Epithelioid Cutaneous Mesenchymal Neoplasms: A Practical Diagnostic Approach



Abstract
1. Introduction
2. Practical Diagnostic Approach to Epithelioid Cutaneous Mesenchymal Neoplasms
2.1. Skin Anatomy
2.2. Clinical Considerations
2.3. Histologic Evaluation
2.4. Immunohistochemistry
2.5. Molecular Diagnostic Advancements
3. Epithelioid Fibroblastic/Myofibroblastic and So-Called Fibrohistiocytic Tumors
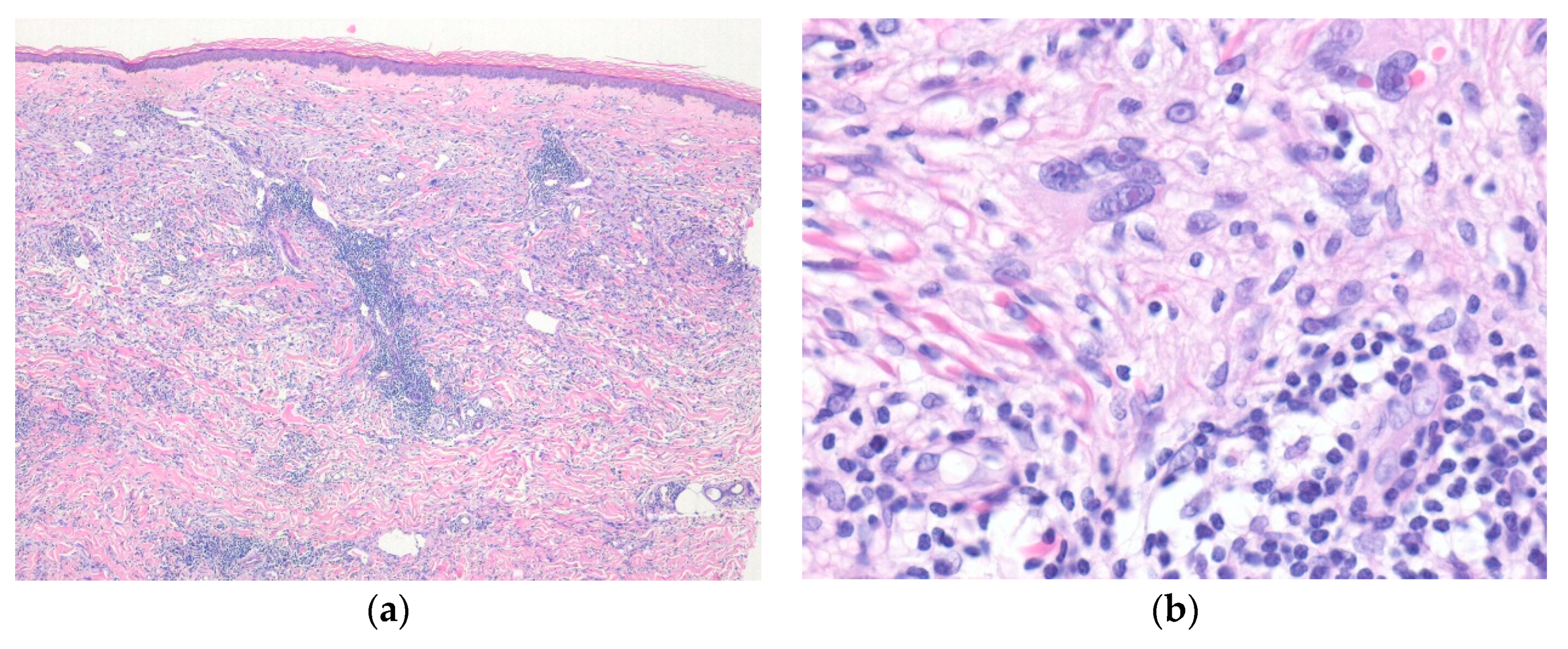
3.1. Epithelioid Fibrous Histiocytoma
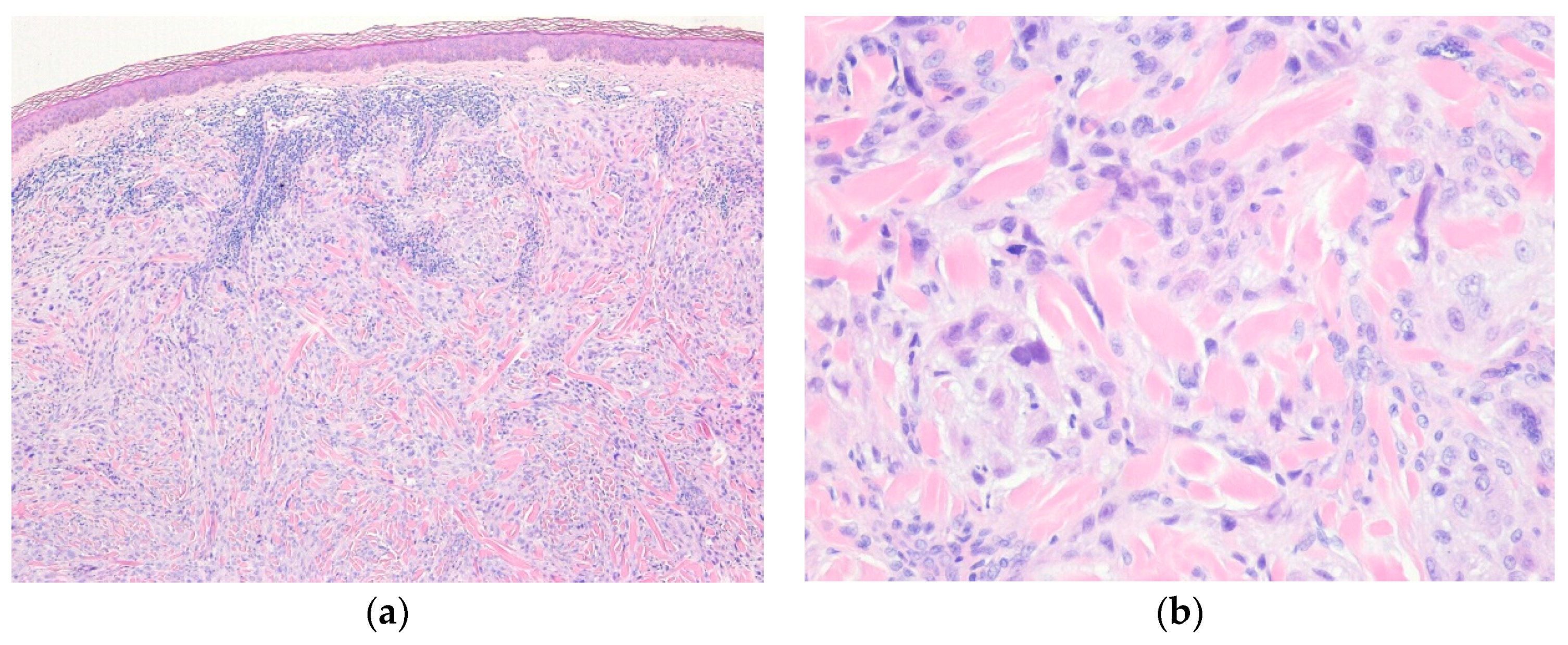
3.2. Myxoinflammatory Fibroblastic Sarcoma
4. Epithelioid Smooth Muscle, Perivascular, and Vascular Tumors
4.1. Cutaneous Smooth Muscle Tumors
4.2. Glomus Tumor
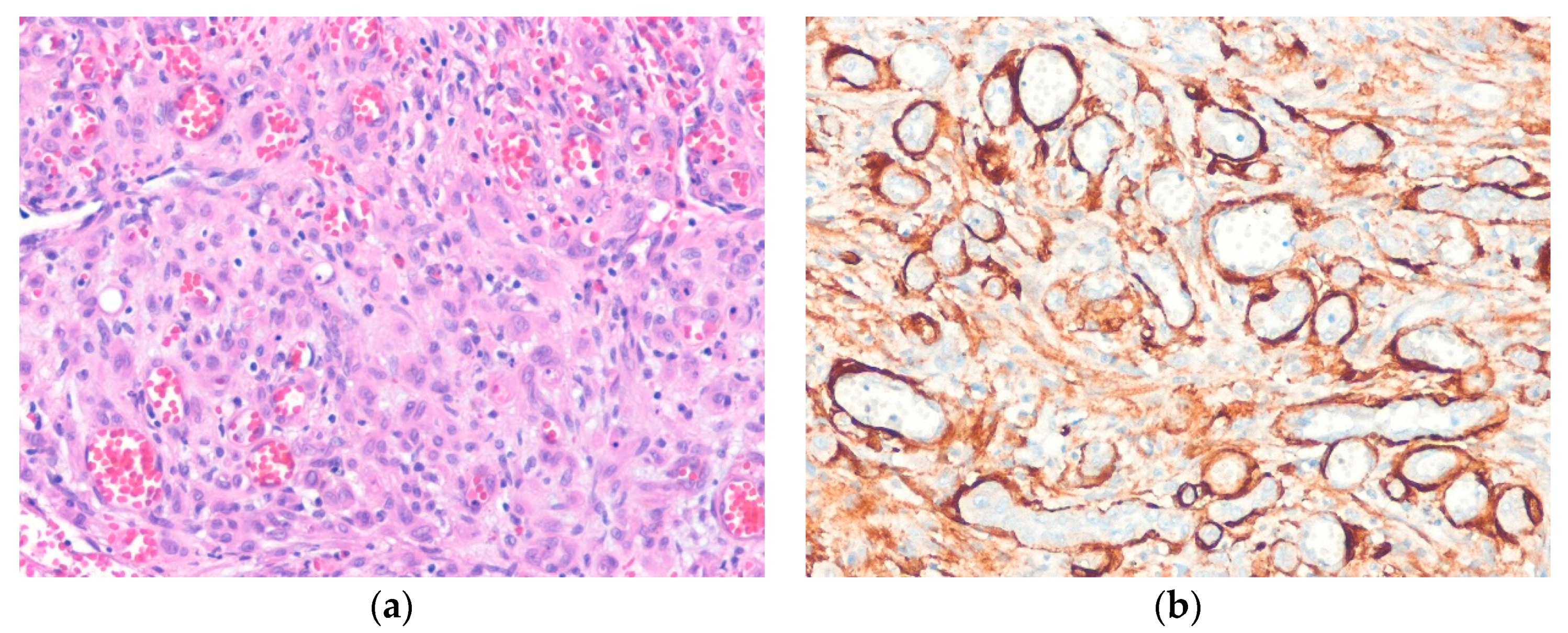
4.3. Epithelioid Hemangioma
4.4. Pseudomyogenic Hemangioendothelioma
4.5. Epithelioid Hemangioendothelioma
5. Epithelioid Nerve Sheath Tumors
5.1. Epithelioid Schwannoma
5.2. Granular Cell Tumor
5.3. Ectopic Meningioma
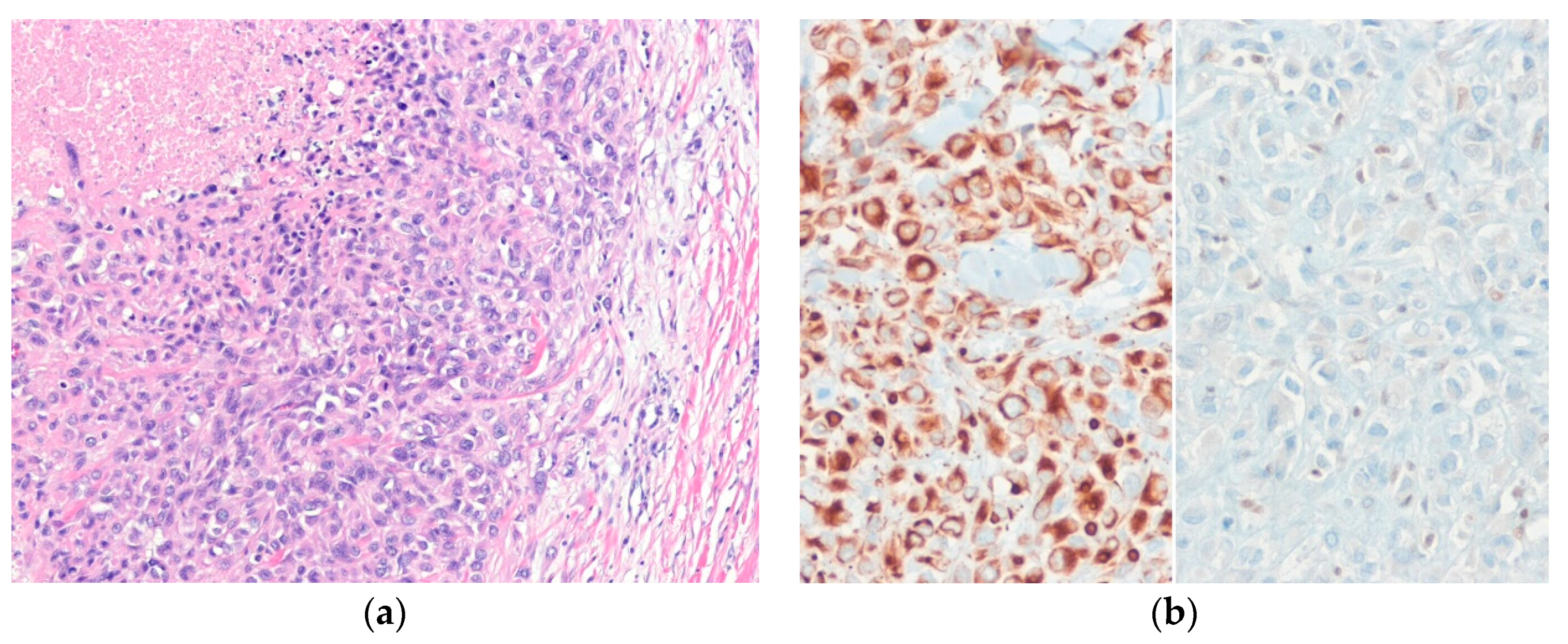
5.4. Epithelioid Malignant Peripheral Nerve Sheath Tumor
6. Epithelioid Mesenchymal Tumors of Uncertain Differentiation
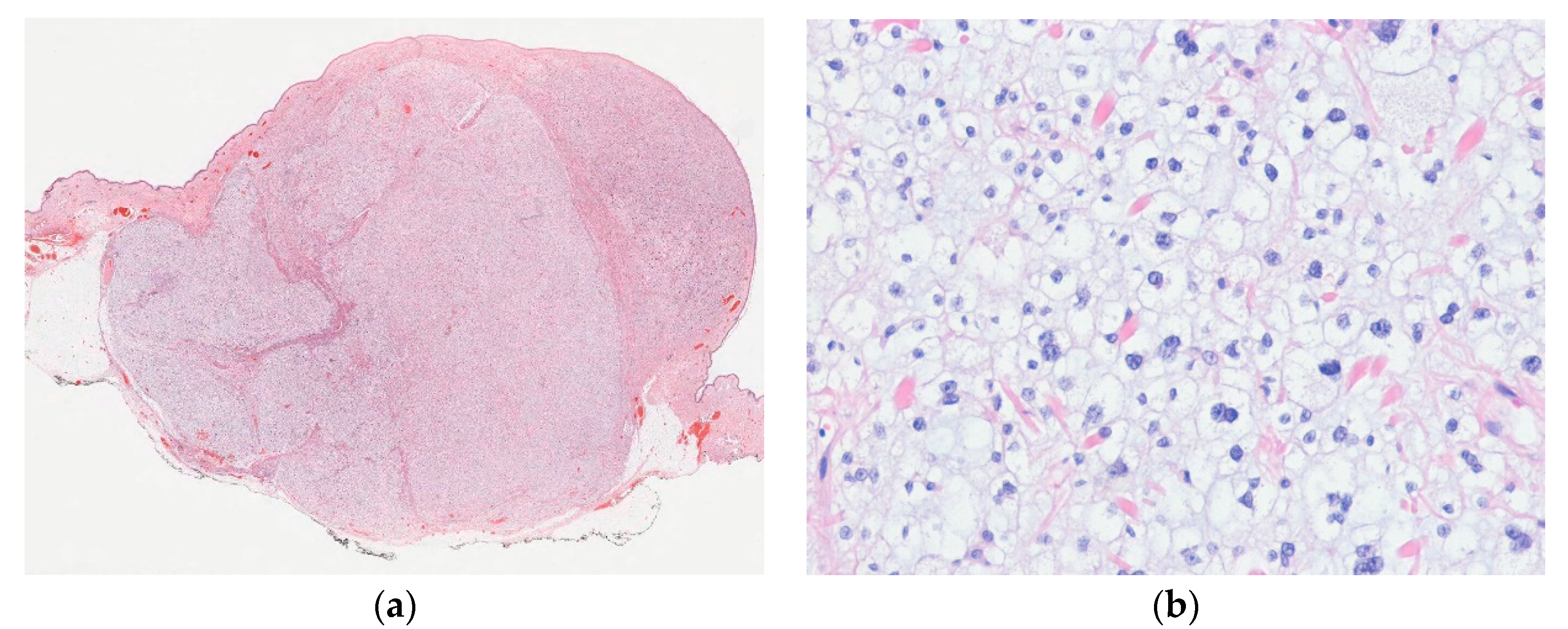
6.1. Distinctive Dermal Clear Cell Mesenchymal Neoplasm
6.2. Cellular Neurothekeoma
6.3. Angiomatoid Fibrous Histiocytoma
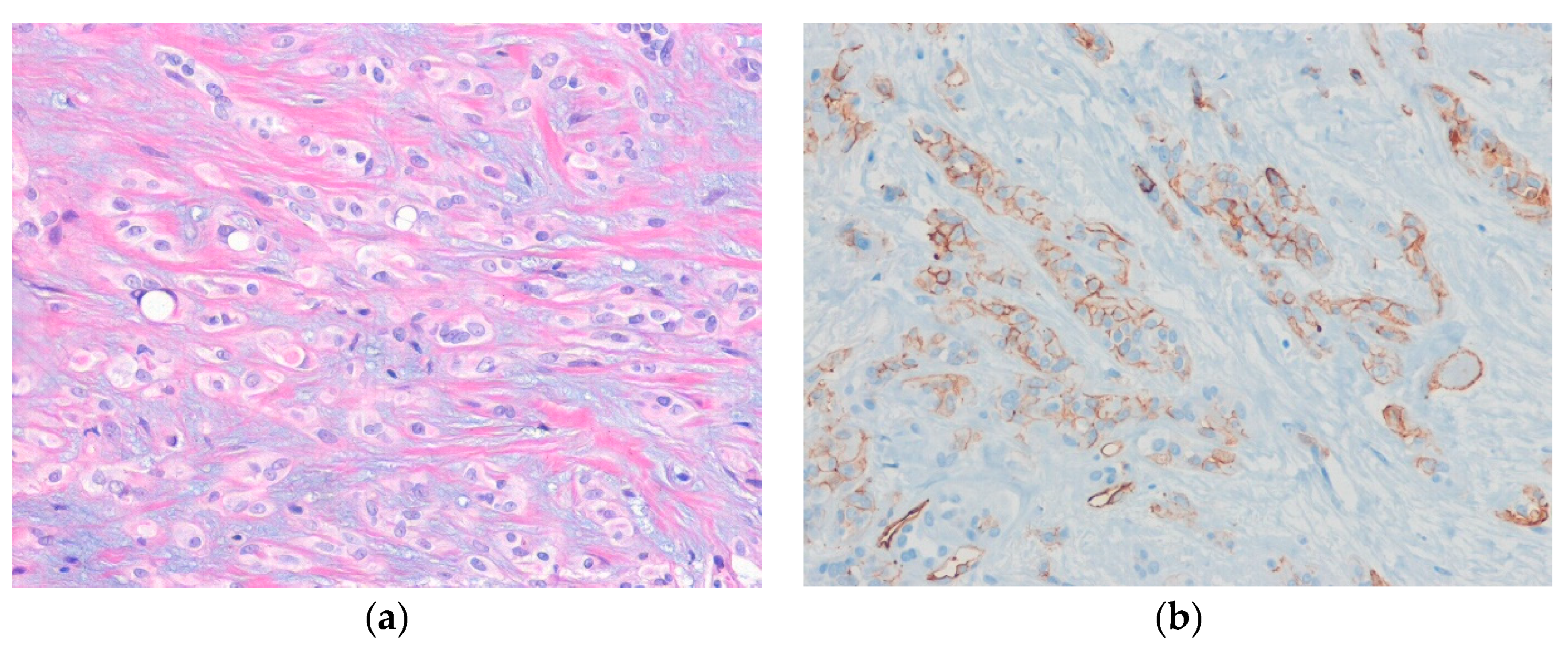
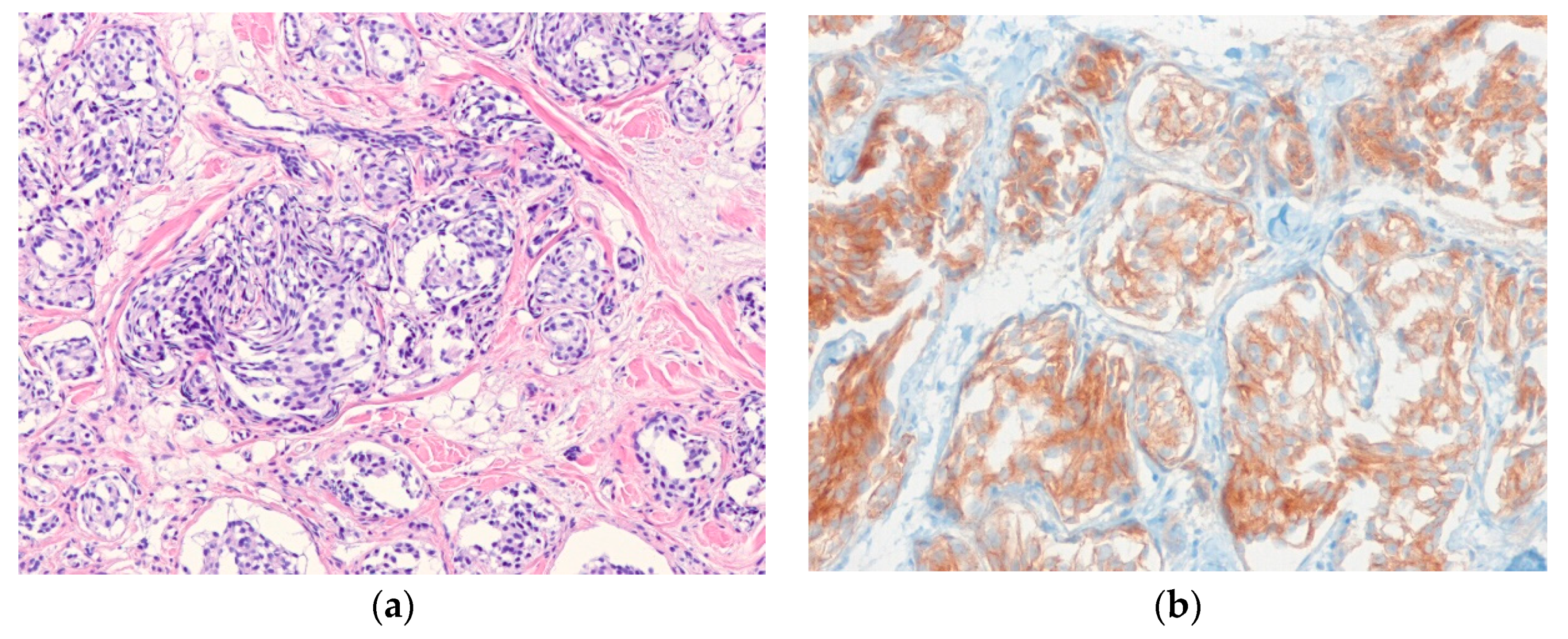
6.4. Myoepithelioma
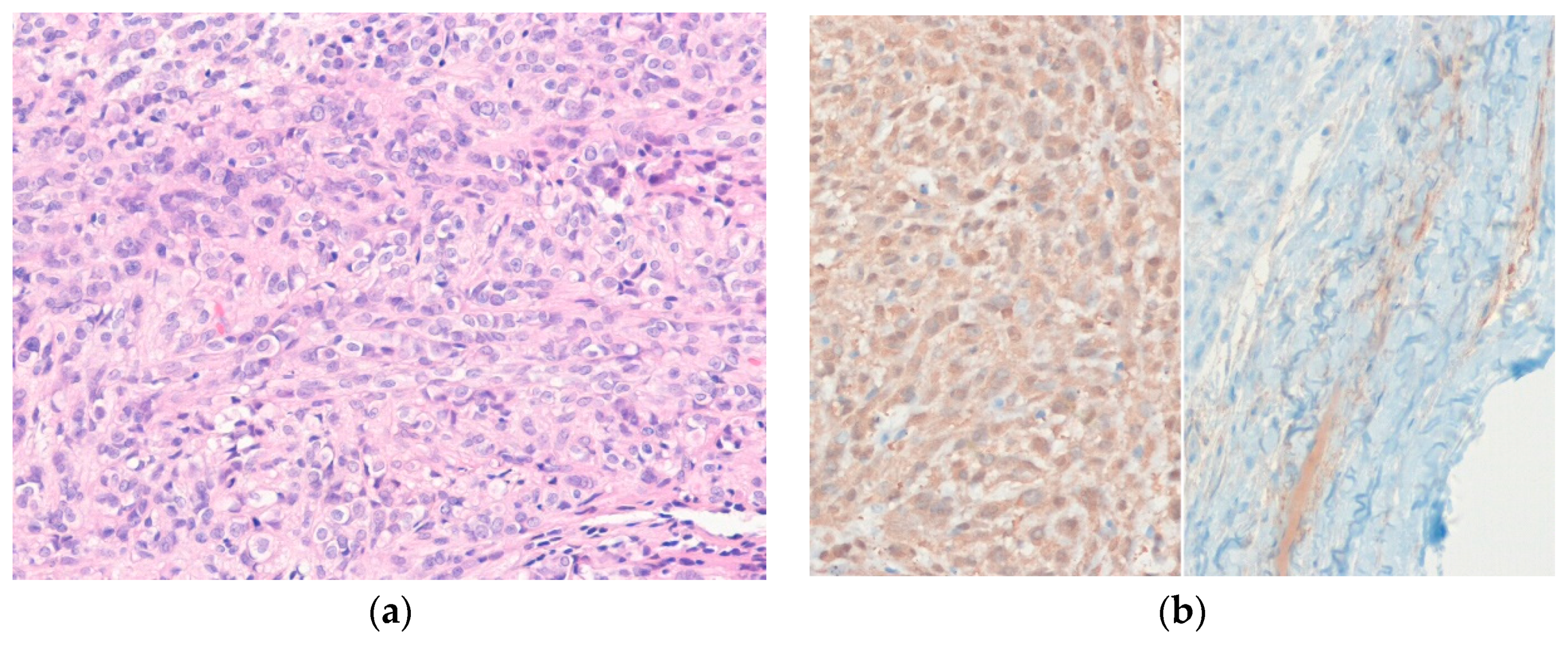
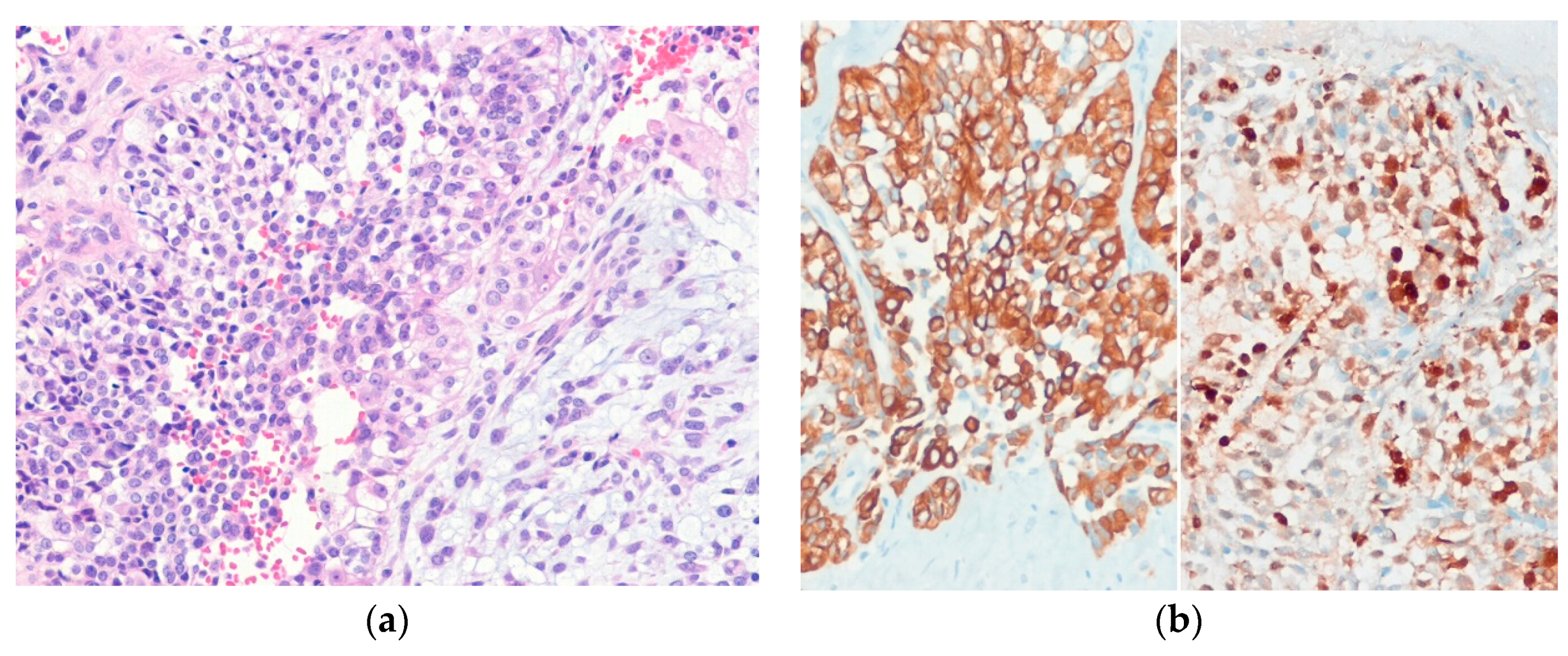
6.5. Epithelioid Sarcoma
6.6. Dermal Clear Cell Sarcoma
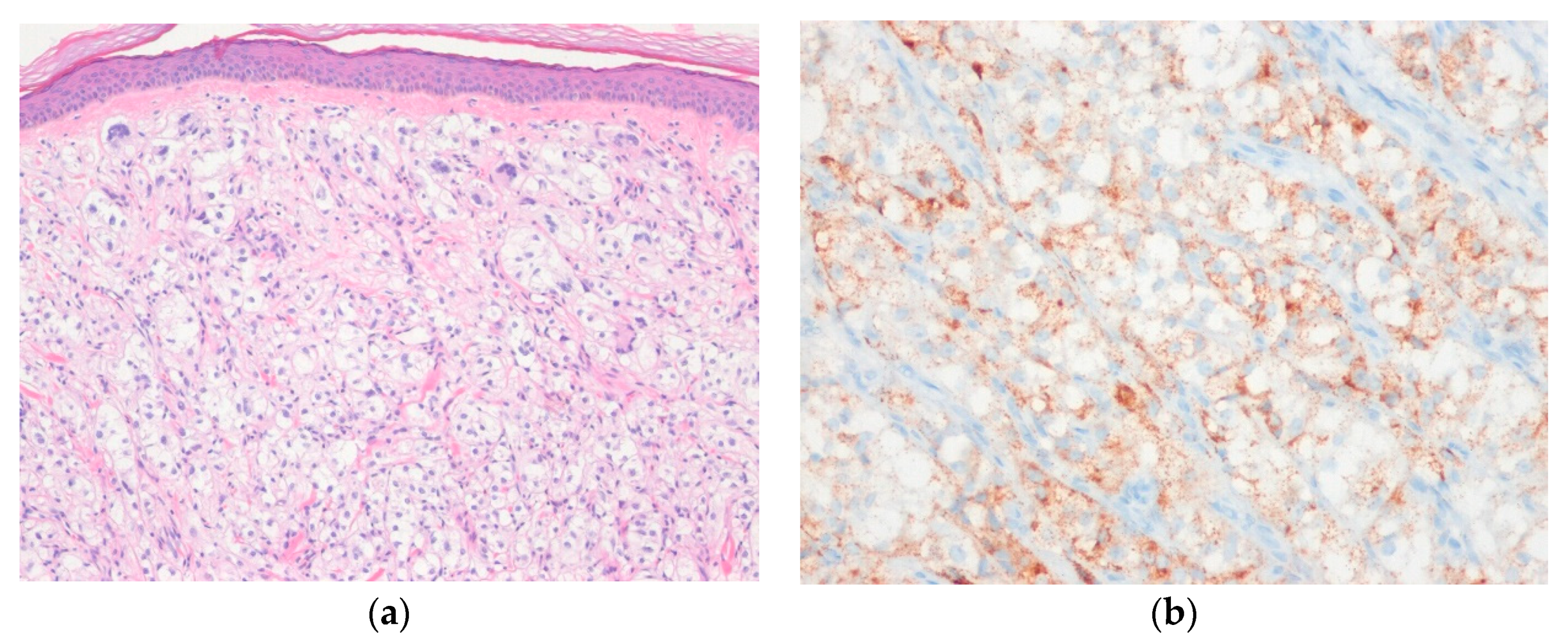
6.7. PEComa
7. Other Tumors with Epithelioid Features
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AFH | angiomatoid fibrous histiocytoma |
| AFX | atypical fibroxanthoma |
| ALK | anaplastic lymphoma kinase |
| DDCCMN | distinctive dermal clear cell mesenchymal neoplasm |
| DFSP | dermatofibrosarcoma protuberans |
| EFH | epithelioid fibrous histiocytoma |
| EMA | epithelial membrane antigen |
| ERG | erythroblast transformation specific-related gene |
| EMPNST | epithelioid malignant peripheral nerve sheath tumor |
| FLI1 | friend leukemia integration 1 |
| GFAP | glial fibrillary acid protein |
| H&E | hematoxylin and eosin |
| HFLT | hemosiderotic fibrolipomatous tumor |
| HMB-45 | human melanoma black-45 |
| HPF | high power field |
| INI1 | integrase interactor 1 |
| MIFS | myxoinflammatory fibroblastic sarcoma |
| MPNST | malignant peripheral nerve sheath tumor |
| MyoD1 | myogenic differentiation 1 |
| OFMT | ossifying fibromyxoid tumor |
| PDS | pleomorphic dermal sarcoma |
| PEComa | perivascular epithelioid cell tumor |
| PGP9.5 | protein gene product 9.5 |
| SMA | smooth muscle actin |
| SMARCB1 | SWI/SNF-related, matrix associated, actin-dependent regulator of chromatin, subfamily B, member 1 |
| SWI/SNF | switch/sucrose non-fermentable |
| TLE1 | transducing-like enhancer of split 1 |
References
- Doyle, L.A.; Hornick, J.L. Epithelioid and epithelial-like tumors. In Practical Soft Tissue Pathology: A Diagnostic Approach, 2nd ed.; Hornick, J.L., Ed.; Elsevier: Philadelphia, PA, USA, 2018; pp. 165–167. [Google Scholar]
- Kanik, L.; Li, M.; Urmacher, C.D. Normal skin. In Histology for Pathologists, 4th ed.; Mills, S.E., Ed.; Elsevier: Philadelphia, PA, USA, 2012; pp. 3–18. [Google Scholar]
- Billings, S.D.; Brenn, T.; Hornick, J.L. Cutaneous angiosarcoma. In WHO Classification of Skin Tumours, 4th ed.; Elder, D.E., Massi, D., Scolyer, R.A., Willemze, R., Eds.; IARC Press: Lyon, France, 2018; pp. 335–336. [Google Scholar]
- Lazar, A.J.; Billings, S.D.; Calonje, E.; Elder, D.E.; Hornick, J.L.; Massi, D.; Requena, L.; Scolyer, R.A. Soft tissue tumours: Introduction. In WHO Classification of Skin Tumours, 4th ed.; Elder, D.E., Massi, D., Scolyer, R.A., Willemze, R., Eds.; IARC Press: Lyon, France, 2018; pp. 292–293. [Google Scholar]
- Dangoor, A.; Seddon, B.; Gerrand, C.; Grimer, R.; Whelan, J.; Judson, I. UK guidelines for the management of soft tissue sarcomas. Clin. Sarcoma Res. 2016, 6, 20. [Google Scholar] [CrossRef]
- Rouhani, P.; Fletcher, C.D.; Devesa, S.S.; Toro, J.R. Cutaneous soft tissue sarcoma incidence patterns in the U.S.: An analysis of 12,114 cases. Cancer 2008, 113, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Mentzel, T. Sarcomas of the skin in the elderly. Clin. Dermatol. 2011, 29, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Goldblum, J.R.; Folpe, A.L.; Weiss, S.W. Approach to the diagnosis of soft tissue tumors. In Enzinger and Weiss’s Soft Tissue Tumors, 7th ed.; Goldblum, J.R., Folpe, A.L., Weiss, S.W., Eds.; Elsevier: Philadelphia, PA, USA, 2018; pp. 121–128. [Google Scholar]
- Choi, J.H.; Ro, J.Y. Cutaneous spindle cell neoplasms: Pattern-based diagnostic approach. Arch. Pathol. Lab. Med. 2018, 142, 958–972. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, I.M.; Hornick, J.L. Diagnostic immunohistochemistry for soft tissue and bone tumors: An update. Adv. Anat. Pathol. 2018, 25, 400–412. [Google Scholar] [CrossRef]
