CRMS/CFSPID Subjects Carrying D1152H CFTR Variant: Can the Second Variant Be a Predictor of Disease Development?


 ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Diagnostic Test and Clinical Data
- Group A: compound heterozygous for D1152H and a CF causing variant;
- Group B: compound heterozygous for D1152H and a: (i) non-CF causing variant; (ii) variant with varying clinical consequences; (iii) variant of unknown significance.
2.2. Statistical Analysis
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Elborn, J.S. Cystic fibrosis. Lancet 2016, 388, 2519–2531. [Google Scholar] [CrossRef]
- Southern, K.W.; Munck, A.; Pollitt, R.; Travert, G.; Zanolla, L.; Dankert-Roelse, J.; Castellani, C. A survey of newborn screening for cystic fibrosis in Europe. J. Cyst. Fibros. 2007, 6, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Massie, J.; Curnow, L.; Glazner, J.; Armstrong, D.S.; Francis, I. Lessons learned from 20 years of newborn screening for cystic fibrosis. Med, J. Aust. 2012, 196, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Taccetti, G.; Botti, M.; Terlizzi, V.; Cavicchi, M.C.; Neri, A.S.; Galici, V.; Mergni, G.; Centrone, C.; Peroni, D.G.; Festini, F. Clinical and Genotypical Features of False-Negative Patients in 26 Years of Cystic Fibrosis Neonatal Screening in Tuscany, Italy. Diagnostics 2020, 10, 446. [Google Scholar] [CrossRef] [PubMed]
- Griesenbach, U. Faculty Opinions recommendation of Cystic Fibrosis Foundation practice guidelines for the management of infants with cystic fibrosis transmembrane conductance regulator-related metabolic syndrome during the first two years of life and beyond. Fac. Opin. Post-Publ. Peer Rev. Biomed. Lit. 2015, 155, S106–S16. [Google Scholar] [CrossRef]
- Ren, C.; Borowitz, D.; Gonska, T.; Howenstine, M.S.; Levy, H.L.; Massie, J.; Milla, C.; Munck, A.; Southern, K.W. Cystic Fibrosis Transmembrane Conductance Regulator-Related Metabolic Syndrome and Cystic Fibrosis Screen Positive, Inconclusive Diagnosis. J. Pediatrics 2017, 181, S45–S51. [Google Scholar] [CrossRef]
- Munck, A.; Mayell, S.; Winters, V.; Shawcross, A.; Derichs, N.; Parad, R.; Barben, J.; Southern, K. Cystic Fibrosis Screen Positive, Inconclusive Diagnosis (CFSPID): A new designation and management recommendations for infants with an inconclusive diagnosis following newborn screening. J. Cyst. Fibros. 2015, 14, 706–713. [Google Scholar] [CrossRef]
