Myocardial Function, Heart Failure and Arrhythmia in Marfan Syndrome: A Systematic Literature Review


Abstract
:1. Introduction
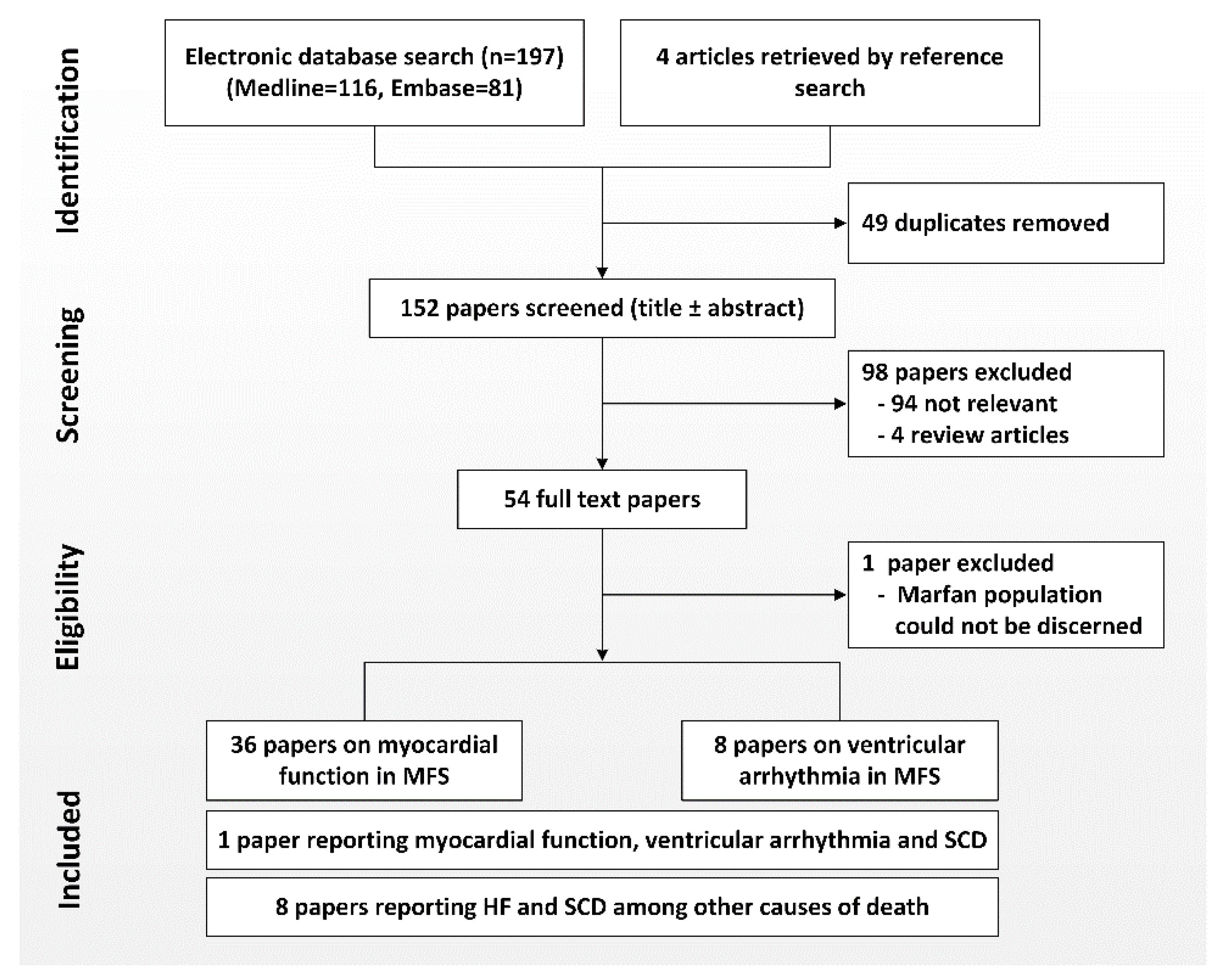
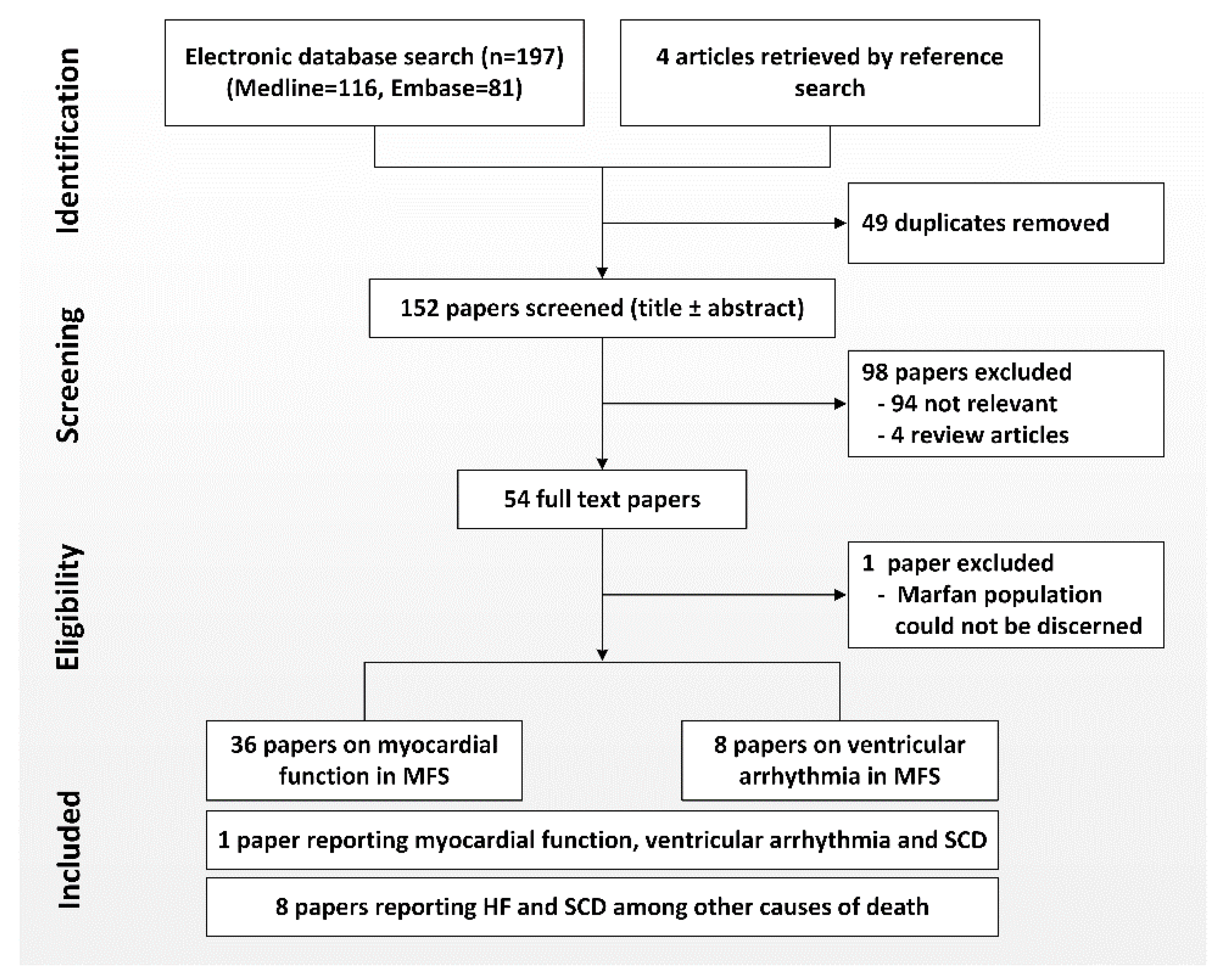
2. Methods
3. Myocardial Involvement
3.1. Left Ventricular Dimensions and Function: Evidence Obtained from Echocardiographic Studies
3.2. Evidence Obtained from Advanced Imaging Techniques
3.3. Involvement of the Right Ventricle and Atria
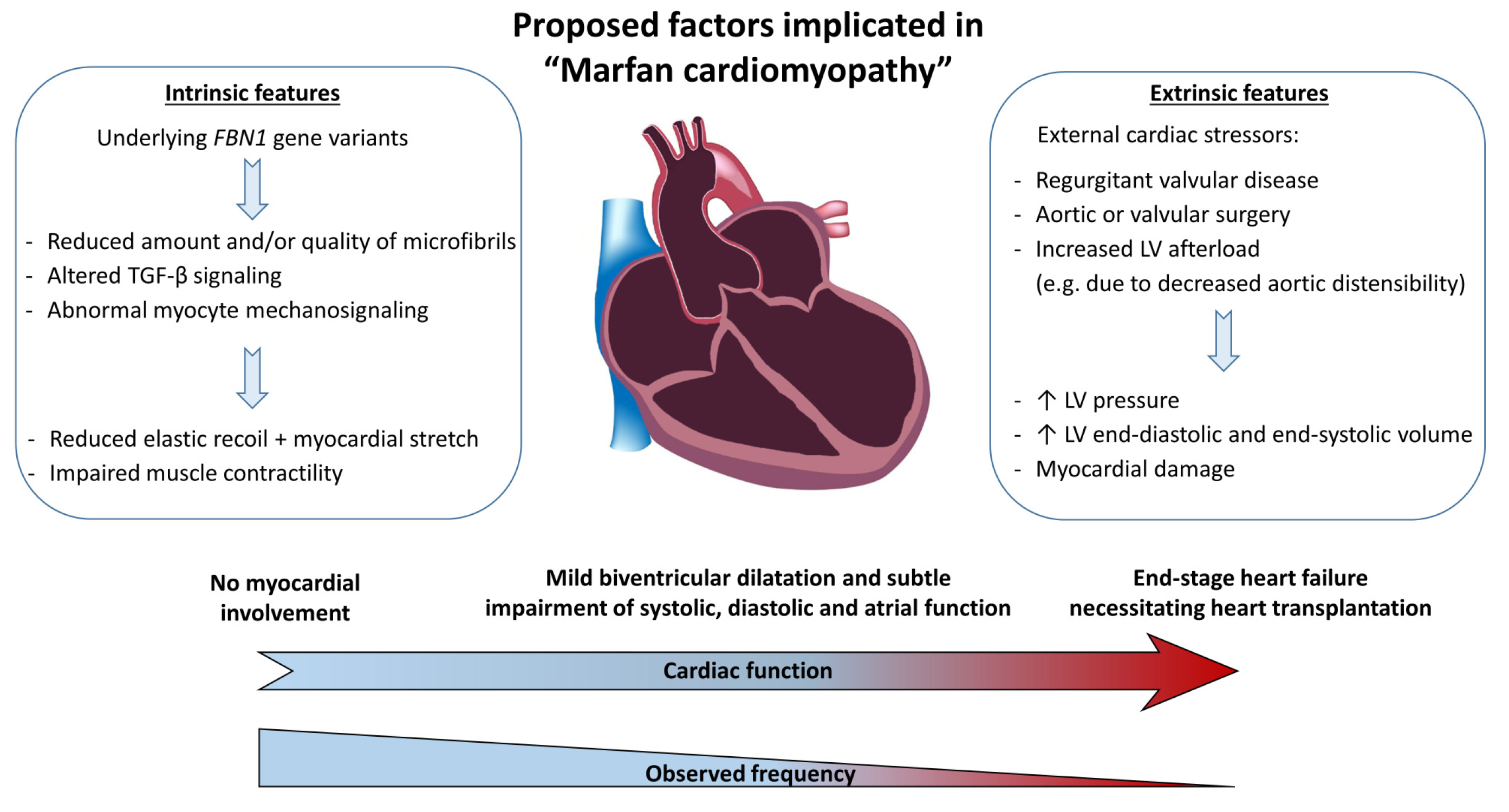
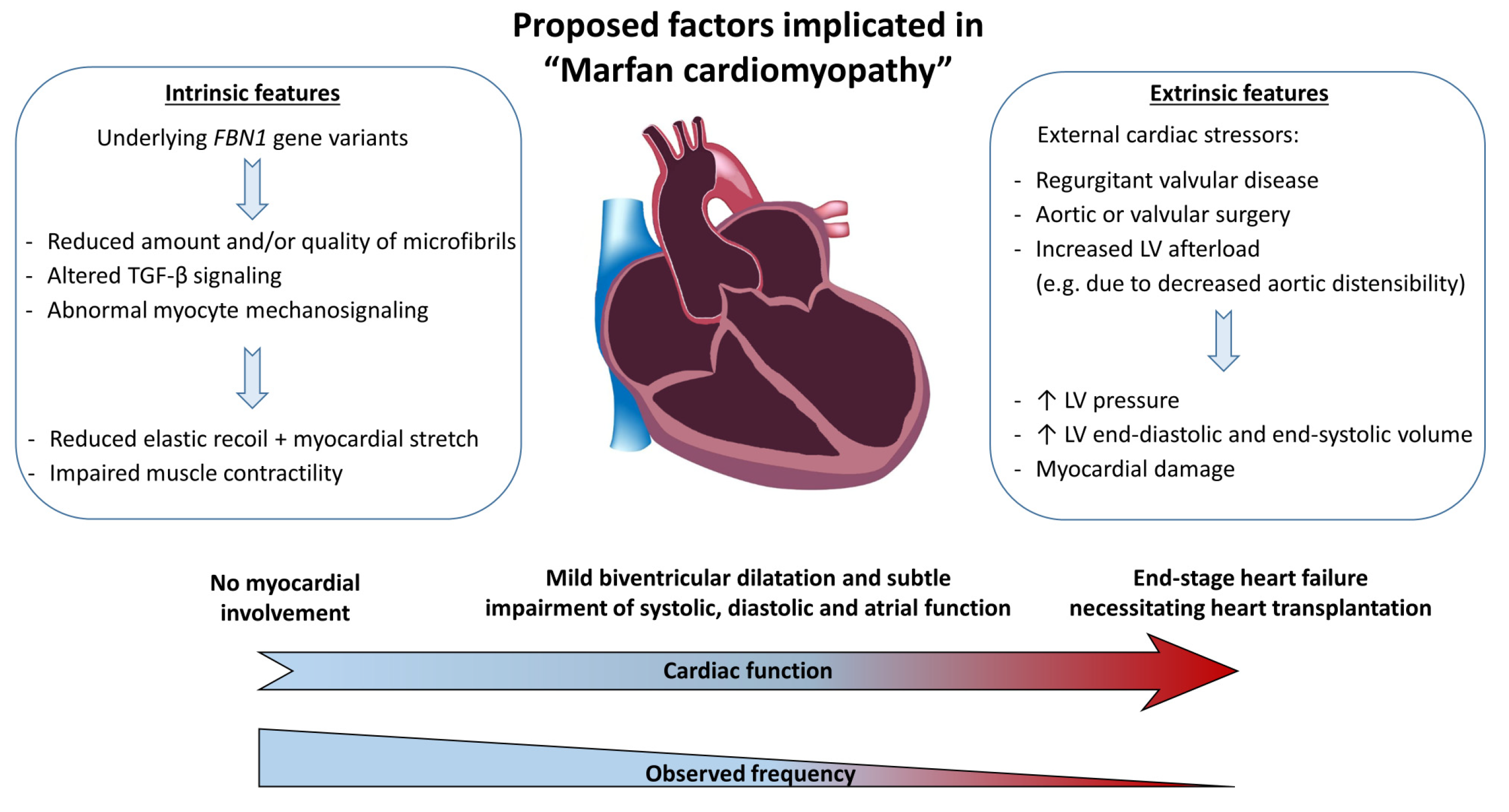
4. Pathophysiology of Marfan Cardiomyopathy
4.1. Intrinsic vs. Stressor-Induced Problem
4.2. Valvular Disease, Surgery and Genotype-Phenotype Relation
4.3. Evidence Obtained from Mouse Models
4.4. Proposed Hypothesis
5. Association with Arrhythmia
Arrhythmia in Marfan Syndrome
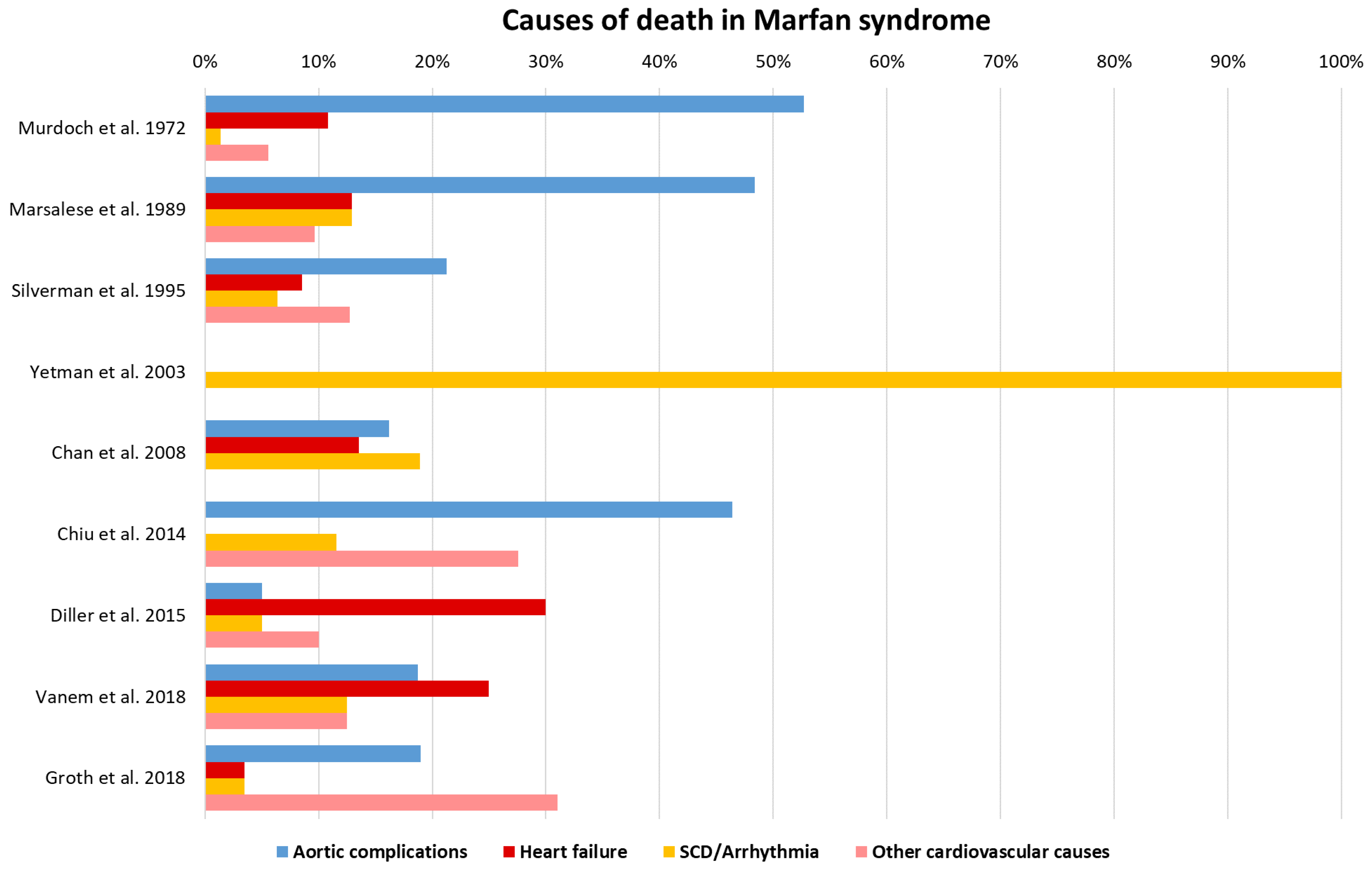
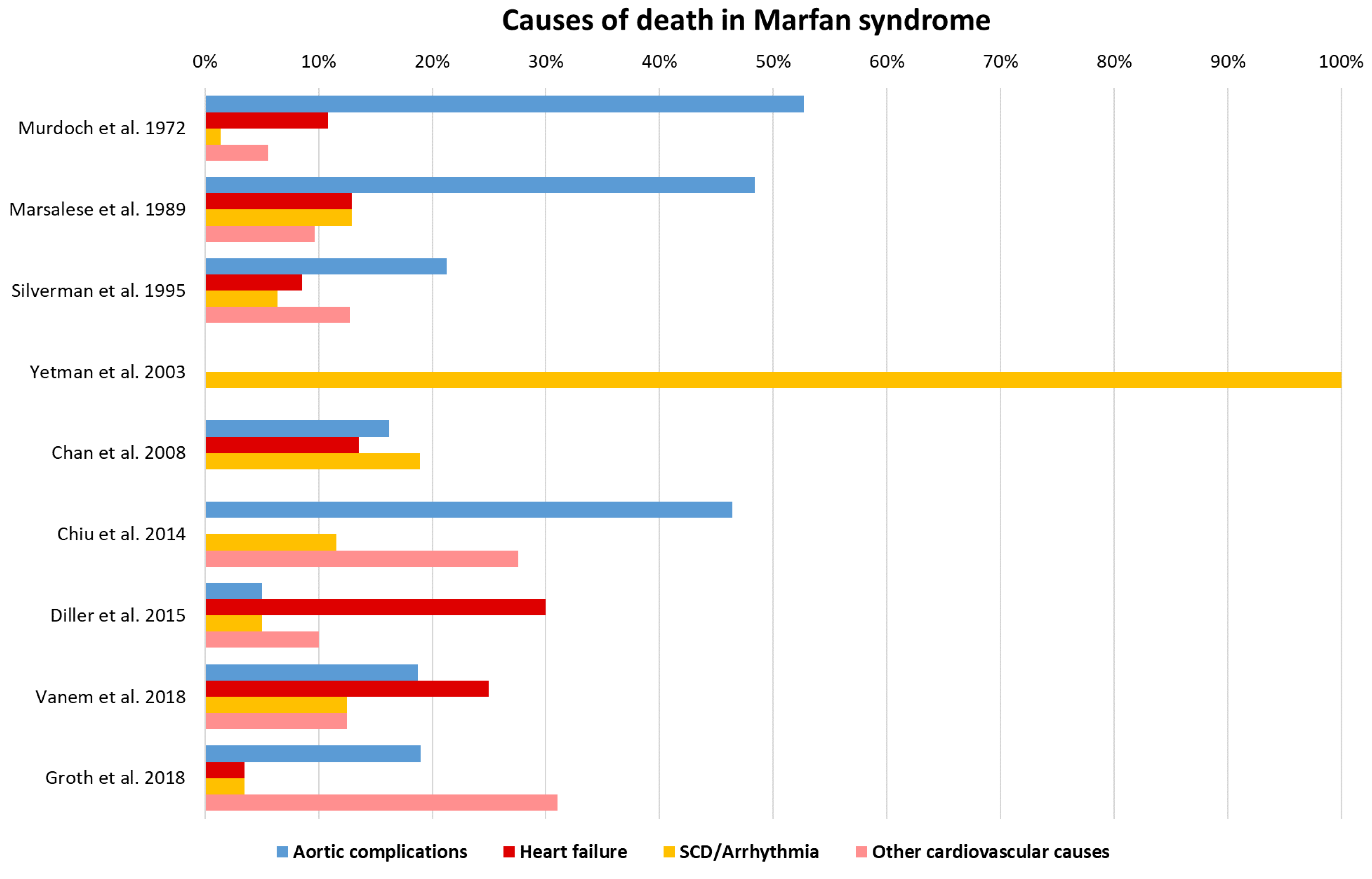
6. Heart Failure and Arrhythmia as Additional Causes of Death
7. Discussion
7.1. Current View on Marfan Cardiomyopathy
7.2. The Intertwined Mechanism of Marfan Cardiomyopathy and Ventricular Arrhythmia
8. Current Limitations and Evidence Gaps
9. Conclusions
Funding
Conflicts of Interest
Abbreviations
| BSA | Body surface area |
| CMR | Cardiac magnetic resonance imaging |
| DN | Dominant negative |
| EF | Ejection fraction |
| FBN1 | Fibrillin-1 |
| FS | Fractional shortening |
| HF | Heart failure |
| HI | Haploinsufficiency |
| LV | Left ventricular |
| LVEDD | Left ventricular end-diastolic diameter |
| LVESD | Left ventricular end-systolic diameter |
| MFS | Marfan syndrome |
| MVP | Mitral valve prolapse |
| NSVT | Non-sustained ventricular tachycardia |
| NTproBNP | N-terminal pro b-type natriuretic peptide |
| PAC | Premature atrial complex |
| PVC | Premature ventricular complex |
| RV | Right ventricular |
| SCD | Sudden cardiac death |
| TDI | Tissue Doppler imaging |
| TGF-β | Transforming growth factor beta |
| VF | Ventricular fibrillation |
| VT | Ventricular tachycardia |
References
- Von Kodolitsch, Y.; De Backer, J.; Schüler, H.; Bannas, P.; Behzadi, C.; Bernhardt, A.M.; Hillebrand, M.; Fuisting, B.; Sheikhzadeh, S.; Rybczynski, M.; et al. Perspectives on the revised Ghent criteria for the diagnosis of Marfan syndrome. Appl. Clin. Genet. 2015, 137–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loeys, B.; Dietz, H.C.; Braverman, A.C.; Callewaert, B.; De Backer, J.; Devereux, R.B.; Hilhorst-Hofstee, Y.; Jondeau, G.; Faivre, L.; Milewicz, D.M.; et al. The revised Ghent nosology for the Marfan syndrome. J. Med. Genet. 2010, 47, 476–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez, F.; Caescu, C.; Wondimu, E.; Galatioto, J. Marfan syndrome; A connective tissue disease at the crossroads of mechanotransduction, TGFβ signaling and cell stemness. Matrix Boil. 2018, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, J.D.; Schwartz, M.A.; Tellides, G.; Milewicz, D.M. Role of mechanotransduction in vascular biology: Focus on thoracic aortic aneurysms and dissections. Circ. Res. 2015, 116, 1448–1461. [Google Scholar] [CrossRef] [Green Version]
- Chaudhry, S.S.; Cain, S.A.; Morgan, A.; Dallas, S.L.; Shuttleworth, C.A.; Kielty, C.M. Fibrillin-1 regulates the bioavailability of TGFβ1. J. Cell Boil. 2007, 176, 355–367. [Google Scholar] [CrossRef]
- Child, A.H. Diagnosis and Management of Marfan Syndrome; Springer: London, UK, 2016; ISBN 9781447154426. [Google Scholar]
- Mariucci, E.; Lovato, L.; Rosati, M.; Palena, L.M.; Bonvicini, M.; Fattori, R. Dilation of peripheral vessels in Marfan syndrome: Importance of thoracoabdominal MR angiography. Int. J. Cardiol. 2013, 167, 2928–2931. [Google Scholar] [CrossRef]
- Schönhoff, F.; Yildiz, M.; Langhammer, B.; Jungi, S.; Wyss, T.R.; Makaloski, V.; Schmidli, J.; Carrel, T. The fate of nonaortic arterial segments in Marfan patients. J. Thorac. Cardiovasc. Surg. 2019, 157, 2150–2156. [Google Scholar] [CrossRef]
