Molecular Characterization of a Novel Splicing Mutation Underlying Mucopolysaccharidosis (MPS) Type VI—Indirect Proof of Principle on Its Pathogenicity

 ,
,  ,
, 
 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Next-Generation Sequence Analysis
2.3. Molecular Diagnosis
2.4. RT-PCR Analysis
2.5. Bioinformatic Analysis
2.6. Minigene Construction
2.7. In Vitro Splicing Assays
2.8. Mutation Nomenclature
3. Results
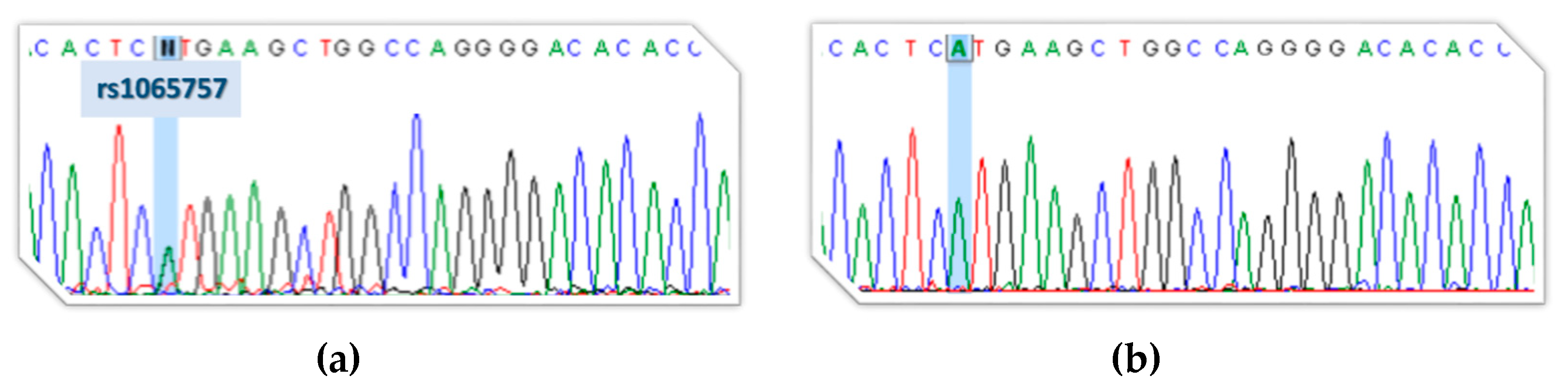
3.1. Next-Generation Sequencing
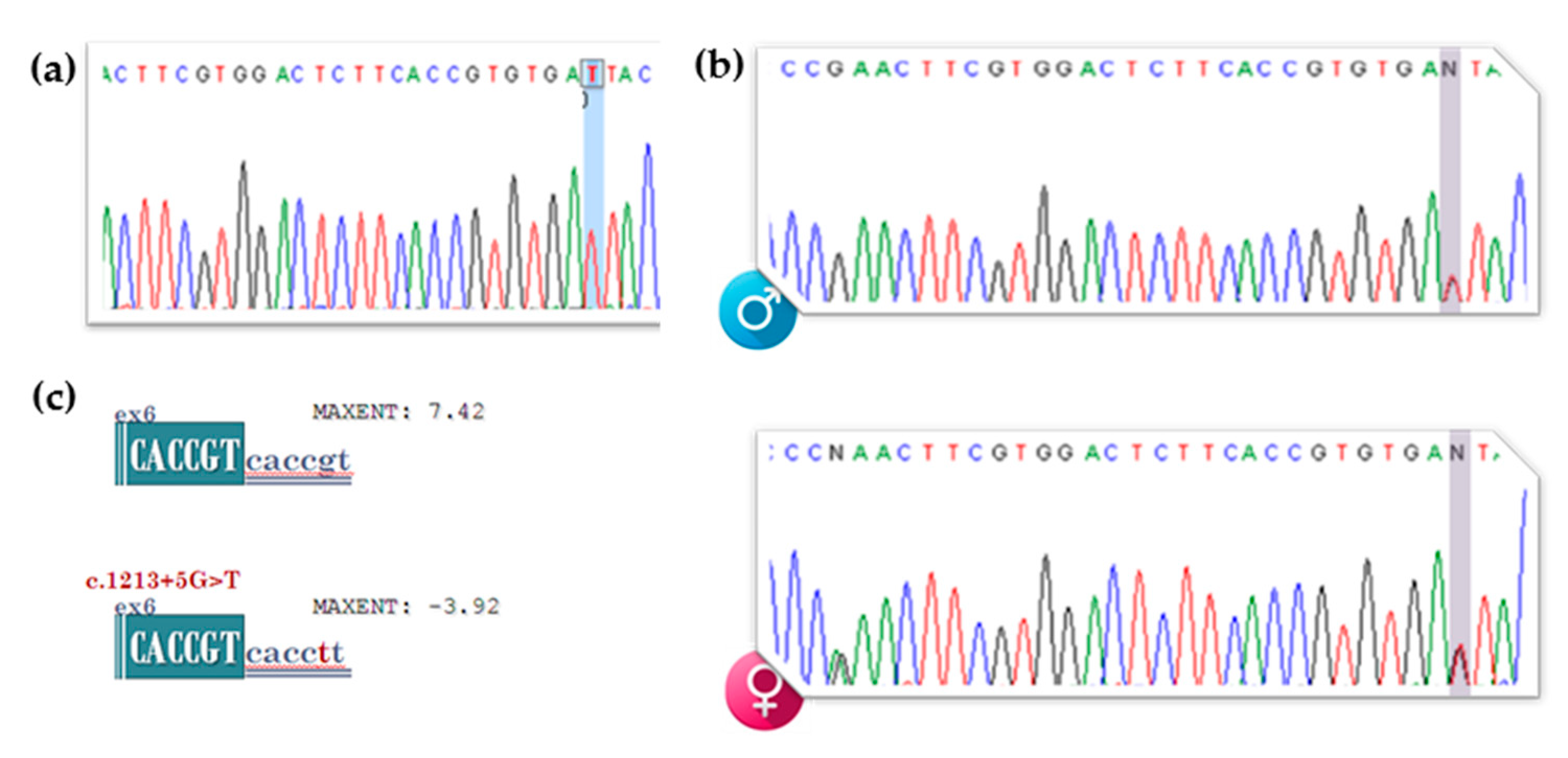
3.2. gDNA Screening of the Proband, Segregation Studies and In Silico Analysis of the Newly Identified VUS (c.1213+5G>T [IVS6+5G>T])
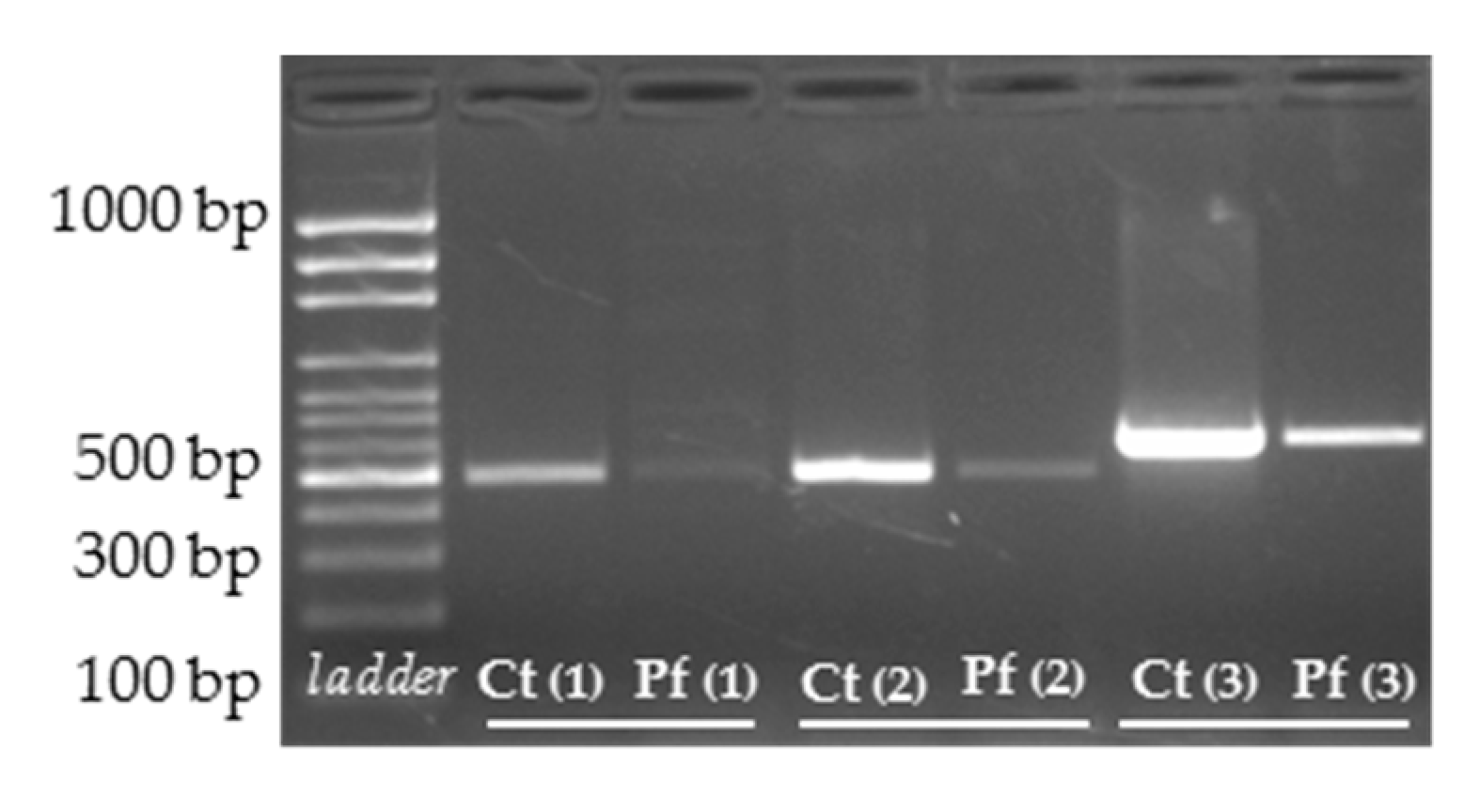
3.3. cDNA Screening and Splicing Pattern Analysis
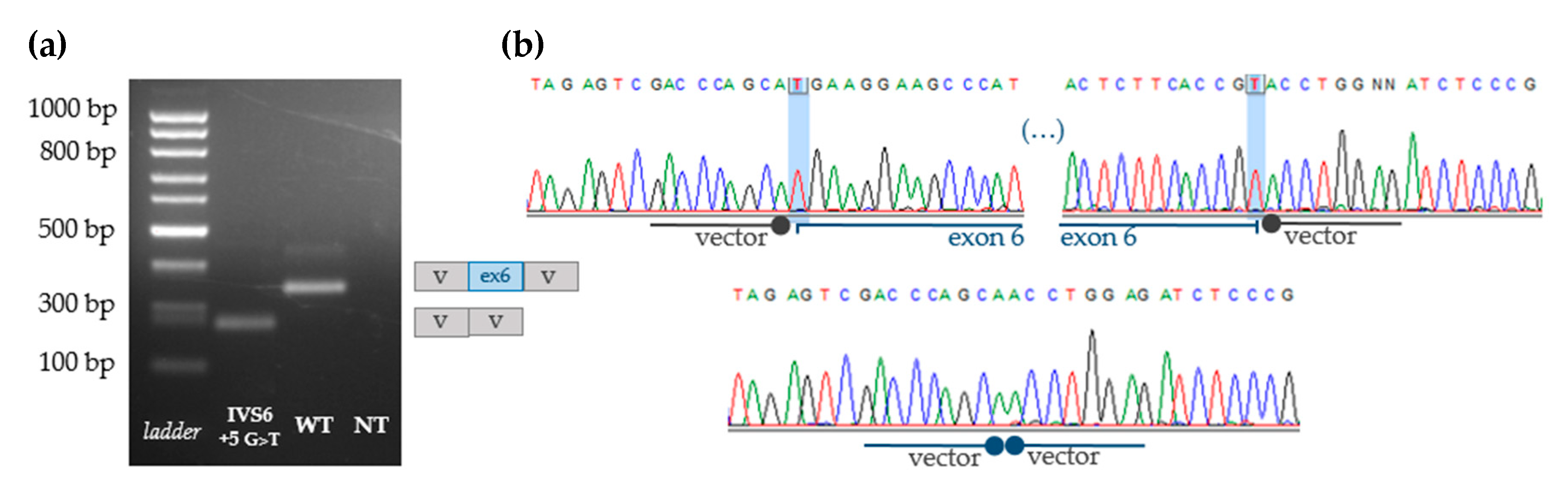
3.4. Indirect Proof of Principle on c.1213+5G>T [IVS6+5G>T] Pathogenicity
3.5. Functional Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Author Correction: Lysosomal storage diseases. Nat. Rev. Dis. Primers 2018, 4, 36. [Google Scholar] [CrossRef] [PubMed]
- Filocamo, M.; Morrone, A. Lysosomal storage disorders: Molecular basis and laboratory testing. Hum. Genom. 2011, 5, 156–169. [Google Scholar] [CrossRef] [PubMed]
- Winchester, B. Lysosomal diseases: Diagnostic update. J. Inherit. Metab. Dis. 2014, 37, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M. Emptying the stores: Lysosomal diseases and therapeutic strategies. Nat. Rev. Drug Discov. 2018, 17, 133–150. [Google Scholar] [CrossRef]
- Rehm, H.L. Disease-targeted sequencing: A cornerstone in the clinic. Nat. Rev. Genet. 2013, 14, 295–300. [Google Scholar] [CrossRef]