- Southern, K.; Barben, J.; Gartner, S.; Munck, A.; Castellani, C.; Mayell, S.; Davies, J.; Winters, V.; Murphy, J.; Salinas, D.; et al. Inconclusive diagnosis after a positive newborn bloodspot screening result for cystic fibrosis; clarification of the harmonised international definition. J. Cyst. Fibros. 2019, 18, 778–780. [Google Scholar] [CrossRef]
- Ooi, C.Y.; Castellani, C.; Keenan, K.; Avolio, J.; Volpi, S.; Boland, M.; Kovesi, T.; Bjornson, C.; Chilvers, M.A.; Morgan, L.; et al. Inconclusive diagnosis of cystic fibrosis after newborn screening. Pediatrics 2015, 135, e1377–e1385. [Google Scholar] [CrossRef]
- Levy, H.L.; Nugent, M.; Schneck, K.; Stachiw-Hietpas, D.; Laxova, A.; Lakser, O.; Rock, M.J.; Dahmer, M.K.; Biller, J.; Nasr, S.Z.; et al. Refining the continuum of CFTR-associated disorders in the era of newborn screening. Clin. Genet. 2016, 89, 539–549. [Google Scholar] [CrossRef]
- Terlizzi, V.; Mergni, G.; Buzzetti, R.; Centrone, C.; Zavataro, L.; Braggion, C. Cystic fibrosis screen positive inconclusive diagnosis (CFSPID): Experience in Tuscany, Italy. J. Cyst. Fibros. 2019, 18, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Castaldo, A.; Cimbalo, C.; Castaldo, R.J.; D’Antonio, M.; Scorza, M.; Salvadori, L.; Sepe, A.; Raia, V.; Tosco, A. Cystic Fibrosis-Screening Positive Inconclusive Diagnosis: Newborn Screening and Long-Term Follow-Up Permits to Early Identify Patients with CFTR-Related Disorders. Diagnostics 2020, 10, 570. [Google Scholar] [CrossRef] [PubMed]
- Kharrazi, M.; Yang, J.; Bishop, T.; Lessing, S.; Young, S.; Graham, S.; Pearl, M.; Chow, H.; Ho, T.; Currier, R.; et al. California Cystic Fibrosis Newborn Screening Consortium. Newborn Screening for Cystic Fibrosis in California. Pediatrics 2015, 136, 1062–1072. [Google Scholar] [CrossRef] [PubMed]
- Munck, A. Inconclusive Diagnosis after Newborn Screening for Cystic Fibrosis. Int J Neonatal Screen. 2020, 6, 19. [Google Scholar] [CrossRef] [PubMed]
- Terlizzi, V.; Carnovale, V.; Castaldo, G.; Castellani, C.; Cirilli, N.; Colombo, C.; Corti, F.; Cresta, F.; D’Adda, A.; Lucarelli, M.; et al. Clinical expression of patients with the D1152H CFTR mutation. J. Cyst. Fibros. 2015, 14, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Giordani, B.; Amato, A.; Majo, F.; Ferrari, G.; Quattrucci, S.; Minicucci, L.; Padoan, R.; Floridia, G.; Salvatore, D.; Carnovale, V.; et al. Gruppo di lavoro RIFC. Report 2015–2016. Epidemiol. Prev. 2019, 43, 1–36. [Google Scholar] [PubMed]