- Stheneur, C.; Faivre, L.; Collod-Beroud, G.; Gautier, E.; Binquet, C.; Bonithon–Kopp, C.; Claustres, M.; Child, A.H.; Arbustini, E.; Adès, L.C.; et al. Prognosis Factors in Probands with an FBN1 Mutation Diagnosed Before the Age of 1 Year. Pediatr. Res. 2011, 69, 265–270. [Google Scholar] [CrossRef] [Green Version]
- Roman, M.J.; Devereux, R.B.; Preiss, L.R.; Asch, F.M.; Eagle, K.A.; Holmes, K.W.; Lemaire, S.A.; Maslen, C.L.; Milewicz, D.M.; Morris, S.A.; et al. Associations of Age and Sex with Marfan Phenotype. Circ. Cardiovasc. Genet. 2017, 10. [Google Scholar] [CrossRef] [Green Version]
- Milleron, O.; Arnoult, F.; Delorme, G.; Detaint, D.; Pellenc, Q.; Raffoul, R.; Tchitchinadze, M.; Langeois, M.; Guien, C.; Beroud, C.; et al. Pathogenic FBN1 Genetic Variation and Aortic Dissection in Patients with Marfan Syndrome. J. Am. Coll. Cardiol. 2020, 75, 843–853. [Google Scholar] [CrossRef]
- Von Kodolitsch, Y.; Demolder, A.; Girdauskas, E.; Kaemmerer, H.; Kornhuber, K.; Mosquera, L.M.; Morris, S.; Neptune, E.; Pyeritz, R.; Rand-Hendriksen, S.; et al. Features of Marfan syndrome not listed in the Ghent nosology—The dark side of the disease. Expert Rev. Cardiovasc. Ther. 2019, 17, 883–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiotsekoglou, A.; Sutherland, G.R.; Moggridge, J.C.; Nassiri, D.K.; Camm, A.J.; Child, A.H. The unravelling of primary myocardial impairment in Marfan syndrome by modern echocardiography. Heart 2009, 95, 1561–1566. [Google Scholar] [CrossRef]
- Yetman, A.T.; Bornemeier, R.A.; McCrindle, B.W. Long-term outcome in patients with Marfan syndrome: Is aortic dissection the only cause of sudden death? J. Am. Coll. Cardiol. 2003, 41, 329–332. [Google Scholar] [CrossRef] [Green Version]
- Diller, G.-P.; Kempny, A.; González, R.A.; Swan, L.; Uebing, A.; Li, W.; Babu-Narayan, S.; Wort, S.J.; Dimopoulos, K.; Gatzoulis, M.A. Survival Prospects and Circumstances of Death in Contemporary Adult Congenital Heart Disease Patients Under Follow-Up at a Large Tertiary Centre. Circulation 2015, 132, 2118–2125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silverman, D.I.; Burton, K.J.; Gray, J.; Bosner, M.S.; Kouchoukos, N.T.; Roman, M.J.; Boxer, M.; Devereux, R.B.; Tsipouras, P. Life expectancy in the Marfan syndrome. Am. J. Cardiol. 1995, 75, 157–160. [Google Scholar] [CrossRef]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. BMJ 2009. [Google Scholar] [CrossRef] [Green Version]
- Fujiseki, Y.; Okuno, K.; Tanaka, M.; Shimada, M.; Takahashi, M.; Kawanishi, K. Myocardial involvement in the Marfan syndrome. Jpn. Heart J. 1985, 26, 1043–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roman, M.J.; Devereux, R.B.; Kramer-Fox, R.; Spitzer, M.C. Comparison of cardiovascular and skeletal features of primary mitral valve prolapse and Marfan syndrome. Am. J. Cardiol. 1989, 63, 317–321. [Google Scholar] [CrossRef]
- Savolainen, A.; Nisula, L.; Keto, P.; Hekali, P.; Viitasalo, M.; Kaitila, L.; Kupari, M. Left ventricular function in children with the Marfan syndrome. Eur. Heart J. 1994, 15, 625–630. [Google Scholar] [CrossRef]
- Porciani, M.C.; Giurlani, L.; Chelucci, A.; Pepe, G.; Giusti, B.; Brunelli, T.; Attanasio, M.; Martinucci, P.; Fattori, R.; Abbatea, R.; et al. Diastolic subclinical primary alterations in marfan syndrome and marfan-related disorders. Clin. Cardiol. 2006, 25, 416–420. [Google Scholar] [CrossRef]
- Chatrath, R.; Beauchesne, L.M.; Connolly, H.M.; Michels, V.V.; Driscoll, D. Left ventricular function in the Marfan syndrome without significant valvular regurgitation. Am. J. Cardiol. 2003, 91, 914–916. [Google Scholar] [CrossRef]
- Meijboom, L.J.; Timmermans, J.; Van Tintelen, J.P.; Nollen, G.J.; De Backer, J.; Berg, M.P.V.D.; Boers, G.H.; Mulder, B.J. Evaluation of left ventricular dimensions and function in Marfan’s syndrome without significant valvular regurgitation. Am. J. Cardiol. 2005, 95, 795–797. [Google Scholar] [CrossRef] [PubMed]
