Identification of a De Novo Xq26.2 Microduplication Encompassing FIRRE Gene in a Child with Intellectual Disability

 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
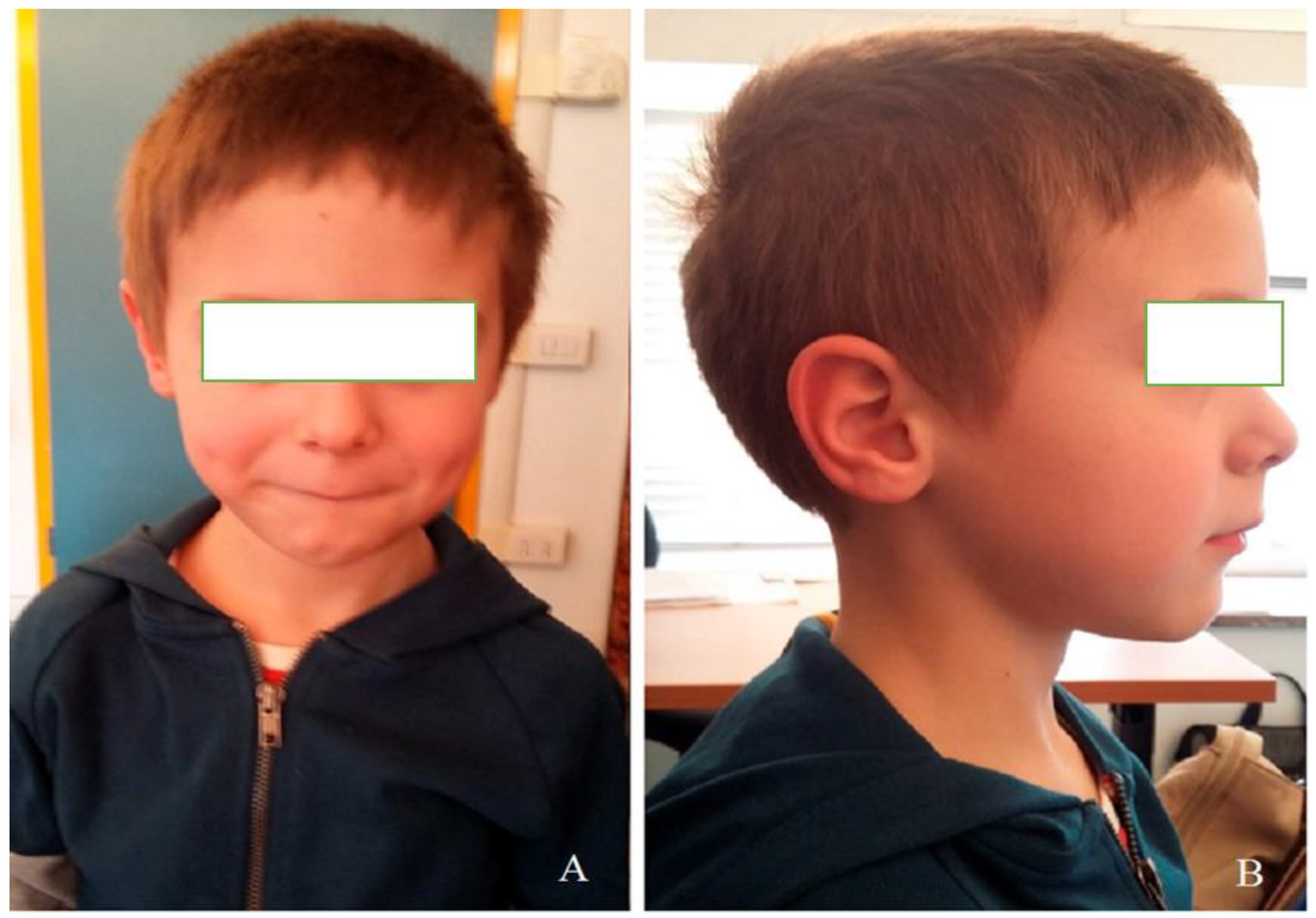
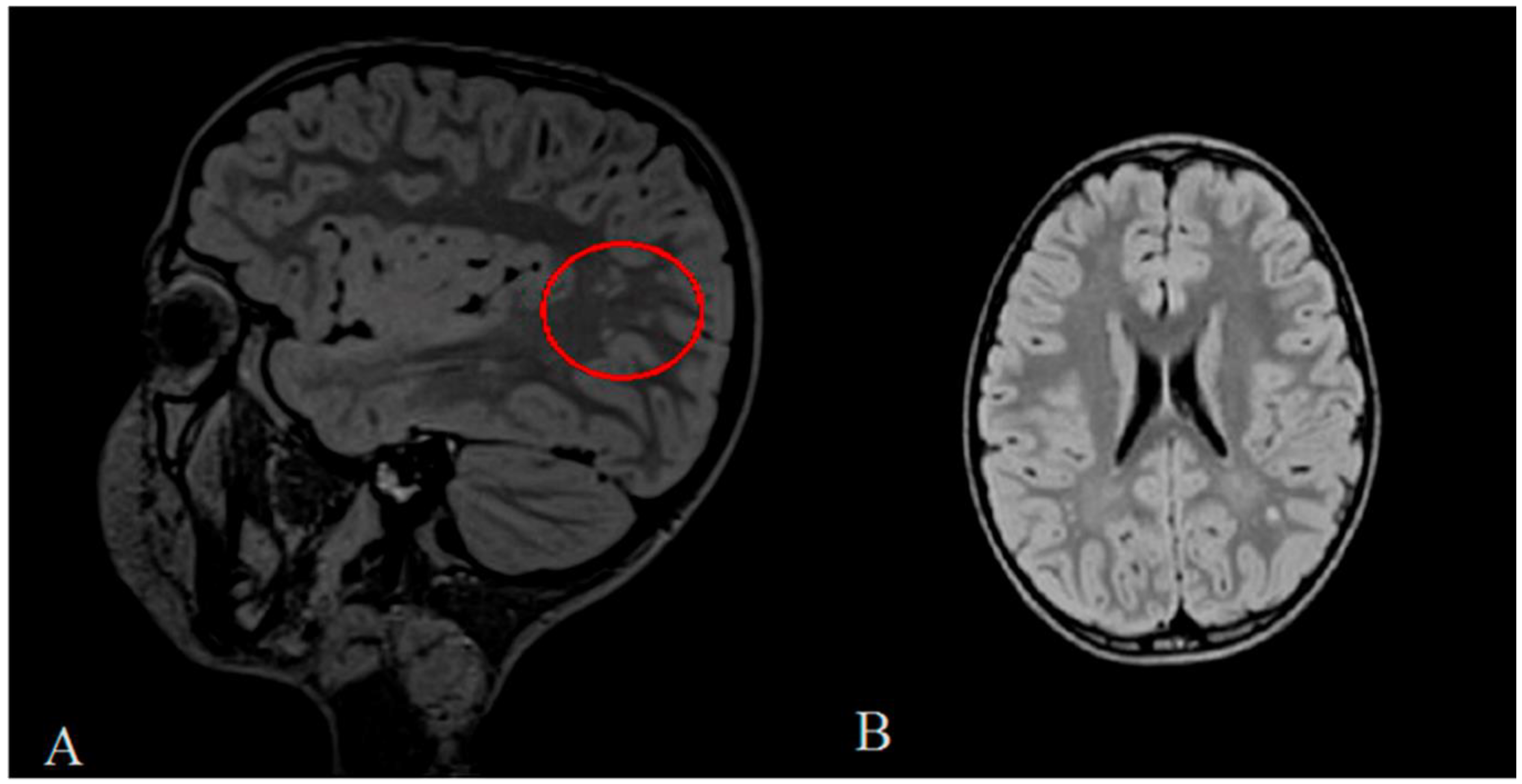
2. Clinical Report
3. Materials and Methods
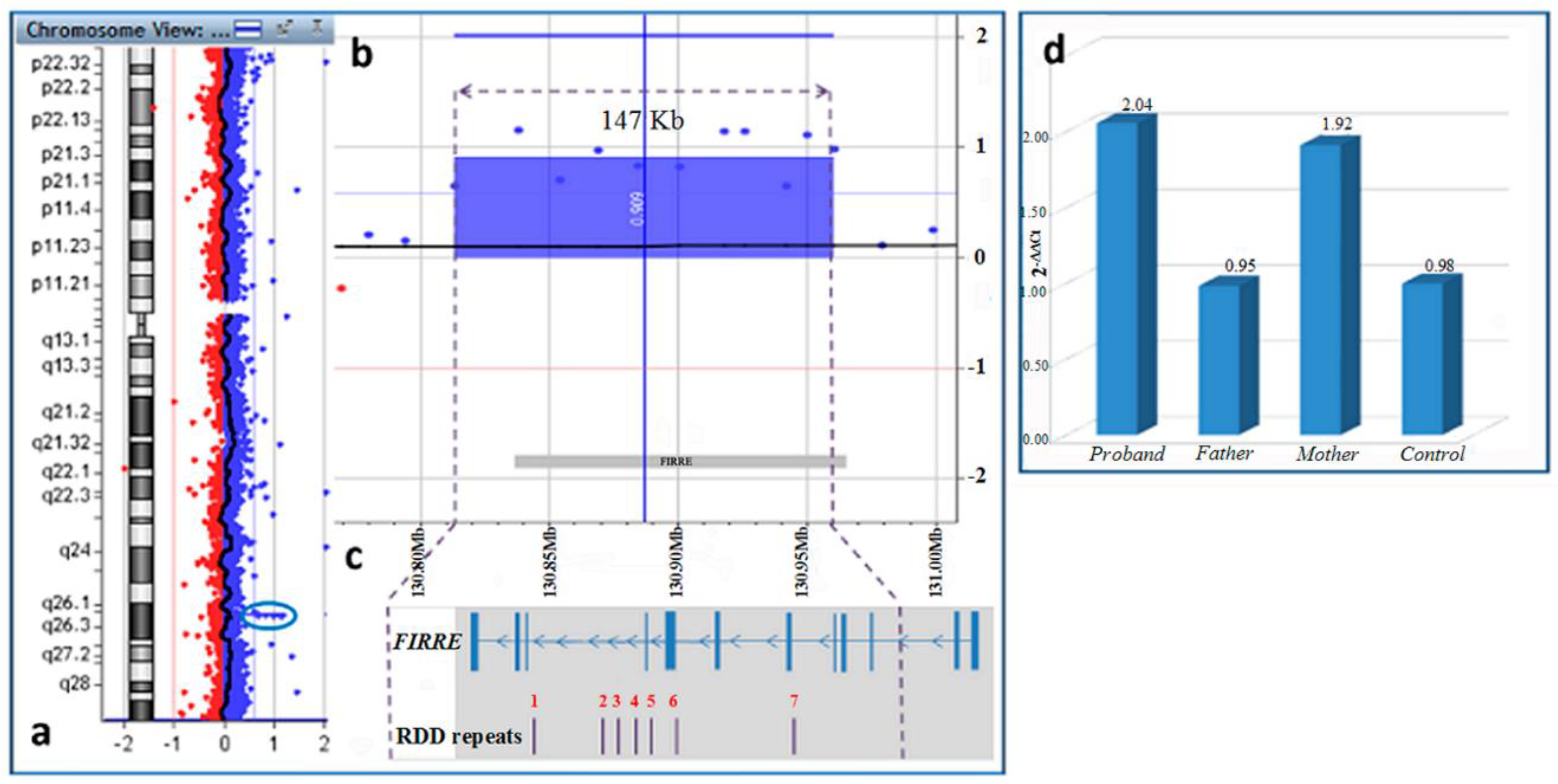
4. Results
5. Discussion
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lee, T.I.; Young, R.A. Transcriptional regulation and its misregulation in disease. Cell 2013, 152, 1237–1251. [Google Scholar] [CrossRef] [PubMed]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Srijyothi, L.; Ponne, S.; Prathama, T.; Ashok, C.; Baluchamy, S. Roles of Non-Coding RNAs in Transcriptional Regulation. In Transcriptional and Post-Transcriptional Regulation, 1st ed.; Ghedirach, K., Ed.; IntechOpen: London, UK, 2018; Chapter 4; pp. 55–75. [Google Scholar]
- Li, L.; Zhuang, Y.; Zhao, X.; Li, X. Long Non-coding RNA in Neuronal Development and Neurological Disorders. Front. Genet. 2019, 9, 744. [Google Scholar] [CrossRef] [PubMed]
- Ang, C.E.; Ma, Q.; Wapinski, O.L.; Fan, S.; Flynn, R.A.; Lee, Q.Y.; Coe, B.; Onoguchi, M.; Olmos, V.H.; Do, B.T.; et al. The novel lncRNA lnc-NR2F1 is pro-neurogenic and mutated in human neurodevelopmental disorders. Elife 2019, 8, e41770. [Google Scholar] [CrossRef] [PubMed]
- van de Vondervoort, I.I.G.M.; Gordebeke, P.M.; Khoshab, N.; Tiesinga, P.H.E.; Buitelaar, J.K.; Kozicz, T.; Aschrafi, A.; Glennon, J.C. Long non-coding RNAs in neurodevelopmental disorders. Front. Mol. Neurosci. 2013, 6, 53. [Google Scholar] [CrossRef]
- D’haene, E.; Jacobs, E.Z.; Volders, P.J.; De Meyer, T.; Menten, B.; Vergult, S. Identification of long non-coding RNAs involved in neuronal development and intellectual disability. Sci Rep. 2016, 6, 28396. [Google Scholar] [CrossRef]