- Lucarelli, M.; Porcaro, L.; Biffignandi, A.; Costantino, L.; Giannone, V.; Alberti, L.; Bruno, S.M.; Corbetta, C.; Torresani, E.; Colombo, C.; et al. A New Targeted CFTR Mutation Panel Based on Next-Generation Sequencing Technology. J. Mol. Diagn. 2017, 19, 788–800. [Google Scholar] [CrossRef]
- LeGrys, V.A.; Yankaskas, J.R.; Quittell, L.M.; Marshall, B.C.; Mogayzel, P.J., Jr. Cystic Fibrosis Foundation. Cystic Fibrosis Foundation. Diagnostic sweat testing: The Cystic Fibrosis Foundation guidelines. J. Pediatrics 2007, 151, 85–89. [Google Scholar] [CrossRef]
- Morinville, V.D.; Husain, S.Z.; Bai, H.; Barth, B.; Alhosh, R.; Durie, P.R.; Freedman, S.D.; Himes, R.; Lowe, M.E.; Pohl, J.; et al. Definitions of Pediatric Pancreatitis and Survey of Present Clinical Practices. J. Pediatrics Gastroenterol. Nutr. 2012, 55, 261–265. [Google Scholar] [CrossRef]
- Lee, T.W.; Brownlee, K.G.; Conway, S.P.; Denton, M.; Littlewood, J.M. Evaluation of a new definition for chronic Pseudomonas aeruginosa infection in cystic fibrosis patients. J. Cyst. Fibros. 2003, 2, 29–34. [Google Scholar] [CrossRef]
- Flume, P.A.; Mogayzel Jr, P.J.; Robinson, K.A.; Goss, C.H.; Rosenblatt, R.L.; Kuhn, R.J.; Bruce, M.C.; The Clinical Practice Guidelines for Pulmonary Therapies Committee. Cystic fibrosis pulmonary guidelines: Treatment of pulmonary exacerbations. Am. J. Respir. Crit. Care Med. 2009, 180, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Farrell, P.M.; White, T.B.; Ren, C.; Hempstead, S.E.; Accurso, F.; Derichs, N.; Howenstine, M.; McColley, S.A.; Rock, M.; Rosenfeld, M.; et al. Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation. J. Pediatrics 2017, 181, S4–S15.e1. [Google Scholar] [CrossRef] [PubMed]
- Bombieri, C.; Claustres, M.; De Boeck, K.; Derichs, N.; Dodge, J.; Girodon, E.; Sermet, I.; Schwarz, M.; Tzetis, M.; Wilschanski, M.; et al. Recommendations for the classification of diseases as CFTR-related disorders. J. Cyst. Fibros. 2011, 10, S86–S102. [Google Scholar] [CrossRef]
- Langfelder-Schwind, E.; Raraigh, K.S.; Parad, R.B. Practive variation of genetic counselor engagement in the cystic fibrosis newborn screen-positive diagnostic resolution process. J. Genet. Couns. 2019, 28, 1178–1188. [Google Scholar] [CrossRef] [PubMed]
- Ferroni, A.; Werkhauser-Bertrand, A.; Le Bourgeois, M.; Beauvais, R.; Vrielynck, S.; Durand, C.; Lenoir, G.; Berche, P.; Sermet-Gaudelus, I. Bacterial contamination in the environment of hospitalized children with cystic fibrosis. J. Cyst. Fibros. 2008, 7, 477–482. [Google Scholar] [CrossRef][Green Version]