- Selmer, K.K.; Gilfillan, G.D.; Strømme, P.; Lyle, R.; Hughes, T.; Hjorthaug, H.S.; Brandal, K.; Nakken, S.; Misceo, D.; Egeland, T.; et al. A mild form of Mucopolysaccharidosis IIIB diagnosed with targeted next-generation sequencing of linked genomic regions. Eur. J. Hum. Genet. 2012, 20, 58–63. [Google Scholar] [CrossRef]
- Sperb-Ludwig, F.; Alegra, T.; Velho, R.V.; Ludwig, N.; Kim, C.A.; Kok, F.; Kitajima, J.P.; van Meel, E.; Kornfeld, S.; Burin, M.G.; et al. Exome sequencing for mucolipidosis III: Detection of a novel GNPTAB Gene Mutation in a Patient With a Very Mild Phenotype. Mol. Genet. Metab. Rep. 2015, 2, 34–37. [Google Scholar] [CrossRef]
- Fernández-Marmiesse, A.; Morey, M.; Pineda, M.; Eiris, J.; Couce, M.L.; Castro-Gago, M.; Fraga, J.M.; Lacerda, L.; Gouveia, S.; Pérez-Poyato, M.S.; et al. Assessment of a targeted resequencing assay as a support tool in the diagnosis of lysosomal storage disorders. Orphanet J. Rare Dis. 2014, 9, 59. [Google Scholar] [CrossRef]
- Yoshida, S.; Kido, J.; Matsumoto, S.; Momosaki, K.; Mitsubuchi, H.; Shimazu, T.; Sugawara, K.; Endo, F.; Nakamura, K. Prenatal diagnosis of Gaucher disease using next-generation sequencing. Pediatr. Int. 2016, 58, 946–949. [Google Scholar] [CrossRef]
- Angelini, C.; Savarese, M.; Fanin, M.; Nigro, V. Next generation sequencing detection of late onset pompe disease. Muscle Nerve 2016, 53, 981–983. [Google Scholar] [CrossRef]
- Di Fruscio, G.; Schulz, A.; De Cegli, R.; Savarese, M.; Mutarelli, M.; Parenti, G.; Banfi, S.; Braulke, T.; Nigro, V.; Ballabio, A. Lysoplex: An efficient toolkit to detect DNA sequence variations in the autophagy-lysosomal pathway. Autophagy 2015, 11, 928–938. [Google Scholar] [CrossRef] [PubMed]
