [PubMed]
- Cassart, M.; Bosson, N.; Garel, C.; Eurin, D.; Avni, F. Fetal intracranial tumors: A review of 27 cases. Eur. Radiol. 2008, 18, 2060–2066. [Google Scholar] [CrossRef]
- Ries, L.A.G.; Smith, M.A.; Gurney, J.G.; Linet, M.; Tamra, T.; Young, J.L.; Bunin, G.R. Cancer Incidence and Survival among Children and Adolescents: United States SEER Program 1975–1995; National Cancer Institute: Bethesda, MD, USA, 1999.
- Larouche, V.; Huang, A.; Bartels, U.; Bouffet, E. Tumors of the central nervous system in the first year of life. Pediatr. Blood Cancer 2007, 49, 1074–1082. [Google Scholar] [CrossRef]
- Guerreiro Stucklin, A.S.; Ryall, S.; Fukuoka, K.; Zapotocky, M.; Lassaletta, A.; Li, C.; Bridge, T.; Kim, B.; Arnoldo, A.; Kowalski, P.E.; et al. Alterations in ALK/ROS1/NTRK/MET drive a group of infantile hemispheric gliomas. Nat. Commun. 2019, 10, 4343. [Google Scholar] [CrossRef]
- Reulecke, B.C.; Erker, C.G.; Fiedler, B.J.; Niederstadt, T.-U.; Kurlemann, G. Brain tumors in children: Initial symptoms and their influence on the time span between symptom onset and diagnosis. J. Child Neurol. 2008, 23, 178–183. [Google Scholar] [CrossRef]
- Epidemiological features of brain tumors in the first 3 years of life. Child’s Nerv. Syst. 1998, 14, 547–550. Available online: https://www.ncbi.nlm.nih.gov/pubmed/9840377 (accessed on 8 December 2018).
- Woodward, P.J.; Sohaey, R.; Kennedy, A.; Koeller, K.K. From the archives of the AFIP: A comprehensive review of fetal tumors with pathologic correlation. Radiographics 2005, 25, 215–242. [Google Scholar] [CrossRef]
- Cavalheiro, S.; Moron, A.F.; Hisaba, W.; Dastoli, P.; Silva, N.S. Fetal brain tumors. Child’s Nerv. Syst. 2003, 19, 529–536. [Google Scholar] [CrossRef]
- Isaacs, H.I. Perinatal brain tumors: A review of 250 cases. Pediatr. Neurol. 2002, 27, 249–261. [Google Scholar] [CrossRef]
- Raisanen, J.M.; Davis, R.L. Congenital brain tumors. Pathology 1993, 2, 103–116. [Google Scholar] [PubMed]
- Toescu, S.M.; James, G.; Phipps, K.; Jeelani, O.; Thompson, D.; Hayward, R.; Aquilina, K. Intracranial Neoplasms in the First Year of Life: Results of a Third Cohort of Patients from a Single Institution. Neurosurgery 2019, 84, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Sgro, M.; Barozzino, T.; Toi, A.; Johnson, J.; Sermer, M.; Chitayat, D. Prenatal detection of cerebral lesions in a fetus with tuberous sclerosis. Ultrasound Obstet. Gynecol. 1999, 14, 356–359. [Google Scholar] [CrossRef]
- Magdum, S.A. Neonatal brain tumours—A review. Early Hum. Dev. 2010, 86, 627–631. [Google Scholar] [CrossRef]
- McGuirt, D. Alternatives to Sedation and General Anesthesia in Pediatric Magnetic Resonance Imaging: A Literature Review. Radiol. Technol. 2016, 88, 18–26. [Google Scholar]
- Haberler, C.; Slavc, I.; Czech, T.; Prayer, D.; Pirker, C.; Budka, H.; Hainfellner, J.A. Malignant predominantly minigemistocytic glioma in two infants: A distinctive glioma variant? Neuropathol. Appl. Neurobiol. 2007, 33, 169–178. [Google Scholar] [CrossRef]
- Macy, M.E.; Birks, D.K.; Barton, V.N.; Chan, M.H.; Donson, A.M.; Kleinschmidt-Demasters, B.K.; Bemis, L.T.; Handler, M.H.; Foreman, N.K. Clinical and molecular characteristics of congenital glioblastoma. Neuro-Oncology 2012, 14, 931–941. [Google Scholar] [CrossRef]