- Schaefer, I.M.; Fletcher, C.D. Recent advances in the diagnosis of soft tissue tumours. Pathology 2018, 50, 37–48. [Google Scholar] [CrossRef]
- Costigan, D.C.; Doyle, L.A. Advances in the clinicopathological and molecular classification of cutaneous mesenchymal neoplasms. Histopathology 2016, 68, 776–795. [Google Scholar] [CrossRef]
- Folpe, A.L. Selected topics in the pathology of epithelioid soft tissue tumors. Mod. Pathol. 2014, 27, S64–S79. [Google Scholar] [CrossRef]
- Agaimy, A. SWI/SNF complex-deficient soft tissue neoplasms: A pattern-based approach to diagnosis and differential diagnosis. Surg. Pathol. Clin. 2019, 12, 149–163. [Google Scholar] [CrossRef]
- Agaimy, A. What is new in epithelioid soft tissue tumors? Virchows Arch. 2020, 476, 81–96. [Google Scholar] [CrossRef]
- Requena, L.; Hornick, J.L. Epithelioid fibrous histiocytoma. In WHO Classification of Skin Tumours, 4th ed.; Elder, D.E., Massi, D., Scolyer, R.A., Willemze, R., Eds.; IARC Press: Lyon, France, 2018; p. 313. [Google Scholar]
- Dickson, B.C.; Swanson, D.; Charames, G.S.; Fletcher, C.D.; Hornick, J.L. Epithelioid fibrous histiocytoma: Molecular characterization of ALK fusion partners in 23 cases. Mod. Pathol. 2018, 31, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Jedrych, J.; Nikiforova, M.; Kennedy, T.F.; Ho, J. Epithelioid cell histiocytoma of the skin with clonal ALK gene rearrangement resulting in VCL-ALK and SQSTM1-ALK gene fusions. Br. J. Dermatol. 2015, 172, 1427–1429. [Google Scholar] [CrossRef] [PubMed]
- Doyle, L.A.; Mariño-Enriquez, A.; Fletcher, C.D.; Hornick, J.L. ALK rearrangement and overexpression in epithelioid fibrous histiocytoma. Mod. Pathol. 2015, 28, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Kazakov, D.V.; Kyrpychova, L.; Martinek, P.; Grossmann, P.; Steiner, P.; Vanecek, T.; Pavlovsky, M.; Bencik, V.; Michal, M.; Michal, M. ALK gene fusions in epithelioid fibrous histiocytoma: A study of 14 cases, with new histopathological findings. Am. J. Dermatopathol. 2018, 40, 805–814. [Google Scholar] [CrossRef]
- Doyle, L.A.; Fletcher, C.D. EMA positivity in epithelioid fibrous histiocytoma: A potential diagnostic pitfall. J. Cutan. Pathol. 2011, 38, 697–703. [Google Scholar] [CrossRef]
- Hornick, J.L.; Laskin, W.B. Myxoinflammatory fibroblastic sarcoma. In WHO Classification of Skin Tumours, 4th ed.; Elder, D.E., Massi, D., Scolyer, R.A., Willemze, R., Eds.; IARC Press: Lyon, France, 2018; pp. 302–303. [Google Scholar]
- Lucas, D.R. Myxoinflammatory fibroblastic sarcoma: Review and update. Arch. Pathol. Lab. Med. 2017, 141, 1503–1507. [Google Scholar] [CrossRef]
- Liu, H.; Sukov, W.R.; Ro, J.Y. The t(1;10)(p22;q24) TGFBR3/MGEA5 translocation in pleomorphic hyalinizing angiectatic tumor, myxoinflammatory fibroblastic sarcoma, and hemosiderotic fibrolipomatous tumor. Arch. Pathol. Lab. Med. 2019, 143, 212–221. [Google Scholar] [CrossRef]
- Hallor, K.H.; Sciot, R.; Staaf, J.; Heidenblad, M.; Rydholm, A.; Bauer, H.C.; Aström, K.; Domanski, H.A.; Meis, J.M.; Kindblom, L.G.; et al. Two genetic pathways, t(1;10) and amplification of 3p11-12, in myxoinflammatory fibroblastic sarcoma, haemosiderotic fibrolipomatous tumour, and morphologically similar lesions. J. Pathol. 2009, 217, 716–727. [Google Scholar] [CrossRef]
- Antonescu, C.R.; Zhang, L.; Nielsen, G.P.; Rosenberg, A.E.; Dal Cin, P.; Fletcher, C.D. Consistent t(1;10) with rearrangements of TGFBR3 and MGEA5 in both myxoinflammatory fibroblastic sarcoma and hemosiderotic fibrolipomatous tumor. Genes Chromosomes Cancer 2011, 50, 757–764. [Google Scholar] [CrossRef]
- Laskin, W.B.; Fetsch, J.F.; Miettinen, M. Myxoinflammatory fibroblastic sarcoma: A clinicopathologic analysis of 104 cases, with emphasis on predictors of outcome. Am. J. Surg. Pathol. 2014, 38, 1–12. [Google Scholar] [CrossRef]
- Boland, J.M.; Folpe, A.L. Hemosiderotic fibrolipomatous tumor, pleomorphic hyalinizing angiectatic tumor, and myxoinflammatory fibroblastic sarcoma: Related or not? Adv. Anat. Pathol. 2017, 24, 268–277. [Google Scholar] [CrossRef]
- Carter, J.M.; Weiss, S.W.; Linos, K.; DiCaudo, D.J.; Folpe, A.L. Superficial CD34-positive fibroblastic tumor: Report of 18 cases of a distinctive low-grade mesenchymal neoplasm of intermediate (borderline) malignancy. Mod. Pathol. 2014, 27, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Folpe, A.; Elston, D.; Kutzner, H. Cutaneous leiomyosarcoma (atypical smooth muscle tumour). In WHO Classification of Skin Tumours, 4th ed.; Elder, D.E., Massi, D., Scolyer, R.A., Willemze, R., Eds.; IARC Press: Lyon, France, 2018; p. 330. [Google Scholar]
- Kraft, S.; Fletcher, C.D. Atypical intradermal smooth muscle neoplasms: Clinicopathologic analysis of 84 cases and a reappraisal of cutaneous “leiomyosarcoma”. Am. J. Surg. Pathol. 2011, 35, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Kazlouskaya, V.; Lai, Y.C.; Khachemoune, A. Leiomyosarcoma of the skin: Review of the literature with an emphasis on prognosis and management. Int. J. Dermatol. 2020, 59, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Suster, S. Epithelioid leiomyosarcoma of the skin and subcutaneous tissue. Clinicopathologic, immunohistochemical, and ultrastructural study of five cases. Am. J. Surg. Pathol. 1994, 18, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Jo, V.Y.; Mariño-Enríquez, A.; Fletcher, C.D. Epithelioid rhabdomyosarcoma: Clinicopathologic analysis of 16 cases of a morphologically distinct variant of rhabdomyosarcoma. Am. J. Surg. Pathol. 2011, 35, 1523–1530. [Google Scholar] [CrossRef]