- Terlizzi, V.; Tosco, A.; Tomaiuolo, R.; Sepe, A.; Amato, N.; Casale, A.; Mercogliano, C.; De Gregorio, F.; Improta, F.; Elce, A.; et al. Prediction of acute pancreatitis risk based on PIP score in children with cystic fibrosis. J. Cyst. Fibros. 2014, 13, 579–584. [Google Scholar] [CrossRef][Green Version]
- Ooi, C.Y.; Sutherland, R.; Castellani, C.; Keenan, K.; Boland, M.; Reisman, J.; Bjornson, C.L.; Chilvers, M.; Bartlett, L.; Kent, S.; et al. Immunoreactive trypsinogen levels in newborn screened infants with an inconclusive diagnosis of cystic fibrosis. BMC Pediatrics 2019, 19, 369. [Google Scholar] [CrossRef]
- Kasi, A.S.; Wee, C.P.; Keens, T.G.; Salinas, D.B. Abnormal Lung Clearance Index in Cystic Fibrosis Screen Positive, Inconclusive Diagnosis (CFSPID) Children with Otherwise Normal FEV1. Lung 2019, 198, 163–167. [Google Scholar] [CrossRef]
- Salinas, D.B.; Azen, C.; Young, S.; Keens, T.G.; Kharrazi, M.; Parad, R.B. Phenotypes of California CF Newborn Screen-Positive Children with CFTR 5T Allele by TG Repeat Length. Genet. Test. Mol. Biomarkers 2016, 20, 496–503. [Google Scholar] [CrossRef]
- Munck, A.; Bourmaud, A.; Bellon, G.; Picq, P.; Farrell, P.M.; on behalf of the DPAM Study Group. Phenotype of children with inconclusive cystic fibrosis diagnosis after newborn screening. Pediatric Pulmonol. 2020, 55, 918–928. [Google Scholar] [CrossRef]
- Terlizzi, V.; Mergni, G.; Centrone, C.; Festini, F.; Taccetti, G. Trend of sweat chloride values in a cohort of patients carrying CFTR mutations of varying clinical consequence: Is there a risk of increasing sweat chloride over time? Pediatric Pulmonol. 2020, 55, 1089–1093. [Google Scholar] [CrossRef] [PubMed]
- Outcomes of Infants with Indeterminate Diagnosis Detected by Cystic Fibrosis Newborn Screening. Pediatric 2015, 135, e1386–e1392. [CrossRef]
- Accurso, F.J.; Van Goor, F.; Zha, J.; Stone, A.J.; Dong, Q.; Ordonez, C.L.; Rowe, S.M.; Clancy, J.P.; Konstan, M.W.; Hoch, H.E.; et al. Sweat chloride as a biomarker of CFTR activity: Proof of concept and ivacaftor clinical trial data. J. Cyst. Fibros. 2014, 13, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Minso, R.; Schulz, A.; Dopfer, C.; Alfeis, N.; Van Barneveld, A.; Makartian-Gyulumyan, L.; Hansen, G.; Junge, S.; Müller, C.; Ringshausen, F.C.; et al. Intestinal current measurement and nasal potential difference to make a diagnosis of cases with inconclusive CFTR genetics and sweat test. BMJ Open Respir. Res. 2020, 7, e000736. [Google Scholar] [CrossRef] [PubMed]