- De Backer, J.; Devos, D.; Segers, P.; Matthys, D.; François, K.; Gillebert, T.; De Paepe, A.M.; De Sutter, J. Primary impairment of left ventricular function in Marfan syndrome. Int. J. Cardiol. 2006, 112, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Das, B.; Taylor, A.; Yetman, A. Left Ventricular Diastolic Dysfunction in Children and Young Adults with Marfan Syndrome. Pediatr. Cardiol. 2006, 27, 256–258. [Google Scholar] [CrossRef]
- Rybczynski, M.; Koschyk, D.H.; Aydin, M.A.; Robinson, P.N.; Brinken, T.; Franzen, O.; Berger, J.; Hofmann, T.; Meinertz, T.; Von Kodolitsch, Y. Tissue Doppler imaging identifies myocardial dysfunction in adults with marfan syndrome. Clin. Cardiol. 2007, 30, 19–24. [Google Scholar] [CrossRef]
- Kiotsekoglou, A.; Bajpai, A.; Bijnens, B.; Kapetanakis, V.; Athanassopoulos, G.; Moggridge, J.C.; Mullen, M.J.; Nassiri, D.K.; Camm, J.; Sutherland, G.R.; et al. Early impairment of left ventricular long-axis systolic function demonstrated by reduced atrioventricular plane displacement in patients with Marfan syndrome. Eur. J. Echocardiogr. 2008, 9, 605–613. [Google Scholar] [CrossRef] [Green Version]
- Alpendurada, F.; Wong, J.; Kiotsekoglou, A.; Banya, W.; Child, A.; Prasad, S.K.; Pennell, D.J.; Mohiaddin, R.H. Evidence for Marfan cardiomyopathy. Eur. J. Heart Fail. 2010, 12, 1085–1091. [Google Scholar] [CrossRef]
- De Witte, P.; Aalberts, J.J.J.; Radonic, T.; Timmermans, J.; Scholte, A.J.; Zwinderman, A.H.; Mulder, B.J.M.; Groenink, M.; Berg, M.P.V.D. Intrinsic biventricular dysfunction in Marfan syndrome. Heart 2011, 97, 2063–2068. [Google Scholar] [CrossRef] [Green Version]
- Winther, S.; Williams, L.K.; Keir, M.; Connelly, K.A.; Bradley, T.J.; Rakowski, H.; Crean, A.M. Cardiovascular Magnetic Resonance Provides Evidence of Abnormal Myocardial Strain and Primary Cardiomyopathy in Marfan syndrome. J. Comput. Assist. Tomogr. 2019, 43, 410–415. [Google Scholar] [CrossRef]
- Kiotsekoglou, A.; Saha, S.; Moggridge, J.C.; Kapetanakis, V.; Govindan, M.; Alpendurada, F.; Mullen, M.J.; Nassiri, D.K.; Camm, J.; Sutherland, G.R.; et al. Impaired Biventricular Deformation in Marfan Syndrome: A Strain and Strain Rate Study in Adult Unoperated Patients. Echocardiography 2011, 28, 416–430. [Google Scholar] [CrossRef]
- Angtuaco, M.J.; Vyas, H.V.; Malik, S.; Holleman, B.N.; Gossett, J.M.; Sachdeva, R. Early detection of cardiac dysfunction by strain and strain rate imaging in children and young adults with marfan syndrome. J. Ultrasound Med. 2012, 31, 1609–1616. [Google Scholar] [CrossRef] [PubMed]
- El Rahman, M.A.; Haase, D.; Rentzsch, A.; Olchvary, J.; Schäfers, H.-J.; Henn, W.; Wagenpfeil, S.; Abdul-Khaliq, H. Left Ventricular Systolic Dysfunction in Asymptomatic Marfan Syndrome Patients Is Related to the Severity of Gene Mutation: Insights from the Novel Three Dimensional Speckle Tracking Echocardiography. PLoS ONE 2015, 10, e0124112. [Google Scholar] [CrossRef] [PubMed]
- Kiotsekoglou, A.; Sutherland, G.R.; Moggridge, J.C.; Kapetanakis, V.; Bajpai, A.; Bunce, N.; Mullen, M.J.; Louridas, G.; Nassiri, D.K.; Camm, J.; et al. Impaired right ventricular systolic function demonstrated by reduced atrioventricular plane displacement in adults with Marfan syndrome. Eur. J. Echocardiogr. 2008, 10, 295–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiotsekoglou, A.; Moggridge, J.C.; Bijnens, B.; Kapetanakis, V.; Alpendurada, F.; Mullen, M.J.; Saha, S.; Nassiri, D.K.; Camm, J.; Sutherland, G.R.; et al. Biventricular and atrial diastolic function assessment using conventional echocardiography and tissue-Doppler imaging in adults with Marfan syndrome. Eur. J. Echocardiogr. 2009, 10, 947–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherptong, R.W.; Vliegen, H.W.; Van Der Wall, E.E.; Hilhorst-Hofstee, Y.; Bax, J.J.; Scholte, A.J.; Delgado, V. Biventricular Performance in Patients with Marfan Syndrome without Significant Valvular Disease: Comparison to Normal Subjects and Longitudinal Follow-Up. J. Am. Soc. Echocardiogr. 2011, 24, 1392–1399.e1. [Google Scholar] [CrossRef] [PubMed]
