Effect of Coronavirus Disease 2019 in Pulmonary Circulation. The Particular Scenario of Precapillary Pulmonary Hypertension


 , ,
, ,  ,
,  and
and
Abstract
1. Introduction
- PAH is a rare, noncurable disease characterized by an aberrant pulmonary vascular remodeling that may be either idiopathic or related to different clinical conditions [6]. PAH-related histological changes include endothelial damage and dysfunction, vasoconstriction, vascular cell proliferation leading to vascular obliteration and wall thickening and, eventually, to the formation of plexiform lesions [7]. Furthermore, there is a marked component of perivascular inflammation and microthrombosis [8].
- CTEPH constitutes a different group of precapillary PH secondary to the obstruction of the pulmonary arteries by organized thrombus after a pulmonary embolism. These pulmonary vascular changes induce small-vessel vasculopathy consisting on altered vascular remodeling initiated or potentiated by a combination of defective angiogenesis, impaired fibrinolysis and endothelial dysfunction [9].
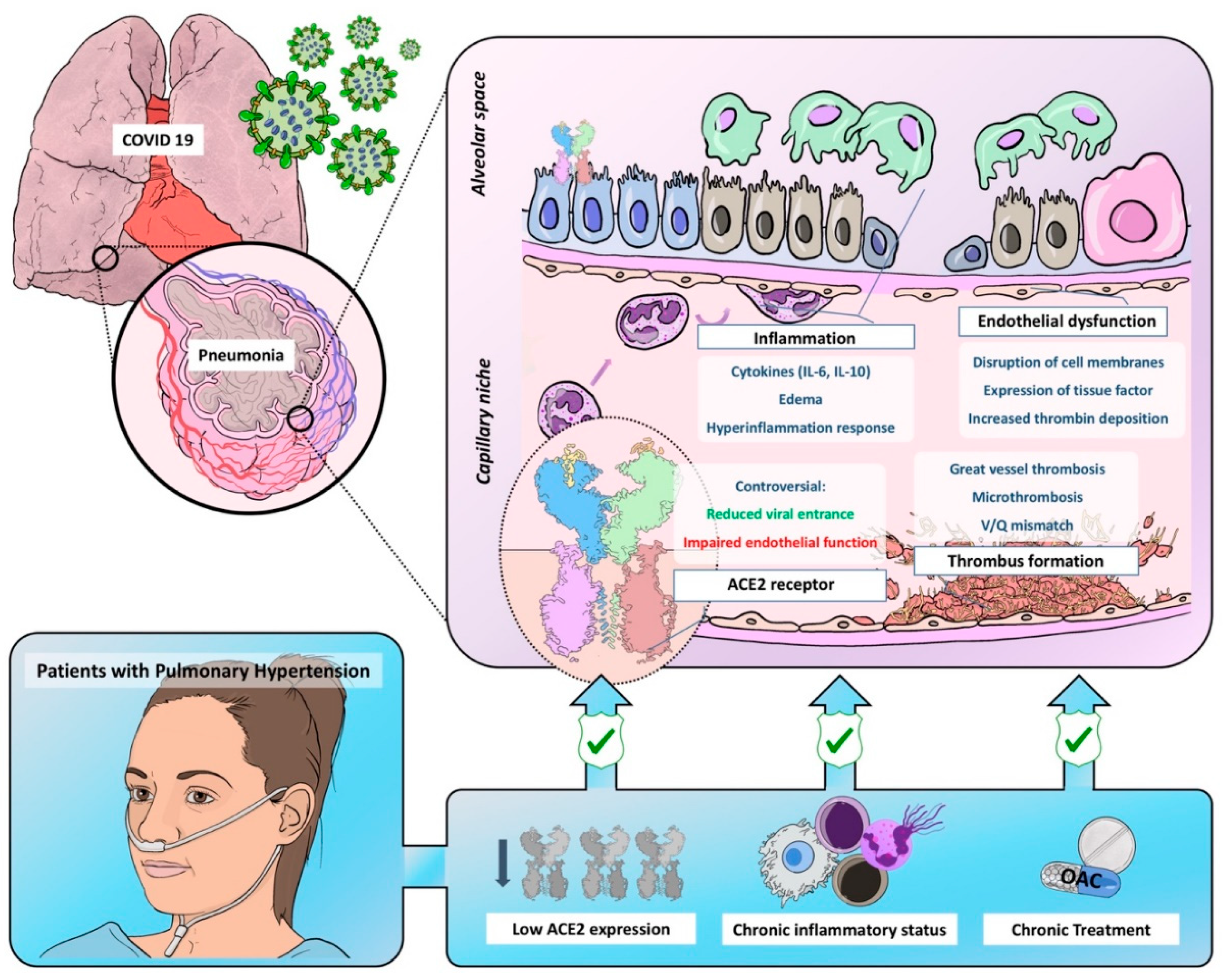
2. Endothelial Dysfunction and COVID-19
3. Thrombosis and COVID-19
4. Inflammatory Response in COVID-19
5. Pulmonary Vasculature Dysfunction
6. Angiotensin Converter Enzyme 2: An Actor in a Leading Role
7. Pulmonary Hypertension: A Paradigm of Endothelial Dysfunction
7.1. Chronic Inflammatory Status
7.2. Low ACE2 Expression in Pulmonary Hypertension: Yin or Yang?
7.3. Hypothetical Protective Effect of Chronic Treatment in PH Patients
7.3.1. Anticoagulation
7.3.2. Specific PH Therapy
8. A Look into the Future: COVID-19 as an Infectious Cause of Chronic PH?
9. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- World Health Organization. WHO Director-General’s Opening Remarks at the Media Briefing on COVID-19–11 March 2020. Available online: https://www.who.int/dg/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19---11-march-2020 (accessed on 12 March 2020).
- Zhai, P.; Ding, Y.; Wu, X.; Long, J.; Zhong, Y.; Li, Y. The epidemiology, diagnosis and treatment of COVID-19. Int. J. Antimicrob. Agents 2020, 55, 105955. [Google Scholar] [CrossRef] [PubMed]
- Wiersinga, W.J.; Rhodes, A.; Cheng, A.C.; Peacock, S.J.; Prescott, H.C. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA 2020. [Google Scholar] [CrossRef] [PubMed]
- Galiè, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Heart J. 2016, 37, 67–119. [Google Scholar] [CrossRef]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53. [Google Scholar] [CrossRef]
- Dai, Z.; Zhu, M.M.; Peng, Y.; Machireddy, N.; Evans, C.E.; Machado, R.; Zhang, X.; Zhao, Y.Y. Therapeutic Targeting of Vascular Remodeling and Right Heart Failure in Pulmonary Arterial Hypertension with a HIF-2α Inhibitor. Am. J. Respir. Crit. Care Med. 2018, 198, 1423–1434. [Google Scholar] [CrossRef]
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmüller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and pathobiology of pulmonary hypertension: State of the art and research perspectives. Eur. Respir. J. 2019, 53. [Google Scholar] [CrossRef]
- Kim, N.H.; Delcroix, M.; Jais, X.; Madani, M.M.; Matsubara, H.; Mayer, E.; Ogo, T.; Tapson, V.F.; Ghofrani, H.A.; Jenkins, D.P. Chronic thromboembolic pulmonary hypertension. Eur. Respir. J. 2019, 53. [Google Scholar] [CrossRef]