- Briggs, J.A.; Wolvetang, E.J.; Mattick, J.S.; Rinn, J.L.; Barry, G. Mechanisms of Long Non-coding RNAs in Mammalian Nervous System Development, Plasticity, Disease, and Evolution. Neuron 2015, 88, 861–877. [Google Scholar] [CrossRef]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef]
- Yang, F.; Babak, T.; Shendure, J.; Disteche, C.M. Global survey of escape from X inactivation by RNA-sequencing in mouse. Genome Res. 2010, 20, 614–622. [Google Scholar] [CrossRef]
- Engreitz, J.M.; Pandya-Jones, A.; McDonel, P.; Shishkin, A.; Sirokman, K.; Surka, C.; Kadri, S.; Xing, J.; Goren, A.; Lander, E.S.; et al. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science 2013, 341, 1237973. [Google Scholar] [CrossRef]
- Froberg, J.E.; Pinter, S.F.; Kriz, A.J.; Jégu, T.; Lee, J.T. Megadomains and superloops form dynamically but are dispensable for X-chromosome inactivation and gene escape. Nat. Commun. 2018, 9, 5004. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.W.; Luo, T.; Zou, S.S.; Wu, A.S. The Role of Long Noncoding RNAs in Central Nervous System and Neurodegenerative Diseases. Front. Behav. Neurosci. 2018, 12, 175. [Google Scholar] [CrossRef] [PubMed]
- Balas, M.M.; Johnson, A.M. Exploring the mechanisms behind longnocoding RNAs and cancer. Non-Coding RNA Res. 2018, 3, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Ramos, A.D.; Andersen, R.E.; Liu, S.J.; Nowakowski, T.J.; Hong, S.J.; Gertz, C.; Salinas, R.D.; Zarabi, H.; Kriegstein, A.R.; Lim, D.A. The long noncoding RNA Pnky regulates neuronal differentiation of embryonic and postnatal neural stem cells. Cell Stem Cell 2015, 16, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Bian, S.; Sun, T. Functions of noncoding RNAs in neural development and neurological diseases. Mol. Neurobiol. 2011, 44, 359–373. [Google Scholar] [CrossRef]