- Campens, L.; Renard, M.; Trachet, B.; Segers, P.; Mosquera, L.M.; De Sutter, J.; Sakai, L.; De Paepe, A.; De Backer, J. Intrinsic cardiomyopathy in Marfan syndrome: Results from in-vivo and ex-vivo studies of the Fbn1C1039G/+ model and longitudinal findings in humans. Pediatr. Res. 2015, 78, 256–263. [Google Scholar] [CrossRef] [Green Version]
- Gehle, P.; Robinson, P.N.; Heinzel, F.; Edelmann, F.; Yigitbasi, M.; Berger, F.; Falk, V.; Pieske, B.; Wellnhofer, E. NT-proBNP and diastolic left ventricular function in patients with Marfan syndrome. IJC Heart Vasc. 2016, 12, 15–20. [Google Scholar] [CrossRef] [Green Version]
- Loeper, F.; Oosterhof, J.; Dorpel, M.V.D.; Van Der Linde, D.; Lu, Y.; Robertson, E.N.; Hambly, B.D.; Jeremy, R.W. Ventricular-Vascular Coupling in Marfan and Non-Marfan Aortopathies. J. Am. Heart Assoc. 2016, 5, e003705. [Google Scholar] [CrossRef]
- Rao, P.; Keenan, J.B.; Rajab, T.K.; Kim, S.; Smith, R.; Amabile, O.; Khalpey, Z. Total artificial heart implantation in a young Marfan syndrome patient. Int. J. Artif. Organs 2018, 41, 175–177. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, Y.; Choi, C.W.; Shudo, Y.; Woo, Y.-P.J. Successful orthotopic heart transplantation in a patient with Marfan syndrome. J. Card. Surg. 2019, 34, 875–876. [Google Scholar] [CrossRef]
- Kesler, K.A.; Hanosh, J.J.; O’Donnell, J.; Faust, S.; Turrentine, M.W.; Mahomed, Y.; Brown, J.W. Heart transplantation in patients with Marfan’s syndrome: A survey of attitudes and results. J. Heart Lung Transplant. 1994, 13, 899–904. [Google Scholar]
- Mullen, J.C.; Lemermeyer, G.; Bentley, M.J. Recurrent Aortic Dissection after Orthotopic Heart Transplantation. Ann. Thorac. Surg. 1996, 62, 1830–1831. [Google Scholar] [CrossRef]
- Varghese, J.; Gilani, S.; Arentzen, C.; Jennison, S. Rupture of chronic thoracic aortic dissection in a Marfan syndrome patient after heart transplantation. J. Heart Lung Transplant. 2006, 25, 610–611. [Google Scholar] [CrossRef] [PubMed]
- Botta, L.; Russo, V.; Grigioni, F.; Arpesella, G.; Rocchi, G.; Di Bartolomeo, R.; Fattori, R. Unusual Rapid Evolution of Type B Aortic Dissection in a Marfan Patient Following Heart Transplantation: Successful Endovascular Treatment. Eur. J. Vasc. Endovasc. Surg. 2006, 32, 358–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knosalla, C.; Weng, Y.-G.; Hammerschmidt, R.; Pasic, M.; Schmitt-Knosalla, I.; Grauhan, O.; Dandel, M.; Lehmkuhl, H.B.; Hetzer, R. Orthotopic Heart Transplantation in Patients with Marfan Syndrome. Ann. Thorac. Surg. 2007, 83, 1691–1695. [Google Scholar] [CrossRef]
- Rajagopal, K.; Rogers, J.G.; Lodge, A.J.; Gaca, J.G.; McCann, R.L.; Milano, C.A.; Hughes, G.C. Two-Stage Total Cardioaortic Replacement for End-Stage Heart and Aortic Disease in Marfan Syndrome: Case Report and Review of the Literature. J. Heart Lung Transplant. 2009, 28, 958–963. [Google Scholar] [CrossRef]
- Hetzer, R.; Siegel, G.; Walter, E.M.D. Cardiomyopathy in Marfan syndrome. Eur. J. Cardio-Thorac. Surg. 2015, 49, 561–568. [Google Scholar] [CrossRef] [Green Version]
- Audenaert, T.; De Pauw, M.; François, K.; De Backer, J. Type B aortic dissection triggered by heart transplantation in a patient with Marfan syndrome. BMJ Case Rep. 2015, 2015. [Google Scholar] [CrossRef] [Green Version]
- Porciani, M.C.; Attanasio, M.; Lepri, V.; Lapini, I.; Demarchi, G.; Padeletti, L.; Pepe, G.; Abbate, R.; Gensini, G.F. Prevalence of cardiovascular manifestations in Marfan syndrome. Ital. Heart J. Suppl. Off. J. Ital. Fed. Cardiol. 2004, 5. [Google Scholar]
- Rybczynski, M.; Mir, T.S.; Sheikhzadeh, S.; Bernhardt, A.M.; Schad, C.; Treede, H.; Veldhoen, S.; Groene, E.F.; Kühne, K.; Koschyk, D.; et al. Frequency and Age-Related Course of Mitral Valve Dysfunction in the Marfan Syndrome. Am. J. Cardiol. 2010, 106, 1048–1053. [Google Scholar] [CrossRef]
- Gott, V.L.; Greene, P.S.; Alejo, D.E.; Cameron, D.E.; Naftel, D.C.; Miller, D.C.; Gillinov, A.M.; Laschinger, J.C.; Borst, H.G.; Cabrol, C.E.; et al. Replacement of the Aortic Root in Patients with Marfan’s Syndrome. N. Engl. J. Med. 1999, 340, 1307–1313. [Google Scholar] [CrossRef] [PubMed]