- Hoeper, M.M.; Kramer, T.; Pan, Z.; Eichstaedt, C.A.; Spiesshoefer, J.; Benjamin, N.; Olsson, K.M.; Meyer, K.; Vizza, C.D.; Vonk-Noordegraaf, A.; et al. Mortality in pulmonary arterial hypertension: Prediction by the 2015 European pulmonary hypertension guidelines risk stratification model. Eur. Respir. J. 2017, 50. [Google Scholar] [CrossRef]
- Harder, E.M.; Small, A.M.; Fares, W.H. Primary cardiac hospitalizations in pulmonary arterial hypertension: Trends and outcomes from 2001 to 2014. Respir. Med. 2020, 161, 105850. [Google Scholar] [CrossRef]
- Sztrymf, B.; Souza, R.; Bertoletti, L.; Jaïs, X.; Sitbon, O.; Price, L.C.; Simonneau, G.; Humbert, M. Prognostic factors of acute heart failure in patients with pulmonary arterial hypertension. Eur. Respir. J. 2010, 35, 1286–1293. [Google Scholar] [CrossRef] [PubMed]
- Driggin, E.; Madhavan, M.V.; Bikdeli, B.; Chuich, T.; Laracy, J.; Bondi-Zoccai, G.; Brown, T.S.; Nigoghossian, C.; Zidar, D.A.; Haythe, J.; et al. Cardiovascular Considerations for Patients, Health Care Workers, and Health Systems During the Coronavirus Disease 2019 (COVID-19) Pandemic. J. Am. Coll. Cardiol. 2020, 75, 2352–2371. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Fan, Y.; Chen, M.; Wu, X.; Zhang, L.; He, T.; Wang, H.; Wan, J.; Wang, X.; Lu, Z. Cardiovascular Implications of Fatal Outcomes of Patients With Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Nuche, J.; Pérez-Olivares, C.; Segura de la Cal, T.; Jiménez López-Guarch, C.; Arribas Ynsaurriaga, F.; Escribano Subías, P. Clinical course of COVID-19 in pulmonary arterial hypertension patients. Rev. Esp. Cardiol. (Engl. Ed.) 2020. [Google Scholar] [CrossRef]
- Horn, E.M.; Chakinala, M.; Oudiz, R.; Joseloff, E.; Rosenzweig, E.B. Could pulmonary arterial hypertension patients be at a lower risk from severe COVID-19? Pulm. Circ. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Cal, T.S.d.l.; Nuche, J.; López-Guarch, C.J.; Pérez-Olivares, C.; Velázquez, M.; Medrano, F.L.; Gude, M.J.L.; Charterina, S.A.; Ynsaurriaga, F.A.; Subías, P.E. Unexpected favourable course of Coronavirus Disease 2019 in chronic thromboembolic pulmonary hypertension patients. Arch. Bronconeumol. 2020. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Narasaraju, T.; Tang, B.M.; Herrmann, M.; Muller, S.; Chow, V.T.K.; Radic, M. Neutrophilia and NETopathy as Key Pathologic Drivers of Progressive Lung Impairment in Patients With COVID-19. Front. Pharmacol. 2020, 11, 870. [Google Scholar] [CrossRef]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Voicu, S.; Bonnin, P.; Stépanian, A.; Chousterman, B.G.; Le Gall, A.; Malissin, I.; Deye, N.; Siguret, V.; Mebazaa, A.; Mégarbane, B. High prevalence of deep vein thrombosis in mechanically ventilated COVID-19 patients. J. Am. Coll. Cardiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Helms, J.; Tacquard, C.; Severac, F.; Leonard-Lorant, I.; Ohana, M.; Delabranche, X.; Merdji, H.; Clere-Jehl, R.; Schenck, M.; Fagot Gandet, F.; et al. High risk of thrombosis in patients with severe SARS-CoV-2 infection: A multicenter prospective cohort study. Intensive Care Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA 2020. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.; Li, D.; Wang, X.; Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 844–847. [Google Scholar] [CrossRef]