- Sofia, V.M.; Surace, C.; Terlizzi, V.; Da Sacco, L.; Alghisi, F.; Angiolillo, A.; Braggion, C.; Cirilli, N.; Colombo, C.; Di Lullo, A.; et al. Trans-heterozygosity for mutations enhances the risk of recurrent/chronic pancreatitis in patients with Cystic Fibrosis. Mol. Med. 2018, 24, 38. [Google Scholar] [CrossRef] [PubMed]
- Terlizzi, V.; De Gregorio, F.; Sepe, A.; Amato, N.; Arduino, C.; Casale, A.; Majo, F.; Tomaiuolo, R.; Castaldo, G.; Raia, V. Brand new SPINK1 and CFTR mutations in a child with acute recurrent pancreatitis: A case report. Minerva Pediatr. 2013, 65, 669–672. [Google Scholar] [PubMed]
- Lucarelli, M.; Narzi, L.; Pierandrei, S.; Bruno, S.M.; Stamato, A.; d’Avanzo, M.; Strom, R.; Quattrucci, S. A new complex allele of the CFTR gene partially explains the variable phenotype of the L997F mutation. Genet Med. 2010, 12, 548–555. [Google Scholar] [CrossRef]
- Salvatore, D.; Padoan, R.; Buzzetti, R.; Amato, A.; Giordani, B.; Ferrari, G.; Majo, F. Patients with cystic fibrosis having a residual function mutation: Data from the Italian registry. Pediatric Pulmonol. 2019, 54, 150–157. [Google Scholar] [CrossRef]
- McKone, E.F.; Emerson, S.S.; Edwards, K.L.; Aitken, M.L. Effect of genotype on phenotype and mortality in cystic fibrosis: A retrospective cohort study. Lancet 2003, 361, 1671–1676. [Google Scholar] [CrossRef]
- Salvatore, D.; Terlizzi, V.; Francalanci, M.; Taccetti, G.; Messore, B.; Biglia, C.; Pisi, G.; Calderazzo, M.A.; Caloiero, M.; Pizzamiglio, G.; et al. Ivacaftor improves lung disease in patients with advanced CF carrying CFTR mutations that confer residual function. Respir. Med. 2020, 171. [Google Scholar] [CrossRef]


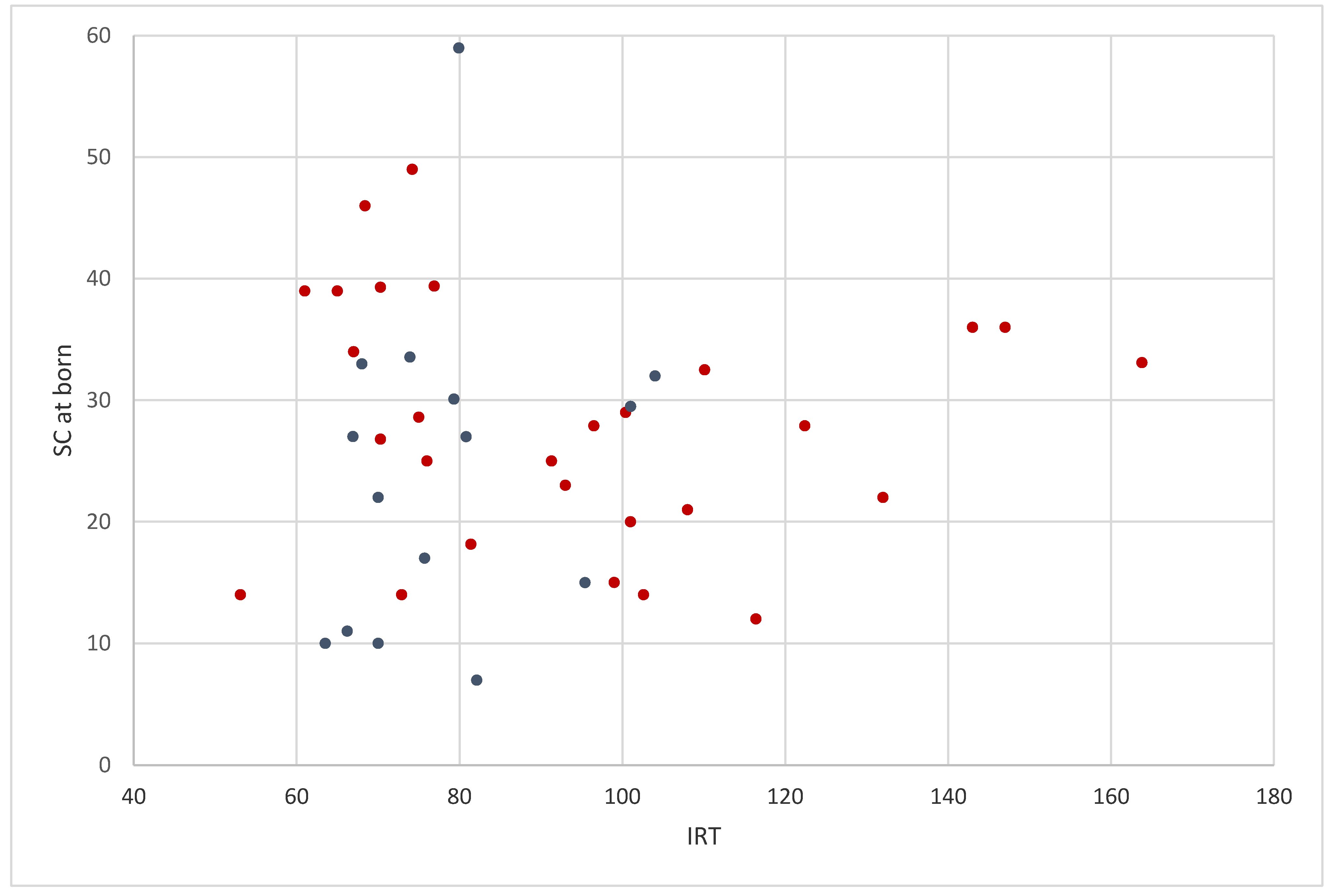{kind=link}
| n | Group A | nr | Group B | p | |
|---|---|---|---|---|---|
| Mean IRT (ng/mL) | 28 | 96.6 | 15 | 78.4 | 0.007 |
| IQR§ 25–75 | 74.2–110.1 | 68–82.1 | |||
| Mean Sweat chloride (mmol/L) | 28 | 27.8 | 15 | 24.2 | 0.328 |
| IQR 25–75 | 21–34 | 11–32 |
| Group A | Group B | ||||
|---|---|---|---|---|---|
| N | % | N | % | p | |
| A. Clinical symptoms | |||||
| Pancreatic sufficiency | 28/28 | 100% | 15/15 | 100% | |
| Pancreatitis | 3/26 | 11.5% | 0/15 | 0.0% | 0.110 |
| Metabolic hypochloremic alkalosis | 1/26 | 3.8% | 0/15 | 0.0% | 0.442 |
| B. Respiratory Infection | |||||