- Gott, V.L.; Cameron, D.E.; Alejo, D.E.; Greene, P.S.; Shake, J.G.; Caparrelli, D.J.; Dietz, H.C. Aortic root replacement in 271 Marfan patients: A 24-year experience11This manuscript was adapted in part from the William, W.L. Glenn Lecture presented by Dr Gott at the American Heart Association Meeting, New Orleans, LA, Nov 13, 2000. Ann. Thorac. Surg. 2002, 73, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Cameron, D.E.; Alejo, D.E.; Patel, N.D.; Nwakanma, L.U.; Weiss, E.S.; Vricella, L.A.; Dietz, H.C.; Spevak, P.J.; Williams, J.A.; Bethea, B.T.; et al. Aortic Root Replacement in 372 Marfan Patients: Evolution of Operative Repair Over 30 Years. Ann. Thorac. Surg. 2009, 87, 1344–1350. [Google Scholar] [CrossRef] [PubMed]
- Ikonomidis, I.; Aboyans, V.; Blacher, J.; Brodmann, M.; Brutsaert, D.L.; Chirinos, J.A.; De Carlo, M.; Delgado, V.; Lancellotti, P.; Lekakis, J.; et al. The role of ventricular–arterial coupling in cardiac disease and heart failure: Assessment, clinical implications and therapeutic interventions. A consensus document of the European Society of Cardiology Working Group on Aorta & Peripheral Vascular Diseases, European Association of Cardiovascular Imaging, and Heart Failure Association. Eur. J. Heart Fail. 2019, 21, 402–424. [Google Scholar] [CrossRef] [Green Version]
- Vitarelli, A.; Conde, Y.; Cimino, E.; D’Angeli, I.; D’Orazio, S.; Stellato, S.; Padella, V.; Caranci, F. Aortic Wall Mechanics in the Marfan Syndrome Assessed by Transesophageal Tissue Doppler Echocardiography. Am. J. Cardiol. 2006, 97, 571–577. [Google Scholar] [CrossRef]
- Adams, J.N.; Brooks, M.; Redpath, T.W.; Smith, F.W.; Dean, J.C.; Gray, J.; Walton, S.; Trent, R.J. Aortic distensibility and stiffness index measured by magnetic resonance imaging in patients with Marfan’s syndrome. Heart 1995, 73, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Yin, F.C.; Brin, K.P.; Ting, C.T.; Pyeritz, R.E. Arterial hemodynamic indexes in Marfan’s syndrome. Circulation 1989, 79, 854–862. [Google Scholar] [CrossRef] [Green Version]
- Kiotsekoglou, A.; Saha, S.K.; Moggridge, J.C.; Kapetanakis, V.; Bijnens, B.; Mullen, M.J.; Camm, J.; Sutherland, G.R.; Wilkinson, I.B.; Child, A.H. Effect of aortic stiffness on left ventricular long-axis systolic function in adults with Marfan syndrome. Hell. J. Cardiol. HJC = Hell. Kardiol. Ep. 2010, 51. [Google Scholar]
- Nollen, G.J.; Meijboom, L.J.; Groenink, M.; Timmermans, J.; Barentsz, J.O.; Merchant, N.; Webb, G.D.; Lamb, H.J.; Tijssen, J.G.; Van Der Wall, E.E.; et al. Comparison of aortic elasticity in patients with the marfan syndrome with and without aortic root replacement. Am. J. Cardiol. 2003, 91, 637–640. [Google Scholar] [CrossRef]
- Mortensen, K.; Aydin, M.; Bernhardt, A.M.J.; Appenzeller, V.; Robinson, P.N.; Berger, J.; Reichenspurner, H.; Von Kodolitsch, Y.; Kai, M.; Muhammet, A.; et al. Arterial mechanical properties after replacement or reconstruction of the aortic root. World J. Cardiovasc. Dis. 2012, 2, 8–13. [Google Scholar] [CrossRef] [Green Version]
- Franken, R.; Groenink, M.; De Waard, V.; Feenstra, H.M.; Scholte, A.J.; Berg, M.P.V.D.; Pals, G.; Zwinderman, A.H.; Timmermans, J.; Mulder, B.J.M. Genotype impacts survival in Marfan syndrome. Eur. Heart J. 2016, 37, 3285–3290. [Google Scholar] [CrossRef] [PubMed]
- Becerra-Muñoz, V.M.; Gomez-Doblas, J.; Porras, C.; Such, M.; Crespo-Leiro, M.G.; Villa, R.B.; De Teresa-Galván, E.; Jimenez-Navarro, M.; Cabrera-Bueno, F. The importance of genotype-phenotype correlation in the clinical management of Marfan syndrome. Orphanet J. Rare Dis. 2018, 13, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, N.; Inuzuka, R.; Maemura, S.; Morita, H.; Nawata, K.; Fujita, D.; Taniguchi, Y.; Yamauchi, H.; Yagi, H.; Kato, M.; et al. Impact of Pathogenic FBN1 Variant Types on the Progression of Aortic Disease in Patients with Marfan Syndrome. Circ. Genom. Precis. Med. 2018, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baudhuin, L.M.; Kotzer, K.E.; Lagerstedt, S.A. Increased frequency of FBN1 truncating and splicing variants in Marfan syndrome patients with aortic events. Genet. Med. 2014, 17, 177–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Backer, J.; Campens, L.; Mosquera, L.M. Looking for the Missing Links. Circ. Genom. Precis. Med. 2018, 11. [Google Scholar] [CrossRef]