- Valério, E.; Almeida, G.C.; Neotti, T.; Nascimento, A.G.; Bezerra, S.M.; Costa, F.D. Epithelioid rhabdomyosarcoma: Report of a cutaneous case and literature review of a recently described variant of rhabdomyosarcoma. Am. J. Dermatopathol. 2020, 42, 275–279. [Google Scholar] [CrossRef]
- Patterson, J.W.; Folpe, A.; Jackett, L. Glomus tumors and variants. In WHO Classification of Skin Tumours, 4th ed.; Elder, D.E., Massi, D., Scolyer, R.A., Willemze, R., Eds.; IARC Press: Lyon, France, 2018; pp. 331–332. [Google Scholar]
- Mravic, M.; LaChaud, G.; Nguyen, A.; Scott, M.A.; Dry, S.M.; James, A.W. Clinical and histopathological diagnosis of glomus tumor: An institutional experience of 138 cases. Int. J. Surg. Pathol. 2015, 23, 181–188. [Google Scholar] [CrossRef]
- Blume-Peytavi, U.; Adler, Y.D.; Geilen, C.C.; Ahmad, W.; Christiano, A.; Goerdt, S.; Orfanos, C.E. Multiple familial cutaneous glomangioma: A pedigree of 4 generations and critical analysis of histologic and genetic differences of glomus tumors. J. Am. Acad. Dermatol. 2000, 42, 633–639. [Google Scholar] [CrossRef]
- Pulitzer, D.R.; Martin, P.C.; Reed, R.J. Epithelioid glomus tumor. Hum. Pathol. 1995, 26, 1022–1027. [Google Scholar] [CrossRef]
- Folpe, A.L.; Fanburg-Smith, J.C.; Miettinen, M.; Weiss, S.W. Atypical and malignant glomus tumors: Analysis of 52 cases, with a proposal for the reclassification of glomus tumors. Am. J. Surg. Pathol. 2001, 25, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Luzar, B.; Martin, B.; Fisher, C.; Calonje, E. Cutaneous malignant glomus tumours: Applicability of currently established malignancy criteria for tumours occurring in the skin. Pathology 2018, 50, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Bozdogan, N.; Dilek, G.B.; Benzer, E.; Karadeniz, M.; Bozdogan, O. Transducing-Like Enhancer of Split 1: A potential immunohistochemical marker for glomus tumor. Am. J. Dermatopathol. 2017, 39, 524–527. [Google Scholar] [CrossRef] [PubMed]
- Mosquera, J.M.; Sboner, A.; Zhang, L.; Chen, C.L.; Sung, Y.S.; Chen, H.W.; Agaram, N.P.; Briskin, D.; Basha, B.M.; Singer, S.; et al. Novel MIR143-NOTCH fusions in benign and malignant glomus tumors. Genes Chromosomes Cancer 2013, 52, 1075–1087. [Google Scholar] [CrossRef] [PubMed]
- Calonje, E.; Glusac, E.J.; Mihm, M.C., Jr.; North, P.E.; Piris, A.; Requena, L.; Sangűeza, O.P.; Wick, M.R. Haemangioma. In WHO Classification of Skin Tumours, 4th ed.; Elder, D.E., Massi, D., Scolyer, R.A., Willemze, R., Eds.; IARC Press: Lyon, France, 2018; pp. 349–350. [Google Scholar]
- Huang, S.C.; Zhang, L.; Sung, Y.S.; Chen, C.L.; Krausz, T.; Dickson, B.C.; Kao, Y.C.; Agaram, N.P.; Fletcher, C.D.; Antonescu, C.R. Frequent FOS gene rearrangements in epithelioid hemangioma: A molecular study of 58 cases with morphologic reappraisal. Am. J. Surg. Pathol. 2015, 39, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Antonescu, C.R.; Chen, H.W.; Zhang, L.; Sung, Y.S.; Panicek, D.; Agaram, N.P.; Dickson, B.C.; Krausz, T.; Fletcher, C.D. ZFP36-FOSB fusion defines a subset of epithelioid hemangioma with atypical features. Genes Chromosomes Cancer 2014, 53, 951–959. [Google Scholar] [CrossRef]
- Fetsch, J.F.; Sesterhenn, I.A.; Miettinen, M.; Davis, C.J., Jr. Epithelioid hemangioma of the penis: A clinicopathologic and immunohistochemical analysis of 19 cases, with special reference to exuberant examples often confused with epithelioid hemangioendothelioma and epithelioid angiosarcoma. Am. J. Surg. Pathol. 2004, 28, 523–533. [Google Scholar] [CrossRef]
- Llamas-Velasco, M.; Kempf, W.; Cota, C.; Fernández-Figueras, M.T.; Lee, J.; Ferrara, G.; Sander, C.; Shapiro, P.E.; Requena, L.; Kutzner, H. Multiple eruptive epithelioid hemangiomas: A subset of cutaneous cellular epithelioid hemangioma with expression of FOS-B. Am. J. Surg. Pathol. 2019, 43, 26–34. [Google Scholar] [CrossRef]
- Kung, I.T.; Gibson, J.B.; Bannatyne, P.M. Kimura’s disease: A clinico-pathological study of 21 cases and its distinction from angiolymphoid hyperplasia with eosinophilia. Pathology 1984, 16, 39–44. [Google Scholar] [CrossRef]
- Hornick, J.L.; Billings, S.D.; Requena, L. Haemangioenothelioma. In WHO Classification of Skin Tumours, 4th ed.; Elder, D.E., Massi, D., Scolyer, R.A., Willemze, R., Eds.; IARC Press: Lyon, France, 2018; pp. 338–340. [Google Scholar]
- Walther, C.; Tayebwa, J.; Lilljebjörn, H.; Magnusson, L.; Nilsson, J.; von Steyern, F.V.; Øra, I.; Domanski, H.A.; Fioretos, T.; Nord, K.H.; et al. A novel SERPINE1-FOSB fusion gene results in transcriptional up-regulation of FOSB in pseudomyogenic haemangioendothelioma. J. Pathol. 2014, 232, 534–540. [Google Scholar] [CrossRef]