- Di Fruscio, G.; Banfi, S.; Nigro, V.; Ballabio, A. Next-Generation Sequencing Approaches to Define the Role of the Autophagy Lysosomal Pathway in Human Disease: The Example of LysoPlex. Methods Mol. Biol. 2017, 1594, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Song, H.K.; Sohn, Y.B.; Choi, Y.J.; Chung, Y.S.; Jang, J.H. A case report of pycnodysostosis with atypical femur fracture diagnosed by next-generation sequencing of candidate genes. Medicine (Baltim.) 2017, 96, e6367. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Fan, Y.; Wang, L.; Huang, Z.; Gu, X.; Yu, Y. Molecular defects identified by whole exome sequencing in a child with atypical mucopolysaccharidosis IIIB. J. Pediatr. Endocrinol. Metab. 2017, 30, 463–469. [Google Scholar] [CrossRef]
- Tsai, A.C.; Hung, Y.W.; Harding, C.; Koeller, D.M.; Wang, J.; Wong, L.C. Next generation deep sequencing corrects diagnostic pitfalls of traditional molecular approach in a patient with prenatal onset of Pompe disease. Am. J. Med. Genet. Part A 2017, 173, 2500–2504. [Google Scholar] [CrossRef]
- Zampieri, S.; Cattarossi, S.; Bembi, B.; Dardis, A. GBA Analysis in Next-Generation Era: Pitfalls, Challenges, and Possible Solutions. J. Mol. Diagn. 2017, 19, 733–741. [Google Scholar] [CrossRef]
- Li, X.; Xiao, R.; Chen, B.; Yang, G.; Zhang, X.; Fu, Z.; Fu, J.; Zhuang, M.; Huang, Y. A novel mutation of SGSH and clinical features analysis of mucopolysaccharidosis type IIIA. Medicine (Baltim.) 2018, 97, e13758. [Google Scholar] [CrossRef]
- Málaga, D.R.; Brusius-Facchin, A.C.; Siebert, M.; Pasqualim, G.; Saraiva-Pereira, M.L.; Souza, C.F.M.; Schwartz, I.V.D.; Matte, U.; Giugliani, R. Sensitivity, advantages, limitations, and clinical utility of targeted next-generation sequencing panels for the diagnosis of selected lysosomal storage disorders. Genet. Mol. Biol. 2019, 42, 197–206. [Google Scholar] [CrossRef]