- Tang, N.; Bai, H.; Chen, X.; Gong, J.; Li, D.; Sun, Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J. Thromb. Haemost. 2020. [Google Scholar] [CrossRef] [PubMed]
- Paranjpe, I.; Fuster, V.; Lala, A.; Russak, A.; Glicksberg, B.S.; Levin, M.A.; Charney, A.W.; Narula, J.; Fayad, Z.A.; Bagiella, E.; et al. Association of Treatment Dose Anticoagulation with In-Hospital Survival Among Hospitalized Patients with COVID-19. J. Am. Coll. Cardiol. 2020. [Google Scholar] [CrossRef]
- Thachil, J.; Tang, N.; Gando, S.; Falanga, A.; Cattaneo, M.; Levi, M.; Clark, C.; Iba, T. ISTH interim guidance on recognition and management of coagulopathy in COVID-19. J. Thromb. Haemost. 2020, 18, 1023–1026. [Google Scholar] [CrossRef]
- Bompard, F.; Monnier, H.; Saab, I.; Tordjman, M.; Abdoul, H.; Fournier, L.; Sanchez, O.; Lorut, C.; Chassagnon, G.; Revel, M.P. Pulmonary embolism in patients with Covid-19 pneumonia. Eur. Respir. J. 2020. [Google Scholar] [CrossRef]
- van Dam, L.F.; Kroft, L.J.M.; van der Wal, L.I.; Cannegieter, S.C.; Eikenboom, J.; de Jonge, E.; Huisman, M.V.; Klok, F.A. Clinical and computed tomography characteristics of COVID-19 associated acute pulmonary embolism: A different phenotype of thrombotic disease? Thromb. Res. 2020, 193, 86–89. [Google Scholar] [CrossRef]
- Huertas, A.; Montani, D.; Savale, L.; Pichon, J.; Tu, L.; Parent, F.; Guignabert, C.; Humbert, M. Endothelial cell dysfunction: A major player in SARS-CoV-2 infection (COVID-19)? Eur. Respir. J. 2020. [Google Scholar] [CrossRef]
- Levi, M.; van der Poll, T. Coagulation and sepsis. Thromb. Res. 2017, 149, 38–44. [Google Scholar] [CrossRef]
- Connors, J.M.; Levy, J.H. Thromboinflammation and the hypercoagulability of COVID-19. J. Thromb. Haemost. 2020. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Geng, Y.J.; Wei, Z.Y.; Qian, H.Y.; Huang, J.; Lodato, R.; Castriotta, R.J. Pathophysiological characteristics and therapeutic approaches for pulmonary injury and cardiovascular complications of coronavirus disease 2019. Cardiovasc. Pathol. 2020, 47, 107228. [Google Scholar] [CrossRef]
- Diao, B.; Wang, C.; Tan, Y.; Chen, X.; Liu, Y.; Ning, L.; Chen, L.; Li, M.; Wang, G.; Yuan, Z.; et al. Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (COVID-19). Front. Immunol. 2020, 11, 827. [Google Scholar] [CrossRef] [PubMed]
- Kronbichler, A.; Effenberger, M.; Eisenhut, M.; Lee, K.H.; Shin, J.I. Seven recommendations to rescue the patients and reduce the mortality from COVID-19 infection: An immunological point of view. Autoimmun. Rev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Chen, L.; Liu, Z.; Pan, J.; Zhou, D.; Wang, H.; Gong, H.; Fu, Z.; Song, Q.; Min, Q.; et al. Clinical Features and Short-term Outcomes of Elderly Patients With COVID-19. Int. J. Infect. Dis. 2020, 10, 245–250. [Google Scholar] [CrossRef]
- Mc Gonagle, D.; O’Donnell, J.S.; Sharif, K.; Emery, P.; Bridgewood, C. Immune mechanisms of pulmonary intravascular coagulopathy in COVID-19 pneumonia. Lancet Rheumatol. 2020. [Google Scholar] [CrossRef]
- Becker, R.C. COVID-19 update: Covid-19-associated coagulopathy. J. Thromb. Thrombolysis 2020. [Google Scholar] [CrossRef]
- La Rosée, P.; Horne, A.; Hines, M.; von Bahr Greenwood, T.; Machowicz, R.; Berliner, N.; Birndt, S.; Gil-Herrera, J.; Girschikofsky, M.; Jordan, M.B.; et al. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood 2019, 133, 2465–2477. [Google Scholar] [CrossRef]