- Hornick, J.L.; Fletcher, C.D. Pseudomyogenic hemangioendothelioma: A distinctive, often multicentric tumor with indolent behavior. Am. J. Surg. Pathol. 2011, 35, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Hung, Y.P.; Fletcher, C.D.; Hornick, J.L. FOSB is a useful diagnostic marker for pseudomyogenic hemangioendothelioma. Am. J. Surg. Pathol. 2017, 41, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Sugita, S.; Hirano, H.; Kikuchi, N.; Kubo, T.; Asanuma, H.; Aoyama, T.; Emori, M.; Hasegawa, T. Diagnostic utility of FOSB immunohistochemistry in pseudomyogenic hemangioendothelioma and its histological mimics. Diagn. Pathol. 2016, 11, 75. [Google Scholar] [CrossRef]
- Mentzel, T.; Beham, A.; Calonje, E.; Katenkamp, D.; Fletcher, C.D. Epithelioid hemangioendothelioma of skin and soft tissues: Clinicopathologic and immunohistochemical study of 30 cases. Am. J. Surg. Pathol. 1997, 21, 363–374. [Google Scholar] [CrossRef]
- Quante, M.; Patel, N.K.; Hill, S.; Merchant, W.; Courtauld, E.; Newman, P.; McKee, P.H. Epithelioid hemangioendothelioma presenting in the skin: A clinicopathologic study of eight cases. Am. J. Dermatopathol. 1998, 20, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Errani, C.; Zhang, L.; Sung, Y.S.; Hajdu, M.; Singer, S.; Maki, R.G.; Healey, J.H.; Antonescu, C.R. A novel WWTR1-CAMTA1 gene fusion is a consistent abnormality in epithelioid hemangioendothelioma of different anatomic sites. Genes Chromosomes Cancer 2011, 50, 644–653. [Google Scholar] [CrossRef]
- Antonescu, C.R.; Le Loarer, F.; Mosquera, J.M.; Sboner, A.; Zhang, L.; Chen, C.L.; Chen, H.W.; Pathan, N.; Krausz, T.; Dickson, B.C.; et al. Novel YAP1-TFE3 fusion defines a distinct subset of epithelioid hemangioendothelioma. Genes Chromosomes Cancer 2013, 52, 775–784. [Google Scholar] [CrossRef]
- Doyle, L.A.; Fletcher, C.D.; Hornick, J.L. Nuclear expression of CAMTA1 distinguishes epithelioid hemangioendothelioma from histologic mimics. Am. J. Surg. Pathol. 2016, 40, 94–102. [Google Scholar] [CrossRef]
- Massina, J.; Yeh, I. Schwannoma. In WHO Classification of Skin Tumours, 4th ed.; Elder, D.E., Massi, D., Scolyer, R.A., Willemze, R., Eds.; IARC Press: Lyon, France, 2018; pp. 365–366. [Google Scholar]
- Kindblom, L.G.; Meis-Kindblom, J.M.; Havel, G.; Busch, C. Benign epithelioid schwannoma. Am. J. Surg. Pathol. 1998, 22, 762–770. [Google Scholar] [CrossRef]
- Hart, J.; Gardner, J.M.; Edgar, M.; Weiss, S.W. Epithelioid schwannomas: An analysis of 58 cases including atypical variants. Am. J. Surg. Pathol. 2016, 40, 704–713. [Google Scholar] [CrossRef]
- Jo, V.Y.; Fletcher, C.D. SMARCB1/INI1 Loss in epithelioid schwannoma: A clinicopathologic and immunohistochemical study of 65 cases. Am. J. Surg. Pathol. 2017, 41, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, M.; Finnell, V.; Fetsch, J.F. Ossifying fibromyxoid tumor of soft parts—A clinicopathologic and immunohistochemical study of 104 cases with long-term follow-up and a critical review of the literature. Am. J. Surg. Pathol. 2008, 32, 996–1005. [Google Scholar] [CrossRef]
- Lazar, A.J.; Argeny, Z.B. Granular cell tumor. In WHO Classification of Skin Tumours, 4th ed.; Elder, D.E., Massi, D., Scolyer, R.A., Willemze, R., Eds.; IARC Press: Lyon, France, 2018; p. 364. [Google Scholar]
- Stemm, M.; Suster, D.; Wakely, P.E., Jr.; Suster, S. Typical and atypical granular cell tumors of soft tissue: A clinicopathologic study of 50 patients. Am. J. Clin. Pathol. 2017, 148, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Pareja, F.; Brandes, A.H.; Basili, T.; Selenica, P.; Geyer, F.C.; Fan, D.; Da Cruz Paula, A.; Kumar, R.; Brown, D.N.; Gularte-Mérida, R.; et al. Loss-of-function mutations in ATP6AP1 and ATP6AP2 in granular cell tumors. Nat. Commun. 2018, 9, 3533. [Google Scholar] [CrossRef] [PubMed]
- Schrader, K.A.; Nelson, T.N.; De Luca, A.; Huntsman, D.G.; McGillivray, B.C. Multiple granular cell tumors are an associated feature of LEOPARD syndrome caused by mutation in PTPN11. Clin. Genet. 2009, 75, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, P.V.; Storm, C.A.; Filiano, J.J.; Dinulos, J.G. Multiple granular cell tumors in a child with Noonan syndrome. Pediatric Dermatol. 2010, 27, 209–211. [Google Scholar] [CrossRef]
- Mirza, F.N.; Tuggle, C.T.; Zogg, C.K.; Mirza, H.N.; Narayan, D. Epidemiology of malignant cutaneous granular cell tumors: A US population-based cohort analysis using the surveillance, epidemiology, and end results (SEER) database. J. Am. Acad. Dermatol. 2018, 78, 490–497. [Google Scholar] [CrossRef]
- Fanburg-Smith, J.C.; Meis-Kindblom, J.M.; Fante, R.; Kindblom, L.G. Malignant granular cell tumor of soft tissue: Diagnostic criteria and clinicopathologic correlation. Am. J. Surg. Pathol. 1998, 22, 779–794. [Google Scholar] [CrossRef]
- Liu, Y.; Zheng, Q.; Wang, C.; Wang, J.; Ming, J.; Zhang, Y.; Li, X.; Cho, W.C.; Wang, L.; Li, Q.C.; et al. Granular cell tumors overexpress TFE3 without gene rearrangement: Evaluation of immunohistochemistry and break-apart FISH in 45 cases. Oncol. Lett. 2019, 18, 6355–6360. [Google Scholar] [CrossRef]