- Kobayashi, H. Recent trends in mucopolysaccharidosis research. J. Hum. Genet. 2019, 64, 127–137. [Google Scholar] [CrossRef]
- Whitley, C.B.; Ridnour, M.D.; Draper, K.A.; Dutton, C.M.; Neglia, J.P. Diagnostic test for mucopolysaccharidosis. I. Direct method for quantifying excessive urinary glycosaminoglycan excretion. Clin. Chem. 1989, 35, 374–379. [Google Scholar] [CrossRef]
- Gray, G.; Claridge, P.; Jenkinson, L.; Green, A. Quantitation of urinary glycosaminoglycans using dimethylene blue as a screening technique for the diagnosis of mucopolysaccharidoses: An evaluation. Ann. Clin. Biochem. 2007, 44, 360–363. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, R.; Brown, J.R.; Al-Mafraji, K.; Lamanna, W.C.; Beitel, J.R.; Boons, G.J.; Esko, J.D.; Crawford, B.E. Disease-specific non-reducing end carbohydrate biomarkers for mucopolysaccharidoses. Nat. Chem. Biol. 2012, 8, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Kubaski, F.; Osago, H.; Mason, R.W.; Yamaguchi, S.; Kobayashi, H.; Tsuchiya, M.; Orii, T.; Tomatsu, S. Glycosaminoglycans detection methods: Applications of mass spectrometry. Mol. Genet. Metab. 2017, 120, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Saville, J.T.; McDermott, B.K.; Fletcher, J.M.; Fuller, M. Disease and subtype specific signatures enable precise diagnosis of the mucopolysaccharidoses. Genet. Med. 2019, 21, 753–757. [Google Scholar] [CrossRef] [PubMed]