- Rurali, E.; Perrucci, G.L.; Pilato, C.A.; Pini, A.; Gaetano, R.; Nigro, P.; Pompilio, G. Precise Therapy for Thoracic Aortic Aneurysm in Marfan Syndrome: A Puzzle Nearing Its Solution. Prog. Cardiovasc. Dis. 2018, 61, 328–335. [Google Scholar] [CrossRef]
- Aalberts, J.J.J.; Van Tintelen, J.P.; Meijboom, L.J.; Polko, A.; Jongbloed, J.D.; Van Der Wal, H.; Pals, G.; Osinga, J.; Timmermans, J.; De Backer, J.; et al. Relation between genotype and left-ventricular dilatation in patients with Marfan syndrome. Gene 2014, 534, 40–43. [Google Scholar] [CrossRef]
- Cook, J.R.; Carta, L.; Benard, L.; Chemaly, E.R.; Chiu, E.; Rao, S.K.; Hampton, T.G.; Yurchenco, P.D.; Consortium, G.R.; Costa, K.D.; et al. Abnormal muscle mechanosignaling triggers cardiomyopathy in mice with Marfan syndrome. J. Clin. Investig. 2014, 124, 1329–1339. [Google Scholar] [CrossRef] [Green Version]
- Tae, H.-J.; Petrashevskaya, N.N.; Marshall, S.; Krawczyk, M.; Talan, M. Cardiac remodeling in the mouse model of Marfan syndrome develops into two distinctive phenotypes. Am. J. Physiol. Circ. Physiol. 2015, 310, H290–H299. [Google Scholar] [CrossRef] [Green Version]
- Rouf, R.; Macfarlane, E.G.; Takimoto, E.; Chaudhary, R.; Nagpal, V.; Rainer, P.P.; Bindman, J.G.; Gerber, E.E.; Bedja, D.; Schiefer, C.; et al. Nonmyocyte ERK1/2 signaling contributes to load-induced cardiomyopathy in Marfan mice. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- Vracko, R.; Thorning, D.; Frederickson, R.G. Spatial arrangements of microfibrils in myocardial scars: Application of antibody to fibrillin. J. Mol. Cell. Cardiol. 1990, 22, 749–757. [Google Scholar] [CrossRef]
- Bouzeghrane, F.; Reinhardt, D.P.; Reudelhuber, T.L.; Thibault, G. Enhanced expression of fibrillin-1, a constituent of the myocardial extracellular matrix in fibrosis. Am. J. Physiol. Circ. Physiol. 2005, 289, H982–H991. [Google Scholar] [CrossRef] [PubMed]
- Lockhart, M.; Wirrig, E.; Phelps, A.; Wessels, A. Extracellular matrix and heart development. Birth Defects Res. Part. A Clin. Mol. Teratol. 2011, 91, 535–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steijns, F.; Van Hengel, J.; Sips, P.Y.; De Backer, J.; Renard, M. A heart for fibrillin: Spatial arrangement in adult wild-type murine myocardial tissue. Histochem. Cell Boil. 2018, 150, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Parker, K.K.; Ingber, D.E. Extracellular matrix, mechanotransduction and structural hierarchies in heart tissue engineering. Philos. Trans. R. Soc. B Boil. Sci. 2007, 362, 1267–1279. [Google Scholar] [CrossRef] [Green Version]
- Aydin, A.; Adsay, B.A.; Sheikhzadeh, S.; Keyser, B.; Rybczynski, M.; Sondermann, C.; Detter, C.; Steven, D.; Robinson, P.N.; Berger, J.; et al. Observational Cohort Study of Ventricular Arrhythmia in Adults with Marfan Syndrome Caused by FBN1 Mutations. PLoS ONE 2013, 8, e81281. [Google Scholar] [CrossRef]
- Mosquera, L.M.; De Wilde, H.; Devos, D.; Babin, D.; Jordaens, L.; Demolder, A.; De Groote, K.; De Wolf, D.; De Backer, J. Myocardial Disease and Ventricullar Arrhythmia in Marfan Syndrome—A Prospective Study. Orphanet J. Rare Dis. 2020. [Google Scholar] [CrossRef]
- Mah, D.Y.; Sleeper, L.A.; Crosson, J.E.; Czosek, R.J.; Love, B.A.; McCrindle, B.W.; Muiño-Mosquera, L.; Olson, A.K.; Pilcher, T.A.; Tierney, E.S.S.; et al. Frequency of Ventricular Arrhythmias and Other Rhythm Abnormalities in Children and Young Adults With the Marfan Syndrome. Am. J. Cardiol. 2018, 122, 1429–1436. [Google Scholar] [CrossRef]