- Cohen, J.N.; Yeh, I.; Jordan, R.C.; Wolsky, R.J.; Horvai, A.E.; McCalmont, T.H.; LeBoit, P.E. Cutaneous non-neural granular cell tumors harbor recurrent ALK gene fusions. Am. J. Surg. Pathol. 2018, 42, 1133–1142. [Google Scholar] [CrossRef]
- Perry, A. Ectopic meningioma/meningothelial hamartoma. In WHO Classification of Tumours of Soft Tissue and Bone, 4th ed.; Fletcher, C.D., Hogendoorn, C.W., Mertens, F., Bridges, J., Eds.; IARC Press: Lyon, France, 2013; pp. 182–183. [Google Scholar]
- Miedema, J.R.; Zedek, D. Cutaneous meningioma. Arch. Pathol. Lab. Med. 2012, 136, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Lopez, D.A.; Silvers, D.N.; Helwig, E.B. Cutaneous meningiomas—A clinicopathologic study. Cancer 1974, 34, 728–744. [Google Scholar] [CrossRef]
- Taher, A.; Pushpanathan, C. Plexiform fibrohistiocytic tumor: A brief review. Arch. Pathol. Lab. Med. 2007, 131, 1135–1138. [Google Scholar] [PubMed]
- Kutzner, H. Malignant peripheral nerve sheath tumor. In WHO Classification of Skin Tumours, 4th ed.; Elder, D.E., Massi, D., Scolyer, R.A., Willemze, R., Eds.; IARC Press: Lyon, France, 2018; p. 367. [Google Scholar]
- Jo, V.Y.; Fletcher, C.D. Epithelioid malignant peripheral nerve sheath tumor: Clinicopathologic analysis of 63 cases. Am. J. Surg. Pathol. 2015, 39, 673–682. [Google Scholar] [CrossRef] [PubMed]
- McMenamin, M.E.; Fletcher, C.D. Expanding the spectrum of malignant change in schwannomas: Epithelioid malignant change, epithelioid malignant peripheral nerve sheath tumor, and epithelioid angiosarcoma: A study of 17 cases. Am. J. Surg. Pathol. 2001, 25, 13–25. [Google Scholar] [CrossRef]
- Schaefer, I.M.; Dong, F.; Garcia, E.P.; Fletcher, C.D.; Jo, V.Y. Recurrent SMARCB1 inactivation in epithelioid malignant peripheral nerve sheath tumors. Am. J. Surg. Pathol. 2019, 43, 835–843. [Google Scholar] [CrossRef]
- Asano, N.; Yoshida, A.; Ichikawa, H.; Mori, T.; Nakamura, M.; Kawai, A.; Hiraoka, N. Immunohistochemistry for trimethylated H3K27 in the diagnosis of malignant peripheral nerve sheath tumours. Histopathology 2017, 70, 385–393. [Google Scholar] [CrossRef]
- Schaefer, I.M.; Fletcher, C.D.; Hornick, J.L. Loss of H3K27 trimethylation distinguishes malignant peripheral nerve sheath tumors from histologic mimics. Mod. Pathol. 2016, 29, 4–13. [Google Scholar] [CrossRef]
- Luzar, B.; Shanesmith, R.; Ramakrishnan, R.; Fisher, C.; Calonje, E. Cutaneous epithelioid malignant peripheral nerve sheath tumour: A clinicopathological analysis of 11 cases. Histopathology 2016, 68, 286–296. [Google Scholar] [CrossRef]
- Lazar, A.J.; Fletcher, C.D. Distinctive dermal clear cell mesenchymal neoplasm: Clinicopathologic analysis of five cases. Am. J. Dermatopathol. 2004, 26, 273–279. [Google Scholar] [CrossRef]
- Gavino, A.C.; Pitha, J.V.; Bakshi, N.A. Atypical distinctive dermal clear cell mesenchymal neoplasm arising in the scalp. J. Cutan. Pathol. 2008, 35, 423–427. [Google Scholar] [CrossRef]
- Cheng, N.; Skupsky, H.; Cassarino, D.S. Clear cell proliferations of the skin. Adv. Anat. Pathol. 2015, 22, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Pletneva, M.A.; Andea, A.; Palanisamy, N.; Betz, B.L.; Carskadon, S.; Wang, M.; Patel, R.M.; Fullen, D.R.; Harms, P.W. Clear cell melanoma: A cutaneous clear cell malignancy. Arch. Pathol. Lab. Med. 2014, 138, 1328–1336. [Google Scholar] [CrossRef] [PubMed]
- Requena, L.; Hornick, J.L. Cellular neurothekeoma. In WHO Classification of Skin Tumours, 4th ed.; Elder, D.E., Massi, D., Scolyer, R.A., Willemze, R., Eds.; IARC Press: Lyon, France, 2018; p. 377. [Google Scholar]
- Hornick, J.L.; Fletcher, C.D. Cellular neurothekeoma: Detailed characterization in a series of 133 cases. Am. J. Surg. Pathol. 2007, 31, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Fetsch, J.F.; Laskin, W.B.; Hallman, J.R.; Lupton, G.P.; Miettinen, M. Neurothekeoma: An analysis of 178 tumors with detailed immunohistochemical data and long-term patient follow-up information. Am. J. Surg. Pathol. 2007, 31, 1103–1114. [Google Scholar] [CrossRef]
- Stratton, J.; Billings, S.D. Cellular neurothekeoma: Analysis of 37 cases emphasizing atypical histologic features. Mod. Pathol. 2014, 27, 701–710. [Google Scholar] [CrossRef]
- Antonescu, C.R.; Rossi, S. Angiomatoid fibrous histiocytoma. In WHO Classification of Tumours of Soft Tissue and Bone, 4th ed.; Fletcher, C.D., Hogendoorn, C.W., Mertens, F., Bridges, J., Eds.; IARC Press: Lyon, France, 2013; pp. 204–205. [Google Scholar]
- Fanburg-Smith, J.C.; Miettinen, M. Angiomatoid “malignant” fibrous histiocytoma: A clinicopathologic study of 158 cases and further exploration of the myoid phenotype. Hum. Pathol. 1999, 30, 1336–1343. [Google Scholar] [CrossRef]