- Rios-Fernández, R.; Callejas-Rubio, J.L.; García-Rodríguez, S.; Sancho, J.; Zubiaur, M.; Ortego-Centeno, N. Tocilizumab as an Adjuvant Therapy for Hemophagocytic Lymphohistiocytosis Associated With Visceral Leishmaniasis. Am. J. Ther. 2016, 23, 1193–1196. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Han, M.; Li, T.; Sun, W.; Wang, D.; Fu, B.; Zhou, Y.; Zheng, X.; Yang, Y.; Li, X.; et al. Effective treatment of severe COVID-19 patients with tocilizumab. Proc. Natl. Acad. Sci. USA 2020, 117, 10970–10975. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Wang, Y.; Colunga-Lozano, L.E.; Prasad, M.; Tangamornsuksan, W.; Rochwerg, B.; Yao, L.; Motaghi, S.; Couban, R.J.; Ghadimi, M.; et al. Efficacy and safety of corticosteroids in COVID-19 based on evidence for COVID-19, other coronavirus infections, influenza, community-acquired pneumonia and acute respiratory distress syndrome: A systematic review and meta-analysis. CMAJ 2020. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Zhao, Y.Y.; Evans, C.E. The stimulation of thrombosis by hypoxia. Thromb. Res. 2019, 181, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Tomar, B.; Anders, H.J.; Desai, J.; Mulay, S.R. Neutrophils and Neutrophil Extracellular Traps Drive Necroinflammation in COVID-19. Cells 2020, 9, 1383. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Coppola, S.; Cressoni, M.; Busana, M.; Rossi, S.; Chiumello, D. Covid-19 Does Not Lead to a “Typical” Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2020. [Google Scholar] [CrossRef]
- Marini, J.J.; Gattinoni, L. Management of COVID-19 Respiratory Distress. JAMA 2020. [Google Scholar] [CrossRef]
- Solaimanzadeh, I. Acetazolamide, Nifedipine and Phosphodiesterase Inhibitors: Rationale for Their Utilization as Adjunctive Countermeasures in the Treatment of Coronavirus Disease 2019 (COVID-19). Cureus 2020, 12, e7343. [Google Scholar] [CrossRef]
- Archer, S.L.; Sharp, W.W.; Weir, E.K. Differentiating COVID-19 Pneumonia from Acute Respiratory Distress Syndrome (ARDS) and High Altitude Pulmonary Edema (HAPE): Therapeutic Implications. Circulation 2020. [Google Scholar] [CrossRef]
- Solaimanzadeh, I. Nifedipine and Amlodipine Are Associated With Improved Mortality and Decreased Risk for Intubation and Mechanical Ventilation in Elderly Patients Hospitalized for COVID-19. Cureus 2020, 12, e8069. [Google Scholar] [CrossRef]
- Luks, A.M.; Swenson, E.R. COVID-19 Lung Injury and High Altitude Pulmonary Edema: A False Equation with Dangerous Implications. Ann. Am. Thorac. Soc. 2020. [Google Scholar] [CrossRef]
- Brugger, H.; Basnyat, B.; Ellerton, J.; Hefti, U.; Strapazzon, G.; Zafren, K. COVID-19 Lung Injury Is Different From High Altitude Pulmonary Edema. High. Alt. Med. Biol. 2020, 21, 204–205. [Google Scholar] [CrossRef]
- Danser, A.H.J.; Epstein, M.; Batlle, D. Renin-Angiotensin System Blockers and the COVID-19 Pandemic: At Present There Is No Evidence to Abandon Renin-Angiotensin System Blockers. Hypertension 2020. [Google Scholar] [CrossRef] [PubMed]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef]