- Savolainen, A.; Kupari, M.; Toivonen, L.; Kaitila, I.; Viitasalo, M. Abnormal ambulatory electrocardiographic findings in patients with the Marfan syndrome. J. Intern. Med. 1997, 241, 225–230. [Google Scholar] [CrossRef]
- Chen, S.-C.; Fagan, L.F.; Nouri, S.; Donahoe, J.L. Ventricular Dysrhythmias in Children With Marfan’s Syndrome. Arch. Pediatr. Adolesc. Med. 1985, 139, 273–276. [Google Scholar] [CrossRef]
- Hoffmann, B.A.; Rybczynski, M.; Rostock, T.; Servatius, H.; Drewitz, I.; Steven, D.; Aydin, A.; Sheikhzadeh, S.; Darko, V.; Von Kodolitsch, Y.; et al. Prospective risk stratification of sudden cardiac death in Marfan’s syndrome. Int. J. Cardiol. 2013, 167, 2539–2545. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, B.; Rybczynski, M.; Sheikhzadeh, S.; Akbulak, R.Ö.; Moser, J.; Jularic, M.; Schreiber, D.; Daubmann, A.; Willems, S.; Von Kodolitsch, Y.; et al. Heart rate turbulence and deceleration capacity for risk prediction of serious arrhythmic events in Marfan syndrome. Clin. Res. Cardiol. 2015, 104, 1054–1063. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Blomstrom-Lundqvist, C.; Mazzanti, A.; Bloma, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death the Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the Europea. Eur. Heart J. 2015, 36, 2793–2867. [Google Scholar]
- Arunamata, A.; Nguyen, C.T.; Ceresnak, S.R.; Dubin, A.M.; Olson, I.L.; Murphy, D.J.; Tierney, E.S.S. Utility of serial 12-lead electrocardiograms in children with Marfan syndrome. Cardiol. Young 2018, 28, 1009–1013. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, J.L.; Walker, B.A.; Halpern, B.L.; Kuzma, J.W.; McKusick, V.A. Life Expectancy and Causes of Death in the Marfan Syndrome. N. Engl. J. Med. 1972, 286, 804–808. [Google Scholar] [CrossRef] [PubMed]
- Marsalese, D.L.; Moodie, D.S.; Vacante, M.; Lytle, B.W.; Gill, C.C.; Sterba, R.; Cosgrove, D.M.; Passalacqua, M.; Goormastic, M.; Kovacs, A. Marfan’s syndrome: Natural history and long-term follow-up of cardiovascular involvement. J. Am. Coll. Cardiol. 1989, 14, 422–428. [Google Scholar] [CrossRef] [Green Version]
- Chiu, H.-H.; Wu, M.-H.; Chen, H.-C.; Kao, F.-Y.; Huang, S.-K. Epidemiological Profile of Marfan Syndrome in a General Population: A National Database Study. Mayo Clin. Proc. 2014, 89, 34–42. [Google Scholar] [CrossRef]
- Vanem, T.T.; Geiran, O.R.; Krohg-Sørensen, K.; Røe, C.; Paus, B.; Rand-Hendriksen, S. Survival, causes of death, and cardiovascular events in patients with Marfan syndrome. Mol. Genet. Genom. Med. 2018, 6, 1114–1123. [Google Scholar] [CrossRef] [Green Version]
- Groth, K.A.; Stochholm, K.; Hove, H.; Andersen, N.H.; Gravholt, C. Causes of Mortality in the Marfan Syndrome(from a Nationwide Register Study). Am. J. Cardiol. 2018, 122, 1231–1235. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| In the Absence of Family History of MFS: |
| (1) Ao * (Z-score ≥ 2) AND EL = MFS |
| (2) Ao * (Z-score ≥ 2) AND causal FBN1 mutation = MFS |
| (3) Ao * (Z-score ≥ 2) AND systemic score ≥ 7 points = MFS |
| (4) EL AND causal FBN1 mutation with known Ao = MFS |
| In the Presence of Family History of MFS: |
| (5) EL AND family history of MFS = MFS |
| (6) Systemic score ≥ 7 points AND family history of MFS = MFS |
| (7) Ao * (Z-score ≥ 2 above 20 years old, ≥ 3 below 20 years) + family history of MFS = MFS |
| Author | Type of Study | Number of Patients with MFS | Controls | Assessment | Findings in MFS |
|---|---|---|---|---|---|
| Roman et al. 1989 [19] | Case-control | 59 children and adults (51% female) | 59 age- and sex-matched controls 59 age- and sex-matched subjects with MVP | M-mode 2D echo | Similar LV diameter and systolic function Increased LV mass |
| Savolainen et al. 1994 [20] | Case-control | 22 children (64% female) | 22 age-matched healthy children | M-mode Doppler CMR | Similar LV diameter and systolic function LV diastolic dysfunction |