- Amatu, A.; Sartore-Bianchi, A.; Siena, S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. ESMO Open 2016, 1, e000023. [Google Scholar] [CrossRef]
- Clarke, M.; Mackay, A.; Ismer, B.; Pickles, J.C.; Tatevossian, R.G.; Newman, S.; Bale, T.A.; Stoler, I.; Izquierdo, E.; Temelso, S.; et al. Infant high grade gliomas comprise multiple subgroups characterized by novel targetable gene fusions and favorable outcomes. Cancer Discov. 2020, 10, 942–963. [Google Scholar] [CrossRef]
- Aghajan, Y.; Levy, M.L.; Malicki, D.M.; Crawford, J.R. Novel PPP1CB-ALK fusion protein in a high-grade glioma of infancy. Case Rep. 2016, 2016, bcr2016217189. [Google Scholar] [CrossRef] [PubMed]
- Ng, A.; Levy, M.L.; Malicki, D.M.; Crawford, J.R. Unusual high-grade and low-grade glioma in an infant with PPP1CB-ALK gene fusion. BMJ Case Rep. 2019, 12, e228248. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.C.; Bababeygy, S.R.; Sarkissian, V.; Fisher, P.G.; Vogel, H.; Barnes, P.; Huhn, S.L. Congenital glioblastoma multiforme: Case report and review of the literature. Pediatr. Neurosurg. 2008, 44, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Seker, A.; Ozek, M.M. Congenital glioblastoma multiforme. Case report and review of the literature. J. Neurosurg. 2006, 105, 473–479. [Google Scholar] [CrossRef]
- Brennan, C.; Momota, H.; Hambardzumyan, D.; Ozawa, T.; Tandon, A.; Pedraza, A.; Holland, E. Glioblastoma subclasses can be defined by activity among signal transduction pathways and associated genomic alterations. PLoS ONE 2009, 4, e7752. [Google Scholar] [CrossRef]
- Gielen, G.H.; Gessi, M.; Buttarelli, F.R.; Baldi, C.; Hammes, J.; zur Muehlen, A.; Doerner, E.; Denkhaus, D.; Warmuth-Metz, M.; Giangaspero, F.; et al. Genetic Analysis of Diffuse High-Grade Astrocytomas in Infancy Defines a Novel Molecular Entity. Brain Pathol. 2015, 25, 409–417. [Google Scholar] [CrossRef]
- Anestis, D.M.; Tsitsopoulos, P.P.; Ble, C.A.; Tsitouras, V.; Tsonidis, C.A. Congenital Glioblastoma Multiforme: An Unusual and Challenging Tumor. Neuropediatrics 2017, 48, 403–412. [Google Scholar] [CrossRef]
- Wang, A.C.; Jones, D.T.W.; Abecassis, I.J.; Cole, B.L.; Leary, S.E.S.; Lockwood, C.M.; Chavez, L.; Capper, D.; Korshunov, A.; Fallah, A.; et al. Desmoplastic Infantile Ganglioglioma/Astrocytoma (DIG/DIA) Are Distinct Entities with Frequent BRAFV600 Mutations. Mol. Cancer Res. 2018, 16, 1491–1498. [Google Scholar] [CrossRef]
- Brat, D.J.; Shehata, B.M.; Castellano-Sanchez, A.A.; Hawkins, C.; Yost, R.B.; Greco, C.; Mazewski, C.; Janss, A.; Ohgaki, H.; Perry, A. Congenital glioblastoma: A clinicopathologic and genetic analysis. Brain Pathol. 2007, 17, 276–281. [Google Scholar] [CrossRef]
- Pollack, I.F.; Hamilton, R.L.; James, C.D.; Finkelstein, S.D.; Burnham, J.; Yates, A.J.; Holmes, E.J.; Zhou, T.; Finlay, J.L.; Children’s Oncology Group. Rarity of PTEN deletions and EGFR amplification in malignant gliomas of childhood: Results from the Children’s Cancer Group 945 cohort. J. Neurosurg. 2006, 105, 418–424. [Google Scholar] [CrossRef]