- Hofmann, H.; Geier, M.; Marzi, A.; Krumbiegel, M.; Peipp, M.; Fey, G.H.; Gramberg, T.; Pöhlmann, S. Susceptibility to SARS coronavirus S protein-driven infection correlates with expression of angiotensin converting enzyme 2 and infection can be blocked by soluble receptor. Biochem. Biophys. Res. Commun. 2004, 319, 1216–1221. [Google Scholar] [CrossRef] [PubMed]
- Igase, M.; Kohara, K.; Nagai, T.; Miki, T.; Ferrario, C.M. Increased expression of angiotensin converting enzyme 2 in conjunction with reduction of neointima by angiotensin II type 1 receptor blockade. Hypertens. Res. 2008, 31, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Furuhashi, M.; Moniwa, N.; Mita, T.; Fuseya, T.; Ishimura, S.; Ohno, K.; Shibata, S.; Tanaka, M.; Watanabe, Y.; Akasaka, H.; et al. Urinary angiotensin-converting enzyme 2 in hypertensive patients may be increased by olmesartan, an angiotensin II receptor blocker. Am. J. Hypertens. 2015, 28, 15–21. [Google Scholar] [CrossRef]
- Shyh, G.I.; Nawarskas, J.J.; Cheng-Lai, A. Angiotensin-Converting Enzyme Inhibitors and Angiotensin Receptor Blockers in Patients With Coronavirus Disease 2019: Friend or Foe? Cardiol. Rev. 2020, 28, 213–216. [Google Scholar] [CrossRef]
- Felice, C.; Nardin, C.; Di Tanna, G.L.; Grossi, U.; Bernardi, E.; Scaldaferri, L.; Romagnoli, M.; Tonon, L.; Cavasin, P.; Novello, S.; et al. Use of RAAS inhibitors and risk of clinical deterioration in COVID-19: Results from an Italian cohort of 133 hypertensives. Am. J. Hypertens. 2020. [Google Scholar] [CrossRef]
- Amat-Santos, I.J.; Santos-Martinez, S.; López-Otero, D.; Nombela-Franco, L.; Gutiérrez-Ibanes, E.; Del Valle, R.; Muñoz-García, E.; Jiménez-Diaz, V.A.; Regueiro, A.; González-Ferreiro, R.; et al. Ramipril in High Risk Patients with COVID-19. J. Am. Coll. Cardiol. 2020. [Google Scholar] [CrossRef]
- Meng, J.; Xiao, G.; Zhang, J.; He, X.; Ou, M.; Bi, J.; Yang, R.; Di, W.; Wang, Z.; Li, Z.; et al. Renin-angiotensin system inhibitors improve the clinical outcomes of COVID-19 patients with hypertension. Emerg. Microbes Infect. 2020, 9, 757–760. [Google Scholar] [CrossRef] [PubMed]
- Fraga-Silva, R.A.; Da Silva, D.G.; Montecucco, F.; Mach, F.; Stergiopulos, N.; da Silva, R.F.; Santos, R.A. The angiotensin-converting enzyme 2/angiotensin-(1-7)/Mas receptor axis: A potential target for treating thrombotic diseases. Thromb. Haemost. 2012, 108, 1089–1096. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.H.; Dong, X.F.; Hao, Q.Q.; Zhou, X.M.; Yu, Q.T.; Li, S.Y.; Chen, X.; Tengbeh, A.F.; Dong, B.; Zhang, Y. ACE2 and Ang-(1-7) protect endothelial cell function and prevent early atherosclerosis by inhibiting inflammatory response. Inflamm. Res. 2015, 64, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Shi, L.; Ma, X.; Su, H.; Ma, G.; Wu, X.; Ying, K.; Zhang, R. RhoA-Rho associated kinase signaling leads to renin-angiotensin system imbalance and angiotensin converting enzyme 2 has a protective role in acute pulmonary embolism. Thromb. Res. 2019, 176, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Fraga-Silva, R.A.; Sorg, B.S.; Wankhede, M.; Dedeugd, C.; Jun, J.Y.; Baker, M.B.; Li, Y.; Castellano, R.K.; Katovich, M.J.; Raizada, M.K.; et al. ACE2 activation promotes antithrombotic activity. Mol. Med. 2010, 16, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zeng, H.; Gu, J.; Li, H.; Zheng, L.; Zou, Q. Progress and Prospects on Vaccine Development against SARS-CoV-2. Vaccines 2020, 8, 153. [Google Scholar] [CrossRef]