- Lewandowski, J.P.; Lee, J.C.; Hwang, T.; Sunwoo, H.; Goldstein, J.M.; Groff, A.F.; Chang, N.P.; Mallard, W.; Williams, A.; Henao-Meija, J.; et al. The Firre locus produces a trans-acting RNA molecule that functions in hematopoiesis. Nat. Commun. 2019, 10, 5137. [Google Scholar] [CrossRef]
- Silva, M.; de Leeuw, N.; Mann, K.; Schuring-Blom, H.; Morgan, S.; Giardino, D.; Rack, K.; Hastings, R. European guidelines for constitutional cytogenomic analysis. Eur. J. Hum. Genet. 2019, 27, 1–16. [Google Scholar] [CrossRef]
- Carbone, A.; Bernardini, L.; Valenzano, F.; Bottillo, I.; De Simone, C.; Capizzi, R.; Capalbo, A.; Romano, F.; Novelli, A.; Dallapiccola, B.; et al. Array-based comparative genomic hybridization in early-stage mycosis fungoides: Recurrent deletion of tumor suppressor genes BCL7A, SMAC/DIABLO, and RHOF. Genes Chromosomes Cancer 2008, 47, 1067–1075. [Google Scholar] [CrossRef]
- Hacisuleyman, E.; Goff, L.A.; Trapnell, C.; Williams, A.; Henao-Mejia, J.; Sun, L.; McClanahan, P.; Hendrickson, D.G.; Sauvageau, M.; Kelley, D.R.; et al. Topological organization of multichromosomal regions by the long intergenic noncoding RNA Firre. Nat. Struct. Mol. Biol. 2014, 21, 198–206. [Google Scholar] [CrossRef]
- Abe, Y.; Kikuchi, A.; Kobayashi, S.; Wakusawa, K.; Tanaka, S.; Inui, T.; Kunishima, S.; Kure, S.; Haginoya, K. Xq26.1-26.2 gain identified on array comparative genomic hybridization in bilateral periventricular nodular heterotopia with overlying polymicrogyria. Dev. Med. Child Neurol. 2014, 56, 1221–1224. [Google Scholar] [CrossRef]