- Ouesleti, S.; Coutinho, M.F.; Ribeiro, I.; Miled, A.; Mosbahi, D.S.; Alves, S. Update of the spectrum of mucopolysaccharidoses type III in Tunisia: Identification of three novel mutations and in silico structural analysis of the missense mutations. World J. Pediatr. 2017, 13, 374–380. [Google Scholar] [CrossRef]
- Yeo, G.; Burge, C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004, 11, 377–394. [Google Scholar] [CrossRef]
- Cartegni, L.; Wang, J.; Zhu, Z.; Zhang, M.Q.; Krainer, A.R. ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003, 31, 3568–3571. [Google Scholar] [CrossRef]
- Fairbrother, W.G.; Yeh, R.F.; Sharp, P.A.; Burge, C.B. Predictive identification of exonic splicing enhancers in human genes. Science 2002, 297, 1007–1013. [Google Scholar] [CrossRef]
- Desmet, F.O.; Hamroun, D.; Lalande, M.; Collod-Béroud, G.; Claustres, M.; Béroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef]
- den Dunnen, J.T.; Dalgleish, R.; Maglott, D.R.; Hart, R.K.; Greenblatt, M.S.; McGowan-Jordan, J.; Roux, A.F.; Smith, T.; Antonarakis, S.E.; Taschner, P.E. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum. Mutat. 2016, 37, 564–569. [Google Scholar] [CrossRef]
- Bach, G.; Moskowitz, S.M.; Tieu, P.T.; Matynia, A.; Neufeld, E.F. Molecular analysis of Hurler syndrome in Druze and Muslim Arab patients in Israel: Multiple allelic mutations of the IDUA gene in a small geographic area. Am. J. Hum. Genet. 1993, 53, 330–338. [Google Scholar] [PubMed]
- Karageorgos, L.; Brooks, D.A.; Harmatz, P.; Ketteridge, D.; Pollard, A.; Melville, E.L.; Parkinson-Lawrence, E.; Clements, P.R.; Hopwood, J.J. Mutational analysis of mucopolysaccharidosis type VI patients undergoing a phase II trial of enzyme replacement therapy. Mol. Genet. Metab. 