- Nicholls, J.; Peiris, M. Good ACE, bad ACE do battle in lung injury, SARS. Nat. Med. 2005, 11, 821–822. [Google Scholar] [CrossRef]
- Guignabert, C.; Tu, L.; Girerd, B.; Ricard, N.; Huertas, A.; Montani, D.; Humbert, M. New molecular targets of pulmonary vascular remodeling in pulmonary arterial hypertension: Importance of endothelial communication. Chest 2015, 147, 529–537. [Google Scholar] [CrossRef]
- Guignabert, C. Dysfunction and Restoration of Endothelial Cell Communications in Pulmonary Arterial Hypertension: Therapeutic Implications; Springer: Singapore, 2020; pp. 147–155. [Google Scholar]
- Farha, S. COVID-19 and pulmonary hypertension. Cleve Clin. J. Med. 2020. [Google Scholar] [CrossRef]
- Fernandes, T.M.; Papamatheakis, D.G.; Poch, D.S.; Kim, N.H. Letter to the Editor regarding “Could pulmonary arterial hypertension patients be at lower risk from severe COVID-19?”. Pulm. Circ. 2020, 10. [Google Scholar] [CrossRef]
- Horn, E.; Chakinala, M.M.; Oudiz, R.; Joseloff, E.; Rosenzweig, E.B. EXPRESS: Author rebuttal to response regarding “Letter to the Editor regarding ‘Could pulmonary arterial hypertension patients be at lower risk from severe COVID-19?’. Pulm. Circ. 2020. [Google Scholar] [CrossRef]
- Provencher, S.; Potus, F.; Bonnet, S. COVID-19 and the pulmonary vasculature. Pulm. Circ. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Magrone, T.; Magrone, M.; Jirillo, E. Focus on Receptors for Coronaviruses with Special Reference to Angiotensin-converting Enzyme 2 as a Potential Drug Target–A Perspective. Endocr. Metab. Immune Disord. Drug Targets 2020. [Google Scholar] [CrossRef] [PubMed]
- Stacher, E.; Graham, B.B.; Hunt, J.M.; Gandjeva, A.; Groshong, S.D.; McLaughlin, V.V.; Jessup, M.; Grizzle, W.E.; Aldred, M.A.; Cool, C.D.; et al. Modern age pathology of pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 261–272. [Google Scholar] [CrossRef]
- Pullamsetti, S.S.; Seeger, W.; Savai, R. Classical IL-6 signaling: A promising therapeutic target for pulmonary arterial hypertension. J. Clin. Invest. 2018, 128, 1720–1723. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Dong, J.; Martin, M.; He, M.; Gongol, B.; Marin, T.L.; Chen, L.; Shi, X.; Yin, Y.; Shang, F.; et al. AMP-activated Protein Kinase Phosphorylation of Angiotensin-Converting Enzyme 2 in Endothelium Mitigates Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2018, 198, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Hemnes, A.R.; Rathinasabapathy, A.; Austin, E.A.; Brittain, E.L.; Carrier, E.J.; Chen, X.; Fessel, J.P.; Fike, C.D.; Fong, P.; Fortune, N.; et al. A potential therapeutic role for angiotensin-converting enzyme 2 in human pulmonary arterial hypertension. Eur. Respir. J. 2018, 51. [Google Scholar] [CrossRef]
- Poterucha, T.J.; Libby, P.; Goldhaber, S.Z. More than an anticoagulant: Do heparins have direct anti-inflammatory effects? Thromb. Haemost. 2017, 117, 437–444. [Google Scholar] [CrossRef]
- Searcy, R.J.; Morales, J.R.; Ferreira, J.A.; Johnson, D.W. The role of inhaled prostacyclin in treating acute respiratory distress syndrome. Ther. Adv. Respir. Dis. 2015, 9, 302–312. [Google Scholar] [CrossRef]
- Spaczyńska, M.; Rocha, S.F.; Oliver, E. Pharmacology of Pulmonary Arterial Hypertension: An overview on current and emerging therapies. ACS Pharmacol. Transl. Sci. 2020. [Google Scholar] [CrossRef]