- Paugh, B.S.; Qu, C.; Jones, C.; Liu, Z.; Adamowicz-Brice, M.; Zhang, J.; Bax, D.A.; Coyle, B.; Barrow, J.; Hargrave, D.; et al. Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J. Clin. Oncol. 2010, 28, 3061–3068. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, S.; Corsinovi, D.; Lessi, F.; Tantillo, E.; Aretini, P.; Menicagli, M.; Scopelliti, C.; Civita, P.; Pasqualetti, F.; Naccarato, A.G.; et al. Mitochondrial enzyme GLUD2 plays a critical role in glioblastoma progression. EBioMedicine 2018, 37, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Guo, S.; Jiang, X.; Bryk, J.; Naumann, R.; Enard, W.; Tomita, M.; Sugimoto, M.; Khaitovich, P.; Pääbo, S. Mice carrying a human GLUD2 gene recapitulate aspects of human transcriptome and metabolome development. Proc. Natl. Acad. Sci. USA 2016, 113, 5358–5363. [Google Scholar] [CrossRef]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef]
- Kameda, M.; Otani, Y.; Ichikawa, T.; Shimada, A.; Ichimura, K.; Date, I. Congenital Glioblastoma with Distinct Clinical and Molecular Characteristics: Case Reports and a Literature Review. World Neurosurg. 2017, 101, 817.e5–817.e14. [Google Scholar] [CrossRef]
- Mackay, A.; Burford, A.; Carvalho, D.; Izquierdo, E.; Fazal-Salom, J.; Taylor, K.R.; Bjerke, L.; Clarke, M.; Vinci, M.; Nandhabalan, M.; et al. Integrated Molecular Meta-Analysis of 1000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017, 32, 520–537.e5. [Google Scholar] [CrossRef]
- Wu, G.; Diaz, A.K.; Paugh, B.S.; Rankin, S.L.; Ju, B.; Li, Y.; Zhu, X.; Qu, C.; Chen, X.; Zhang, J.; et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat. Genet. 2014, 46, 444–450. [Google Scholar] [CrossRef]
- Jones, D.T.W.; Hutter, B.; Jäger, N.; Korshunov, A.; Kool, M.; Warnatz, H.-J.; Zichner, T.; Lambert, S.R.; Ryzhova, M.; Quang, D.A.K.; et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat. Genet. 2013, 45, 927–932. [Google Scholar] [CrossRef]
- Valera, E.T.; Neder, L.; Queiroz, R.G.; Santos, A.C.; Sousa, G.R.; Oliveira, R.S.; Santos, M.V.; Machado, H.R.; Tone, L.G. Perinatal complex low- and high-grade glial tumor harboring a novel GIGYF2-ALK fusion. Pediatr. Blood Cancer 2020, 67, e28015. [Google Scholar] [CrossRef]
- Johanns, T.M.; Ferguson, C.J.; Grierson, P.M.; Dahiya, S.; Ansstas, G. Rapid Clinical and Radiographic Response with Combined Dabrafenib and Trametinib in Adults With BRAF-Mutated High-Grade Glioma. J. Natl. Compr. Cancer Netw. 2018, 16, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Lassaletta, A.; Zapotocky, M.; Mistry, M.; Ramaswamy, V.; Honnorat, M.; Krishnatry, R.; Guerreiro Stucklin, A.; Zhukova, N.; Arnoldo, A.; Ryall, S.; et al. Therapeutic and Prognostic Implications of BRAF V600E in Pediatric Low-Grade Gliomas. J. Clin. Oncol. 2017, 35, 2934–2941. [Google Scholar] [CrossRef] [PubMed]
- Lassaletta, A.; Guerreiro Stucklin, A.; Ramaswamy, V.; Zapotocky, M.; McKeown, T.; Hawkins, C.; Bouffet, E.; Tabori, U. Profound clinical and radiological response to BRAF inhibition in a 2-month-old diencephalic child with hypothalamic/chiasmatic glioma. Pediatr. Blood Cancer 2016, 63, 2038–2041. [Google Scholar] [CrossRef] [PubMed]
- Del Bufalo, F.; Carai, A.; Figà-Talamanca, L.; Pettorini, B.; Mallucci, C.; Giangaspero, F.; Antonelli, M.; Badiali, M.; Moi, L.; Bianco, G.; et al. Response of recurrent BRAFV600E mutated ganglioglioma to Vemurafenib as single agent. J. Transl. Med. 2014, 12, 356. [Google Scholar] [CrossRef] [PubMed]