2007, 90, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Roca, X.; Krainer, A.R.; Eperon, I.C. Pick one, but be quick: 5’ splice sites and the problems of too many choices. Genes Dev. 2013, 27, 129–144. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coutinho, M.F.; Encarnação, M.; Matos, L.; Silva, L.; Ribeiro, D.; Santos, J.I.; Prata, M.J.; Vilarinho, L.; Alves, S. Molecular Characterization of a Novel Splicing Mutation Underlying Mucopolysaccharidosis (MPS) Type VI—Indirect Proof of Principle on Its Pathogenicity. Diagnostics 2020, 10, 58. https://doi.org/10.3390/diagnostics10020058
Coutinho MF, Encarnação M, Matos L, Silva L, Ribeiro D, Santos JI, Prata MJ, Vilarinho L, Alves S. Molecular Characterization of a Novel Splicing Mutation Underlying Mucopolysaccharidosis (MPS) Type VI—Indirect Proof of Principle on Its Pathogenicity. Diagnostics. 2020; 10(2):58. https://doi.org/10.3390/diagnostics10020058
Chicago/Turabian StyleCoutinho, Maria Francisca, Marisa Encarnação, Liliana Matos, Lisbeth Silva, Diogo Ribeiro, Juliana Inês Santos, Maria João Prata, Laura Vilarinho, and Sandra Alves. 2020. "Molecular Characterization of a Novel Splicing Mutation Underlying Mucopolysaccharidosis (MPS) Type VI—Indirect Proof of Principle on Its Pathogenicity" Diagnostics 10, no. 2: 58. https://doi.org/10.3390/diagnostics10020058
APA StyleCoutinho, M. F., Encarnação, M., Matos, L., Silva, L., Ribeiro, D., Santos, J. I., Prata, M. J., Vilarinho, L., & Alves, S. (2020). Molecular Characterization of a Novel Splicing Mutation Underlying Mucopolysaccharidosis (MPS) Type VI—Indirect Proof of Principle on Its Pathogenicity. Diagnostics, 10(2), 58. https://doi.org/10.3390/diagnostics10020058