- Galiè, N.; Ghofrani, H.A.; Torbicki, A.; Barst, R.J.; Rubin, L.J.; Badesch, D.; Fleming, T.; Parpia, T.; Burgess, G.; Branzi, A.; et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N. Engl. J. Med. 2005, 353, 2148–2157. [Google Scholar] [CrossRef] [PubMed]
- Cornet, A.D.; Hofstra, J.J.; Swart, E.L.; Girbes, A.R.; Juffermans, N.P. Sildenafil attenuates pulmonary arterial pressure but does not improve oxygenation during ARDS. Intensive Care Med. 2010, 36, 758–764. [Google Scholar] [CrossRef] [PubMed]
- Isidori, A.M.; Giannetta, E.; Pofi, R.; Venneri, M.A.; Gianfrilli, D.; Campolo, F.; Mastroianni, C.M.; Lenzi, A.; d’Ettorre, G. Targeting the NO-cGMP-PDE5 pathway in COVID-19 infection. Andrology 2020. [Google Scholar] [CrossRef] [PubMed]
- Araz, O. Current Pharmacological Approach to ARDS: The Place of Bosentan. Eurasian J. Med. 2020, 52, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Huang, J.A.; Fraidenburg, D.R. Bosentan as rescue treatment in refractory hypoxemia and pulmonary hypertension in a patient with ARDS and H7N9 influenza virus infection. Lung 2014, 192, 635–636. [Google Scholar] [CrossRef] [PubMed]
- Javor, S.; Salsano, A. Why not consider an endothelin receptor antagonist against SARS-CoV-2? Med. Hypotheses 2020, 141, 109792. [Google Scholar] [CrossRef]
- Chen, L.; Liu, P.; Gao, H.; Sun, B.; Chao, D.; Wang, F.; Zhu, Y.; Hedenstierna, G.; Wang, C.G. Inhalation of nitric oxide in the treatment of severe acute respiratory syndrome: A rescue trial in Beijing. Clin. Infect. Dis. 2004, 39, 1531–1535. [Google Scholar] [CrossRef]
- Zamanian, R.T.; Pollack, C.V.; Gentile, M.A.; Rashid, M.; Fox, J.C.; Mahaffey, K.W.; de Jesus Perez, V. Outpatient Inhaled Nitric Oxide in a Patient with Vasoreactive IPAH and COVID-19 Infection. Am. J. Respir. Crit. Care Med. 2020. [Google Scholar] [CrossRef]
- Akerström, S.; Mousavi-Jazi, M.; Klingström, J.; Leijon, M.; Lundkvist, A.; Mirazimi, A. Nitric oxide inhibits the replication cycle of severe acute respiratory syndrome coronavirus. J. Virol. 2005, 79, 1966–1969. [Google Scholar] [CrossRef]
- Keyaerts, E.; Vijgen, L.; Chen, L.; Maes, P.; Hedenstierna, G.; Van Ranst, M. Inhibition of SARS-coronavirus infection in vitro by S-nitroso-N-acetylpenicillamine, a nitric oxide donor compound. Int. J. Infect. Dis. 2004, 8, 223–226. [Google Scholar] [CrossRef]
- Chung, L.; Liu, J.; Parsons, L.; Hassoun, P.M.; McGoon, M.; Badesch, D.B.; Miller, D.P.; Nicolls, M.R.; Zamanian, R.T. Characterization of connective tissue disease-associated pulmonary arterial hypertension from REVEAL: Identifying systemic sclerosis as a unique phenotype. Chest 2010, 138, 1383–1394. [Google Scholar] [CrossRef] [PubMed]
- Potus, F.; Mai, V.; Lebret, M.; Malenfant, S.; Breton-Gagnon, E.; Lajoie, A.C.; Boucherat, O.; Bonnet, S.; Provencher, S. Novel insights on the pulmonary vascular consequences of COVID-19. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- George, P.M.; Wells, A.U.; Jenkins, R.G. Pulmonary fibrosis and COVID-19: The potential role for antifibrotic therapy. Lancet Respir. Med. 2020. [Google Scholar] [CrossRef]
- Zhang, H.; Li, Y.; Zeng, Y.; Wu, R.; Ou, J. Endothelin-1 downregulates angiotensin-converting enzyme-2 expression in human bronchial epithelial cells. Pharmacology 2013, 91, 297–304. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Drug | NCT | Intervention | Control |