| Pseudomonas aeruginosa * | 10/26 | 38.5 | 1/13 | 7.7% | 0.044 |
| MSSA | 12/26 ° | 46.2% | 6/13 | 46.2% | 0.871 |
| MRSA | 3/26 ˆ | 11.5% | 2/13 | 15.4% | 0.735 |
| C. Pulmonary exacerbations | |||||
| N of antibiotic therapy in the first year | 25 | 1.8 | 13 | 0.6 | 0.027 |
| IQR 25–75 | 0–3 | 0–0 | |||
| D. Diagnostic tools performed | |||||
| Oropharyngeal swab | 26/26 | 100.0% | 13/15 | 86.7% | 0.056 |
| Chest X-ray | 25/26 | 96.2% | 7/15 | 46.7% | <0.001 |
| E. Therapies | |||||
| Chest physiotherapy | 18/26 | 69.2% | 2/15 | 13.3% | 0.001 |
| Saline Supplementation | 15/26 | 57.7% | 3/15 | 20.0% | 0.019 |
| Age of Final Diagnosis (Months) | Final Diagnosis | First Variant | Second Variant | Symptoms | First sweat Chloride (mmol/L) | Last Sweat Chloride (mmol/L) | |
|---|---|---|---|---|---|---|---|
| 1 | 43 | CF | D1152H | G542X | None | 34 | 71 |
| 2 | 7 | CFTR-RD | D1152H | R1158X | Pancreatitis | 20 | 20 |
| 3 | 27 | CFTR-RD | D1152H | L732X | Pancreatitis | 21 | 21 |
| 4 | 11 | CFTR-RD | D1152H | F508del | Pancreatitis | 12 | 24 |
Publisher's Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terlizzi, V.; Padoan, R.; Claut, L.; Colombo, C.; Fabrizzi, B.; Lucarelli, M.; Bruno, S.M.; Castaldo, A.; Bonomi, P.; Taccetti, G.; et al. CRMS/CFSPID Subjects Carrying D1152H CFTR Variant: Can the Second Variant Be a Predictor of Disease Development? Diagnostics 2020, 10, 1080. https://doi.org/10.3390/diagnostics10121080
Terlizzi V, Padoan R, Claut L, Colombo C, Fabrizzi B, Lucarelli M, Bruno SM, Castaldo A, Bonomi P, Taccetti G, et al. CRMS/CFSPID Subjects Carrying D1152H CFTR Variant: Can the Second Variant Be a Predictor of Disease Development? Diagnostics. 2020; 10(12):1080. https://doi.org/10.3390/diagnostics10121080
Chicago/Turabian StyleTerlizzi, Vito, Rita Padoan, Laura Claut, Carla Colombo, Benedetta Fabrizzi, Marco Lucarelli, Sabina Maria Bruno, Alice Castaldo, Paolo Bonomi, Giovanni Taccetti, and et al. 2020. "CRMS/CFSPID Subjects Carrying D1152H CFTR Variant: Can the Second Variant Be a Predictor of Disease Development?" Diagnostics 10, no. 12: 1080. https://doi.org/10.3390/diagnostics10121080
APA StyleTerlizzi, V., Padoan, R., Claut, L., Colombo, C., Fabrizzi, B., Lucarelli, M., Bruno, S. M., Castaldo, A., Bonomi, P., Taccetti, G., & Tosco, A. (2020). CRMS/CFSPID Subjects Carrying D1152H CFTR Variant: Can the Second Variant Be a Predictor of Disease Development? Diagnostics, 10(12), 1080. https://doi.org/10.3390/diagnostics10121080