- Weinreb, I.; Rubin, B.P.; Goldblum, J.R. Pleomorphic angiomatoid fibrous histiocytoma: A case confirmed by fluorescence in situ hybridization analysis for EWSR1 rearrangement. J. Cutan. Pathol. 2008, 35, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, I.M.; Fletcher, C.D. Myxoid variant of so-called angiomatoid “malignant fibrous histiocytoma”: Clinicopathologic characterization in a series of 21 cases. Am. J. Surg. Pathol. 2014, 38, 816–823. [Google Scholar] [CrossRef]
- Thway, K.; Fisher, C. Angiomatoid fibrous histiocytoma: The current status of pathology and genetics. Arch. Pathol. Lab. Med. 2015, 139, 674–682. [Google Scholar] [CrossRef]
- Cheah, A.L.; Zou, Y.; Lanigan, C.; Billings, S.D.; Rubin, B.P.; Hornick, J.L.; Goldblum, J.R. ALK Expression in angiomatoid fibrous histiocytoma: A potential diagnostic pitfall. Am. J. Surg. Pathol. 2019, 43, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Hung, Y.P.; Fletcher, C.D.; Hornick, J.L. Evaluation of NKX2-2 expression in round cell sarcomas and other tumors with EWSR1 rearrangement: Imperfect specificity for Ewing sarcoma. Mod. Pathol. 2016, 29, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Hornick, J.L.; Fisher, C.; LeBoit, P.E. Myoepithelioma. In WHO Classification of Skin Tumours, 4th ed.; Elder, D.E., Massi, D., Scolyer, R.A., Willemze, R., Eds.; IARC Press: Lyon, France, 2018; p. 194. [Google Scholar]
- Jo, V.Y.; Antonescu, C.R.; Zhang, L.; Dal Cin, P.; Hornick, J.L.; Fletcher, C.D. Cutaneous syncytial myoepithelioma: Clinicopathologic characterization in a series of 38 cases. Am. J. Surg. Pathol. 2013, 37, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Jo, V.Y.; Antonescu, C.R.; Dickson, B.C.; Swanson, D.; Zhang, L.; Fletcher, C.D.; Demicco, E.G. Cutaneous syncytial myoepithelioma is characterized by recurrent EWSR1-PBX3 Fusions. Am. J. Surg. Pathol. 2019, 43, 1349–1354. [Google Scholar] [CrossRef] [PubMed]
- Flucke, U.; Palmedo, G.; Blankenhorn, N.; Slootweg, P.J.; Kutzner, H.; Mentzel, T. EWSR1 gene rearrangement occurs in a subset of cutaneous myoepithelial tumors: A study of 18 cases. Mod. Pathol. 2011, 24, 1444–1450. [Google Scholar] [CrossRef]
- Hornick, J.L.; Fletcher, C.D. Cutaneous myoepithelioma: A clinicopathologic and immunohistochemical study of 14 cases. Hum. Pathol. 2004, 35, 14–24. [Google Scholar] [CrossRef]
- Hornick, J.L.; Fletcher, C.D. Myoepithelial tumors of soft tissue: A clinicopathologic and immunohistochemical study of 101 cases with evaluation of prognostic parameters. Am. J. Surg. Pathol. 2003, 27, 1183–1196. [Google Scholar] [CrossRef]
- Meis-Kindblom, J.M.; Bergh, P.; Gunterberg, B.; Kindblom, L.G. Extraskeletal myxoid chondrosarcoma: A reappraisal of its morphologic spectrum and prognostic factors based on 117 cases. Am. J. Surg. Pathol. 1999, 23, 636–650. [Google Scholar] [CrossRef]
- Hornick, J.L.; Patel, R.M. Epithelioid sarcoma. In WHO Classification of Skin Tumours, 4th ed.; Elder, D.E., Massi, D., Scolyer, R.A., Willemze, R., Eds.; IARC Press: Lyon, France, 2018; p. 373. [Google Scholar]
- Miettinen, M.; Fanburg-Smith, J.C.; Virolainen, M.; Shmookler, B.M.; Fetsch, J.F. Epithelioid sarcoma: An immunohistochemical analysis of 112 classical and variant cases and a discussion of the differential diagnosis. Hum. Pathol. 1999, 30, 934–942. [Google Scholar] [CrossRef]
- Laskin, W.B.; Miettinen, M. Epithelioid sarcoma: New insights based on an extended immunohistochemical analysis. Arch. Pathol. Lab. Med. 2003, 127, 1161–1168. [Google Scholar]
- Guillou, L.; Wadden, C.; Coindre, J.M.; Krausz, T.; Fletcher, C.D. “Proximal-type” epithelioid sarcoma, a distinctive aggressive neoplasm showing rhabdoid features. Clinicopathologic, immunohistochemical, and ultrastructural study of a series. Am. J. Surg. Pathol. 1997, 21, 130–146. [Google Scholar] [CrossRef] [PubMed]
- Hornick, J.L.; Dal Cin, P.; Fletcher, C.D. Loss of INI1 expression is characteristic of both conventional and proximal-type epithelioid sarcoma. Am. J. Surg. Pathol. 2009, 33, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, L.M.; Folpe, A.L.; Pawel, B.R.; Judkins, A.R.; Biegel, J.A. Epithelioid sarcoma is associated with a high percentage of SMARCB1 deletions. Mod. Pathol. 2013, 26, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Chbani, L.; Guillou, L.; Terrier, P.; Decouvelaere, A.V.; Grégoire, F.; Terrier-Lacombe, M.J.; Ranchère, D.; Robin, Y.M.; Collin, F.; Fréneaux, P.; et al. Epithelioid sarcoma: A clinicopathologic and immunohistochemical analysis of 106 cases from the French sarcoma group. Am. J. Clin. Pathol. 2009, 131, 222–227. [Google Scholar] [CrossRef]
- Busam, K.J.; Kutzner, H. Dermal clear cell sarcoma. In WHO Classification of Skin Tumours, 4th ed.; Elder, D.E., Massi, D., Scolyer, R.A., Willemze, R., Eds.; IARC Press: Lyon, France, 2018; p. 374. [Google Scholar]
- Chung, E.B.; Enzinger, F.M. Malignant melanoma of soft parts. A reassessment of clear cell sarcoma. Am. J. Surg. Pathol. 1983, 7, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Gonzaga, M.I.; Grant, L.; Curtin, C.; Gootee, J.; Silberstein, P.; Voth, E. The epidemiology and survivorship of clear cell sarcoma: A National Cancer Database (NCDB) review. J. Cancer Res. Clin. Oncol. 2018, 144, 1711–1716. [Google Scholar] [CrossRef] [PubMed]