|---|---|---|---|
| Heparin | 04362085 04345848 04406389 04359277 04367831 04377997 04401293 04444700 04360824 04372589 04344756 | Therapeutic anticoagulation | Prophylactic anticoagulation |
| Bivalirudin | 04445935 | Therapeutic anticoagulation | Standard care (prophylactic heparin) |
| Rivaroxaban | 04416048 04394377 | Therapeutic anticoagulation | Standard care (prophylactic heparin) |
| Drug | NCT | Intervention | Control |
|---|---|---|---|
| Sildenadil | 04304313 | Sildenafil 100 mg od. | NA |
| Ambrisentan | 04393246 | Ambrisentan 5 mg od. | Standard care |
| Iloprost | 04420741 | Intravenous iloprost | Saline |
| 04445246 | Inhaled iloprost | Standard care | |
| Nitric oxide | 04338828 04388683 04383002 04358588 04290858 04305457 04290871 04337918 04397692 04306393 | Inhaled nitric oxide | Standard care |
| 04421508 04398290 03331445 | Inhaled nitric oxide | Pulsed inhaled N2 | |
| VIP analog | 04433546 | Oral VIP analog | Placebo |
| 04360096 | Inhaled VIP analog | Placebo | |
| 04311697 | Intravenous VIP analog | Placebo | |
| Recombinant ACE2 | 04375046 04382950 | Intravenous ACE2 | Standard care |
| Tocilizumab | 04435717 04412291 04346355 04403685 04361552 04322773 04330638 04331808 | Tocilizumab | Standard care |
| 04345445 04377503 | Tocilizumab | Corticosteroids | |
| 04412772 04377750 04335071 04372186 04356937 04320615 04335305 | Tocilizumab | Placebo | |
| 04332094 | Tocilizuman + HCQ + Azytromicin | HCQ + Azytromicin | |
| 04363736 | Tocilizumab 4 mg/Kg | Tocilizumab 8 mg/Kg | |
| 04409262 | Tocilizumab + Remdesivir | Remdesivir | |
| 04310228 | Tocilizumab | Fapiravir |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nuche, J.; Segura de la Cal, T.; Jiménez López Guarch, C.; López-Medrano, F.; Delgado, C.P.-O.; Ynsaurriaga, F.A.; Delgado, J.F.; Ibáñez, B.; Oliver, E.; Subías, P.E. Effect of Coronavirus Disease 2019 in Pulmonary Circulation. The Particular Scenario of Precapillary Pulmonary Hypertension. Diagnostics 2020, 10, 548. https://doi.org/10.3390/diagnostics10080548
Nuche J, Segura de la Cal T, Jiménez López Guarch C, López-Medrano F, Delgado CP-O, Ynsaurriaga FA, Delgado JF, Ibáñez B, Oliver E, Subías PE. Effect of Coronavirus Disease 2019 in Pulmonary Circulation. The Particular Scenario of Precapillary Pulmonary Hypertension. Diagnostics. 2020; 10(8):548. https://doi.org/10.3390/diagnostics10080548
Chicago/Turabian StyleNuche, Jorge, Teresa Segura de la Cal, Carmen Jiménez López Guarch, Francisco López-Medrano, Carmen Pérez-Olivares Delgado, Fernando Arribas Ynsaurriaga, Juan F. Delgado, Borja Ibáñez, Eduardo Oliver, and Pilar Escribano Subías. 2020. "Effect of Coronavirus Disease 2019 in Pulmonary Circulation. The Particular Scenario of Precapillary Pulmonary Hypertension" Diagnostics 10, no. 8: 548. https://doi.org/10.3390/diagnostics10080548
APA StyleNuche, J., Segura de la Cal, T., Jiménez López Guarch, C., López-Medrano, F., Delgado, C. P.-O., Ynsaurriaga, F. A., Delgado, J. F., Ibáñez, B., Oliver, E., & Subías, P. E. (2020). Effect of Coronavirus Disease 2019 in Pulmonary Circulation. The Particular Scenario of Precapillary Pulmonary Hypertension. Diagnostics, 10(8), 548. https://doi.org/10.3390/diagnostics10080548