- Petruzzellis, G.; Valentini, D.; Del Bufalo, F.; Ceglie, G.; Carai, A.; Colafati, G.S.; Agolini, E.; Diomedi-Camassei, F.; Corsetti, T.; Alessi, I.; et al. Vemurafenib Treatment of Pleomorphic Xanthoastrocytoma in a Child with Down Syndrome. Front. Oncol. 2019, 9, 277. [Google Scholar] [CrossRef]
- McGirt, M.J.; Chaichana, K.L.; Gathinji, M.; Attenello, F.J.; Than, K.; Olivi, A.; Weingart, J.D.; Brem, H.; Quiñones-Hinojosa, A.R. Independent association of extent of resection with survival in patients with malignant brain astrocytoma. J. Neurosurg. 2009, 110, 156–162. [Google Scholar] [CrossRef]
- Khalil, E.M. Treatment results of adults and children with medulloblastoma NCI, Cairo University experience. J. Egypt. Natl. Cancer Inst. 2008, 20, 175–186. [Google Scholar]
- Grundy, R.G.; Wilne, S.H.; Robinson, K.J.; Ironside, J.W.; Cox, T.; Chong, W.K.; Michalski, A.; Campbell, R.H.A.; Bailey, C.C.; Thorp, N.; et al. Primary postoperative chemotherapy without radiotherapy for treatment of brain tumours other than ependymoma in children under 3 years: Results of the first UKCCSG/SIOP CNS 9204 trial. Eur. J. Cancer 2010, 46, 120–133. [Google Scholar] [CrossRef] [PubMed]
- Osorio, D.S.; Patel, N.; Ji, L.; Sposto, R.; Stanek, J.; Gardner, S.L.; Allen, J.C.; Cornelius, A.; McCowage, G.B.; Termuhlen, A.; et al. Pre-irradiation intensive induction and marrow-ablative consolidation chemotherapy in young children with newly diagnosed high-grade brainstem gliomas: Report of the “head-start” I and II clinical trials. J. Neuro-Oncol. 2018, 140, 717–725. [Google Scholar] [CrossRef]
- Mulhern, R.K.; Merchant, T.E.; Gajjar, A.; Reddick, W.E.; Kun, L.E. Late neurocognitive sequelae in survivors of brain tumours in childhood. Lancet Oncol. 2004, 5, 399–408. [Google Scholar] [CrossRef]
- Ater, J.L.; van Eys, J.; Woo, S.Y.; Moore, B.; Copeland, D.R.; Bruner, J. MOPP chemotherapy without irradiation as primary postsurgical therapy for brain tumors in infants and young children. J. Neuro-Oncol. 1997, 32, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Doebele, R.C.; Davis, L.E.; Vaishnavi, A.; Le, A.T.; Estrada-Bernal, A.; Keysar, S.; Jimeno, A.; Varella-Garcia, M.; Aisner, D.L.; Li, Y.; et al. An Oncogenic NTRK Fusion in a Patient with Soft-Tissue Sarcoma with Response to the Tropomyosin-Related Kinase Inhibitor LOXO-101. Cancer Discov. 2015, 5, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Laetsch, T.W.; DuBois, S.G.; Mascarenhas, L.; Turpin, B.; Federman, N.; Albert, C.M.; Nagasubramanian, R.; Davis, J.L.; Rudzinski, E.; Feraco, A.M.; et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: Phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol. 2018, 19, 705–714. [Google Scholar] [CrossRef]
- Hong, D.S.; DuBois, S.G.; Kummar, S.; Farago, A.F.; Albert, C.M.; Rohrberg, K.S.; van Tilburg, C.M.; Nagasubramanian, R.; Berlin, J.D.; Federman, N.; et al. Larotrectinib in patients with TRK fusion-positive solid tumours: A pooled analysis of three phase 1/2 clinical trials. Lancet Oncol. 2020, 21, 531–540. [Google Scholar] [CrossRef]
- Geoerger: Larotrectinib Efficacy and Safety in Pediatric Patients with TRK Fusion Cancer. Available online: https://scholar.google.com/scholar_lookup?title=Larotrectinib%20efficacyand%20safety%20in%20pediatric%20patients%20with%20TRK%20fusion%20cancer&publication_year=2019&author=B%20Geoerger&author=C%20vanTilburg&author=S%20DuBois (accessed on 2 June 2020).