- Hisaoka, M.; Ishida, T.; Kuo, T.T.; Matsuyama, A.; Imamura, T.; Nishida, K.; Kuroda, H.; Inayama, Y.; Oshiro, H.; Kobayashi, H.; et al. Clear cell sarcoma of soft tissue: A clinicopathologic, immunohistochemical, and molecular analysis of 33 cases. Am. J. Surg. Pathol. 2008, 32, 452–460. [Google Scholar] [CrossRef]
- Wang, W.L.; Mayordomo, E.; Zhang, W.; Hernandez, V.S.; Tuvin, D.; Garcia, L.; Lev, D.C.; Lazar, A.J.; López-Terrada, D. Detection and characterization of EWSR1/ATF1 and EWSR1/CREB1 chimeric transcripts in clear cell sarcoma (melanoma of soft parts). Mod. Pathol. 2009, 22, 1201–1209. [Google Scholar] [CrossRef]
- Kiuru, M.; Hameed, M.; Busam, K.J. Compound clear cell sarcoma misdiagnosed as a Spitz nevus. J. Cutan. Pathol. 2013, 40, 950–954. [Google Scholar] [CrossRef]
- Luzar, B.; Billings, S.D.; de la Fouchardiere, A.; Pissaloux, D.; Alberti, L.; Calonje, E. Compound clear cell sarcoma of the skin—A potential diagnostic pitfall: Report of a series of 4 new cases and a review of the literature. Am. J. Surg. Pathol. 2020, 44, 21–29. [Google Scholar] [CrossRef]
- Yang, L.; Chen, Y.; Cui, T.; Knösel, T.; Zhang, Q.; Geier, C.; Katenkamp, D.; Petersen, I. Identification of biomarkers to distinguish clear cell sarcoma from malignant melanoma. Hum. Pathol. 2012, 43, 1463–1470. [Google Scholar] [CrossRef] [PubMed]
- Hornick, J.L.; Pan, C.C. PEComa. In WHO Classification of Tumours of Soft Tissue and Bone, 4th ed.; Fletcher, C.D., Hogendoorn, C.W., Mertens, F., Bridges, J., Eds.; IARC Press: Lyon, France, 2013; pp. 230–231. [Google Scholar]
- Liegl, B.; Hornick, J.L.; Fletcher, C.D. Primary cutaneous PEComa: Distinctive clear cell lesions of skin. Am. J. Surg. Pathol. 2008, 32, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Charli-Joseph, Y.; Saggini, A.; Vemula, S.; Weier, J.; Mirza, S.; LeBoit, P.E. Primary cutaneous perivascular epithelioid cell tumor: A clinicopathological and molecular reappraisal. J. Am. Acad. Dermatol. 2014, 71, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- Stuart, L.N.; Tipton, R.G.; DeWall, M.R.; Parker, D.C.; Stelton, C.D.; Morrison, A.O.; Coleman, L.W.; Fosko, S.W.; Vidal, C.I.; Yadira Hurley, M.; et al. Primary cutaneous perivascular epithelioid cell tumor (PEComa): Five new cases and review of the literature. J. Cutan. Pathol. 2017, 44, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Hornick, J.L.; Fletcher, C.D. Sclerosing PEComa: Clinicopathologic analysis of a distinctive variant with a predilection for the retroperitoneum. Am. J. Surg. Pathol. 2008, 32, 493–501. [Google Scholar] [CrossRef]
- Larque, A.B.; Kradin, R.L.; Chebib, I.; Nielsen, G.P.; Selig, M.K.; Thiele, E.A.; Stemmer-Rachamimov, A.; Bredella, M.A.; Kurzawa, P.; Deshpande, V. Fibroma-like PEComa: A tuberous sclerosis complex-related lesion. Am. J. Surg. Pathol. 2018, 42, 500–505. [Google Scholar] [CrossRef]
- Folpe, A.L.; Kwiatkowski, D.J. Perivascular epithelioid cell neoplasms: Pathology and pathogenesis. Hum. Pathol. 2010, 41, 1–15. [Google Scholar] [CrossRef]
- Llamas-Velasco, M.; Mentzel, T.; Requena, L.; Palmedo, G.; Kasten, R.; Kutzner, H. Cutaneous PEComa does not harbour TFE3 gene fusions: Immunohistochemical and molecular study of 17 cases. Histopathology 2013, 63, 122–129. [Google Scholar] [CrossRef]
- Wambacher-Gasser, B.; Zelger, B.; Zelger, B.G.; Steiner, H. Clear cell dermatofibroma. Histopathology 1997, 30, 64–69. [Google Scholar] [CrossRef]
- Beer, T.W.; Calonje, E.; Wick, M.R. Atypical fibroxanthoma and variants. In WHO Classification of Skin Tumours, 4th ed.; Elder, D.E., Massi, D., Scolyer, R.A., Willemze, R., Eds.; IARC Press: Lyon, France, 2018; pp. 368–369. [Google Scholar]
- López, L.; Vélez, R. Atypical fibroxanthoma. Arch. Pathol. Lab. Med. 2016, 140, 376–379. [Google Scholar] [CrossRef]
- Brenn, T.; Wick, M.R. Pleomorphic dermal sarcoma. In WHO Classification of Skin Tumours, 4th ed.; Elder, D.E., Massi, D., Scolyer, R.A., Willemze, R., Eds.; IARC Press: Lyon, France, 2018; pp. 370–371. [Google Scholar]
- Griewank, K.G.; Wiesner, T.; Murali, R.; Pischler, C.; Müller, H.; Koelsche, C.; Möller, I.; Franklin, C.; Cosgarea, I.; Sucker, A.; et al. Atypical fibroxanthoma and pleomorphic dermal sarcoma harbor frequent NOTCH1/2 and FAT1 mutations and similar DNA copy number alteration profiles. Mod. Pathol. 2018, 31, 418–428. [Google Scholar] [CrossRef]
- Miller, K.; Goodlad, J.R.; Brenn, T. Pleomorphic dermal sarcoma: Adverse histologic features predict aggressive behavior and allow distinction from atypical fibroxanthoma. Am. J. Surg. Pathol. 2012, 36, 1317–1326. [Google Scholar] [CrossRef]
- Brenn, T. Soft tissue special issue: Cutaneous pleomorphic spindle cell tumors. Head Neck Pathol. 2020, 14, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Lazar, A.J.; Mahar, A. Dermatofibrosarcoma protuberans and variants. In WHO Classification of Skin Tumours, 4th ed.; Elder, D.E., Massi, D., Scolyer, R.A., Willemze, R., Eds.; IARC Press: Lyon, France, 2018; pp. 304–306. [Google Scholar]