- Zang, Y.; Zhou, X.; Wang, Q.; Li, X.; Huang, H. LncRNA FIRRE/NF-kB feedback loop contributes to OGD/R injury of cerebral microglial cells. Biochem. Biophys. Res. Commun. 2018, 501, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Liu, X.; Xie, M.; Liu, M.; Ye, M.; Li, M.; Chen, X.M.; Li, X.; Zhou, R. The NF-kB responsive long noncoding RNA FIRRE regulates posttranscriptional regulation of inflammatory gene expression through interacting with hnRNPU. J. Immunol. 2017, 199, 3571–3582. [Google Scholar] [CrossRef] [PubMed]
- Andergassen, D.; Smith, Z.D.; Lewandowski, J.P.; Gerhardinger, C.; Meissner, A.; Rinn, J.L. In vivo Firre and Dxz4 deletion elucidates roles for autosomal gene regulation. Elife 2019, 18, e47214. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Deng, X.; Ma, W.; Berletch, J.B.; Rabaia, N.; Wei, G.; Moore, J.M.; Filippova, G.N.; Xu, J.; Liu, Y.; et al. The lncRNA Firre anchors the inactive X chromosome to the nucleolus by binding CTCF and maintains H3K27me3 methylation. Genome Biol. 2015, 16, 52. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Disteche, C.M.; Berletch, J.B. X Inactivation and Escape: Epigenetic and Structural Features. Front. Cell Dev. Biol. 2019, 7, 219. [Google Scholar] [CrossRef]
- Refolo, V.; Stefanova, N. Neuroinflammation and Glial Phenotypic Changes in Alpha-Synucleinopathies. Front. Cell. Neurosci. 2019, 13, 263. [Google Scholar] [CrossRef]
- Morgan, J.T.; Chana, G.; Pardo, C.A.; Achim, C.; Semendeferi, K.; Buckwalter, J.; Courchesne, E.; Everall, I.P. Microglial activation and increased microglial density observed in the dorsolateral prefrontal cortex in autism. Biol. Psychiatry 2010, 68, 368–376. [Google Scholar] [CrossRef]
- Tetreault, N.A.; Hakeem, A.Y.; Jiang, S.; Williams, B.A.; Allman, E.; Wold, B.J.; Allman, J.M. Microglia in the cerebral cortex in autism. J. Autism Dev. Disord. 2012, 42, 2569–2584. [Google Scholar] [CrossRef]