- Iams, W.; Lovly, C. Anaplastic Lymphoma Kinase as a Therapeutic Target in Non–Small Cell Lung Cancer. Cancer J. 2015, 21, 378–382. [Google Scholar] [CrossRef]
- Nagasaka, M.; Ge, Y.; Sukari, A.; Kukreja, G.; Ou, S.-H.I. A user’s guide to lorlatinib. Crit. Rev. Oncol. Hematol. 2020, 151, 102969. [Google Scholar] [CrossRef]
- Smith, M.; Fagan, C.F.; Pomari, E.; Germano, G.; Frasson, F.; Walsh, C.; Silverman, I.; Bonvini, P.; Li, G. Entrectinib Shows Pediatric Potential. Cancer Discov. 2019, 9, OF4. [Google Scholar] [CrossRef]


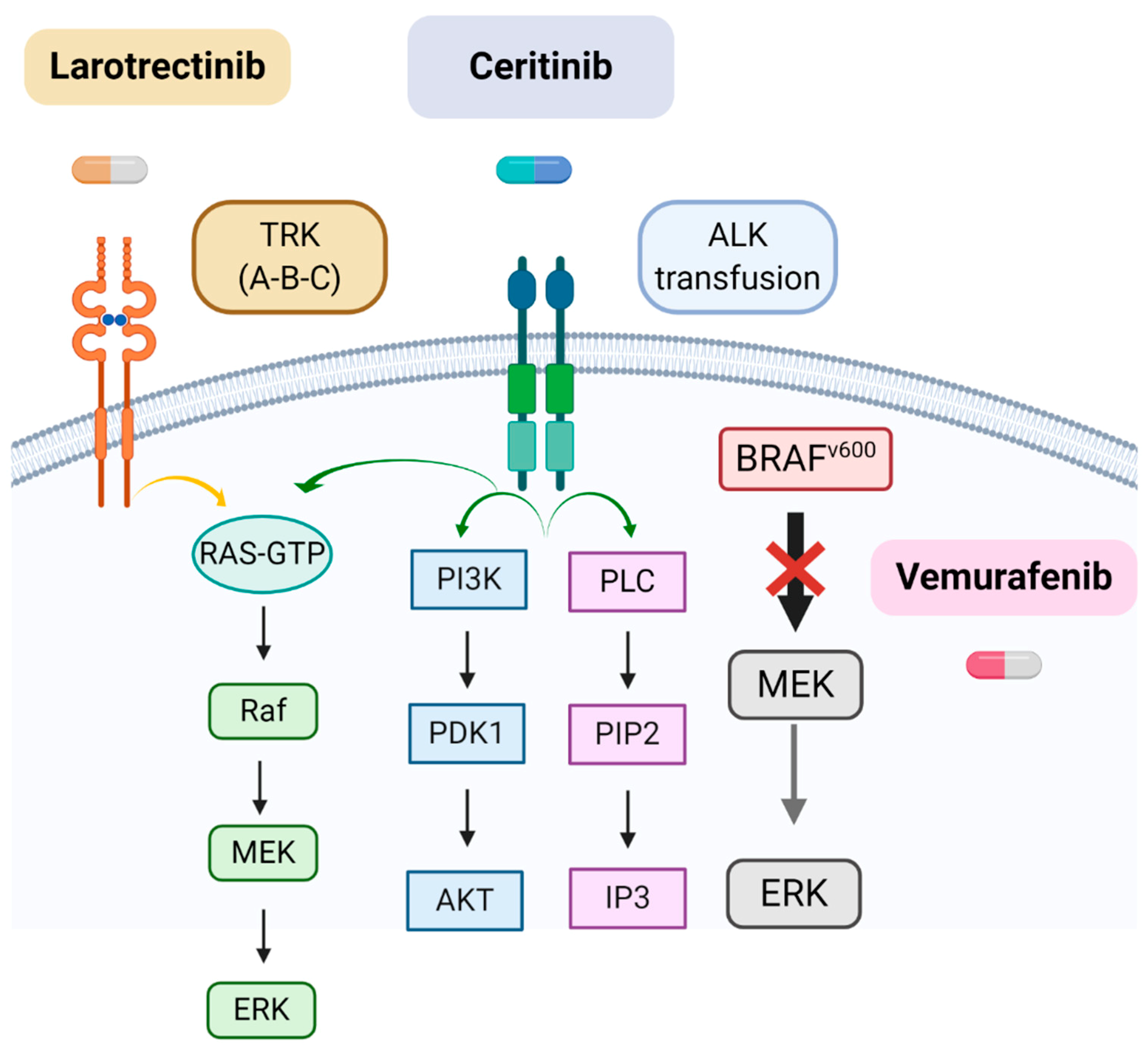{kind=link}
| Molecular Alteration | Type of iHGGs | Target Therapy | Refs |
|---|---|---|---|
| TRK fusions (NTRK1/2/3) | Non-brainstem iHGGs | Larotrectinib | [56,57,58] |
| ALK rearrangements | IHG and LGG-like DIGG/DIA and IHG | Ceritinib, Alectinib, Brigatinib and Ensartinib | [55,59] |
| BRAF V600E | Hypothalamic/chiasmatic glioma | Vemurafenib | [46,47,48] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ceglie, G.; Vinci, M.; Carai, A.; Rossi, S.; Colafati, G.S.; Cacchione, A.; Tornesello, A.; Miele, E.; Locatelli, F.; Mastronuzzi, A. Infantile/Congenital High-Grade Gliomas: Molecular Features and Therapeutic Perspectives. Diagnostics 2020, 10, 648. https://doi.org/10.3390/diagnostics10090648
Ceglie G, Vinci M, Carai A, Rossi S, Colafati GS, Cacchione A, Tornesello A, Miele E, Locatelli F, Mastronuzzi A. Infantile/Congenital High-Grade Gliomas: Molecular Features and Therapeutic Perspectives. Diagnostics. 2020; 10(9):648. https://doi.org/10.3390/diagnostics10090648
Chicago/Turabian StyleCeglie, Giulia, Maria Vinci, Andrea Carai, Sabrina Rossi, Giovanna Stefania Colafati, Antonella Cacchione, Assunta Tornesello, Evelina Miele, Franco Locatelli, and Angela Mastronuzzi. 2020. "Infantile/Congenital High-Grade Gliomas: Molecular Features and Therapeutic Perspectives" Diagnostics 10, no. 9: 648. https://doi.org/10.3390/diagnostics10090648
APA StyleCeglie, G., Vinci, M., Carai, A., Rossi, S., Colafati, G. S., Cacchione, A., Tornesello, A., Miele, E., Locatelli, F., & Mastronuzzi, A. (2020). Infantile/Congenital High-Grade Gliomas: Molecular Features and Therapeutic Perspectives. Diagnostics, 10(9), 648. https://doi.org/10.3390/diagnostics10090648