- Llombart, B.; Serra-Guillén, C.; Monteagudo, C.; López Guerrero, J.A.; Sanmartín, O. Dermatofibrosarcoma protuberans: A comprehensive review and update on diagnosis and management. Semin. Diagn. Pathol. 2013, 30, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Maire, G.; Pédeutour, F.; Coindre, J.M. COL1A1-PDGFB gene fusion demonstrates a common histogenetic origin for dermatofibrosarcoma protuberans and its granular cell variant. Am. J. Surg. Pathol. 2002, 26, 932–937. [Google Scholar] [CrossRef] [PubMed]
- Suchak, R.; Thway, K.; Zelger, B.; Fisher, C.; Calonje, E. Primary cutaneous epithelioid angiosarcoma: A clinicopathologic study of 13 cases of a rare neoplasm occurring outside the setting of conventional angiosarcomas and with predilection for the limbs. Am. J. Surg. Pathol. 2011, 35, 60–69. [Google Scholar] [CrossRef]
- Nascimento, A.F.; Bertoni, F.; Fletcher, C.D. Epithelioid variant of myxofibrosarcoma: Expanding the clinicomorphologic spectrum of myxofibrosarcoma in a series of 17 cases. Am. J. Surg. Pathol. 2007, 31, 99–105. [Google Scholar] [CrossRef]
- Miettinen, M.; Enzinger, F.M. Epithelioid variant of pleomorphic liposarcoma: A study of 12 cases of a distinctive variant of high-grade liposarcoma. Mod. Pathol. 1999, 12, 722–728. [Google Scholar]
- Agaimy, A.; Michal, M.; Hadravsky, L.; Michal, M. Dedifferentiated liposarcoma composed predominantly of rhabdoid/epithelioid cells: A frequently misdiagnosed highly aggressive variant. Hum. Pathol. 2018, 77, 20–27. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lobulated Architecture | |
| Epithelioid hemangioma | |
| Myoepithelioma | |
| Epithelioid MPNST | |
| Nested Architecture | |
| Cellular neurothekeoma | |
| Ectopic meningioma | |
| Dermal clear cell sarcoma | |
| PEComa | |
| Trabecular and Cord-Like Architecture | |
| Myoepithelioma | |
| Epithelioid schwannoma | |
| Epithelioid hemangioendothelioma | |
| Sclerosing PEComa | |
| Sheet-Like Architecture | |
| Epithelioid fibrous histiocytoma | |
| Cutaneous leiomyosarcoma (epithelioid variant) | |
| Glomus tumor | |
| Epithelioid schwannoma | |
| Granular cell tumor | |
| Distinctive dermal clear cell mesenchymal neoplasm | |
| Angiomatoid fibrous histiocytoma | |
| Epithelioid sarcoma | |
| Clear or Granular Cells | |
| Granular cell tumor | |
| Distinctive dermal clear cell mesenchymal neoplasm | |
| Myoepithelioma | |
| Dermal clear cell sarcoma | |
| PEComa | |
| Prominent Inflammatory Cells | |
| Myxoinflammatory fibroblastic sarcoma | |
| Pseudomyogenic hemangioendothelioma (subset) | |
| Epithelioid hemangioma | |
| Epithelioid Fibrous Histiocytoma | Pseudomyogenic Hemangio-endothelioma | Myoepithelioma/Myoepithelial Carcinoma | Epithelioid MPNST | Epithelioid Sarcoma | PEComa | Malignant Melanoma | Carcinoma | |
|---|---|---|---|---|---|---|---|---|
| CD34 | − | − | − | +/− | +/− | − | − | − |
| SMA | − | − | +/− | − | +/− | + | − | − |
| S100 protein | − | − | + | + | + | + | − | |
| ALK | + | − | − | − | − | − | − | − |
| SMARCB1 (INI1) | + | + | +/− | +/− | − | − | + | + |
| Vascular markers (e.g., CD31, ERG) | − | + | − | − | − | − | − | − |
| Melanocytic markers (e.g., HMB45, Melan-A) | − | − | − | − | − | + | + | − |
| Epithelial markers (e.g., Cytokeratin (AE1/AE3), EMA) | − | + | + | +/− | + | − | − | + |
| Neoplasm | Cytogenetic Alterations | Molecular Alterations |
|---|---|---|
| Epithelioid fibrous histiocytoma | 2p23 | VCL-ALK SQSTM1-ALK |
| Myxoinflammatory fibroblastic sarcoma | t(1;10)(p22;q24) 3p11-12 amplification | TGFBR3-MGEA5 VGLL3, CHMP2B amplification |
| Glomus tumor | MIR143-NOTCH NOTCH rearrangement | |
| Epithelioid hemangioma | ZFP36-FOSB WWTR1-FOSB | |
| Pseudomyogenic hemangioendothelioma | t(7;19)(q22;q13) | SERPINE 1-FOSB |
| Epithelioid hemangioendothelioma | t(1;3)(p36;q25) t(X;11)(p11;q22) | WWTR1-CAMTA1 YAP1-TFE3 |
| Granular cell tumor | ATP6AP1, ATP6AP2 mutation | |
| Epithelioid MPNST (subset) | SMARCB1 (INI1) mutation | |
| Angiomatoid fibrous histiocytoma | t(2;22)(q33;q12) t(12;22)(q13;q12) t(12;16)(q13;p11) | EWSR1-CREB1 EWSR1-ATF1 FUS-ATF1 |
| Cutaneous syncytial myoepithelioma | EWSR1-PBX3 | |
| Epithelioid sarcoma | SMARCB1 (INI1) deletion | |
| Dermal clear cell sarcoma | t(12;22)(q13;q12) t(2;22)(q33;q12) | EWSR1-AFT1 EWSR1-CREB1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, J.H.; Ro, J.Y. Epithelioid Cutaneous Mesenchymal Neoplasms: A Practical Diagnostic Approach. Diagnostics 2020, 10, 233. https://doi.org/10.3390/diagnostics10040233
Choi JH, Ro JY. Epithelioid Cutaneous Mesenchymal Neoplasms: A Practical Diagnostic Approach. Diagnostics. 2020; 10(4):233. https://doi.org/10.3390/diagnostics10040233
Chicago/Turabian StyleChoi, Joon Hyuk, and Jae Y. Ro. 2020. "Epithelioid Cutaneous Mesenchymal Neoplasms: A Practical Diagnostic Approach" Diagnostics 10, no. 4: 233. https://doi.org/10.3390/diagnostics10040233
APA StyleChoi, J. H., & Ro, J. Y. (2020). Epithelioid Cutaneous Mesenchymal Neoplasms: A Practical Diagnostic Approach. Diagnostics, 10(4), 233. https://doi.org/10.3390/diagnostics10040233