- Pecorelli, A.; Cordone, V.; Messano, N.; Zhang, C.; Falone, S.; Amicarelli, F.; Hayek, J.; Valacchi, G. Altered inflammasome machinery as a key player in the perpetuation of Rett syndrome oxiinflammation. Redox Biol. 2020, 28, 101334. [Google Scholar] [CrossRef]
- Chiurazzi, P.; Dirozzi, F. Advances in understanding genetic basis of intellectual disability. F1000Research 2016, 5. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miolo, G.; Bernardini, L.; Capalbo, A.; Favia, A.; Goldoni, M.; Pivetta, B.; Tessitori, G.; Corona, G. Identification of a De Novo Xq26.2 Microduplication Encompassing FIRRE Gene in a Child with Intellectual Disability. Diagnostics 2020, 10, 1009. https://doi.org/10.3390/diagnostics10121009
Miolo G, Bernardini L, Capalbo A, Favia A, Goldoni M, Pivetta B, Tessitori G, Corona G. Identification of a De Novo Xq26.2 Microduplication Encompassing FIRRE Gene in a Child with Intellectual Disability. Diagnostics. 2020; 10(12):1009. https://doi.org/10.3390/diagnostics10121009
Chicago/Turabian StyleMiolo, Gianmaria, Laura Bernardini, Anna Capalbo, Anna Favia, Marina Goldoni, Barbara Pivetta, Giovanni Tessitori, and Giuseppe Corona. 2020. "Identification of a De Novo Xq26.2 Microduplication Encompassing FIRRE Gene in a Child with Intellectual Disability" Diagnostics 10, no. 12: 1009. https://doi.org/10.3390/diagnostics10121009
APA StyleMiolo, G., Bernardini, L., Capalbo, A., Favia, A., Goldoni, M., Pivetta, B., Tessitori, G., & Corona, G. (2020). Identification of a De Novo Xq26.2 Microduplication Encompassing FIRRE Gene in a Child with Intellectual Disability. Diagnostics, 10(12), 1009. https://doi.org/10.3390/diagnostics10121009