| Porciani et al. 2002 [21] | Case-control | 20 adult MFS and 8 MASS phenotype (54% female) | 28 healthy, age and gender-matched controls | M-mode 2D echo Doppler | Similar LV diameter and systolic function LV diastolic dysfunction |
| Yetman et al. 2003 [14] | Follow-up: 6 years ^ (1.6–24.5) | 70 children and adults (51% female) | / | 2D echo | 68% had LV dilatation (LVEDD Z-score > 2) 11% had LV systolic dysfunction (FS < 30%) |
| Chatrath et al. 2003 [22] | Follow-up: 10.8 years * (1–29) | 36 children and adults (36% female) | / | M-mode | 19% had LV dilatation (LVEDD > 95% above normal limits) No change in LV dimensions during follow-up No LV systolic dysfunction |
| Meijboom et al. 2005 [23] | Follow-up: 6 years * (0.3–15) | 234 adults (51% female) | / | M-mode 2D echo | 9% had mild LV dysfunction (FS 25–30%) 7% had LV dilatation (LVEDD > 117% (2SD + 5%)) 3% developed LV dilatation during follow-up 1% had LVEDD > 112% and FS < 30% |
| De Backer et al. 2006 [24] | Case-control | 26 adults (54% female) | 26 age and sex-matched controls | 2D echo Doppler TDI CMR | LV systolic and diastolic dysfunction LV dilatation |
| Das et al. 2006 [25] | Case-control | 40 children and adults (68% female) | 40 age and sex-matched controls | M-mode Doppler | Similar systolic function LV diastolic dysfunction LV dilatation |
| Rybczynski et al. 2007 [26] | Case-control | 55 adults (49% female) | 86 healthy controls | 2D echo Doppler TDI | LV diastolic dysfunction LV systolic dysfunction |
| Kiotsekoglou et al. 2008 [27] | Case-control | 66 adults (44% female) | 61 healthy controls | M-mode 2D echo Doppler TDI | 17% had LV dilatation (predicted LVEDD ≥ 112% and FS ≥ 25%) LV diastolic dysfunction LV systolic dysfunction |
| Kiotsekoglou et al. 2009 [34] | Case-control | 66 adults (44% female) | 61 age, sex, height, weight, and BSA-matched normal volunteers | M-mode 2D echo Doppler TDI | RV systolic dysfunction RV dilatation Increased right atrium area |
| Kiotsekoglou et al. 2009 [35] | Case-control | 72 adults (42% female) | 73 age, sex, and BSA-matched controls | 2D echo Doppler TDI | LV diastolic dysfunction RV diastolic dysfunction Atrial systolic and diastolic dysfunction |
| Alpendurada et al. 2010 [28] | Cross-sectional | 68 adults (40% female) | / | CMR | 25% had reduced LV EF (below 95% CI for sex and age decile) 10% had reduced RV EF (below 95% CI for sex and age decile) LV dilatation RV dilatation |
| Kiotsekoglou et al. 2011 [31] | Case-control | 44 adults (41% female) | 49 controls without significant differences in age, sex, height, weight, and BSA | M-mode 2D echo Doppler Strain rate imaging | 20% had LV dilatation (predicted LVEDD ≥ 112% and FS ≥ 25%) LV diastolic dysfunction LV systolic dysfunction |
| de Witte et al. 2011 [29] | Case-control | 144 adults (51% female) | 19 healthy controls | CMR | 9% had reduced LV EF (<45%) LV systolic dysfunction RV systolic dysfunction |
| Scherptong et al. 2011 [36] | Case-control Follow-up: 4 years ^ | 50 adults (50% female) | 50 controls matched for age, sex, and BSA | M-mode 2D echo Doppler Strain rate imaging | Similar LV and RV EF LV systolic dysfunction RV systolic dysfunction No changes in systolic or diastolic function during follow-up |
| Angtuaco et al. 2012 [32] | Case-control | 16 children and adults (56% female) | 26 controls without significant differences in sex, race, age, weight, height, and BSA | M-mode 2D echo Doppler Strain rate imaging | LV systolic dysfunction No significant differences in strain |
| Abd El Rahman et al. 2015 [33] | Case-control | 45 children and adults (42% female) | 40 age-matched healthy controls | M-mode 2D and 3D echo Doppler 3D speckle tracking | LV diastolic dysfunction LV systolic dysfunction Left atrial diastolic dysfunction No differences in left atrial systolic function No differences in M-mode LVEDD, LVESD and FS |
| Campens et al. 2015 [37] | Case-control Follow-up: 6 years * (3.4–10.3) | 19 adults (47% female) | 19 age and sex-matched controls | 2D echo Doppler TDI | No changes in LV dimensions during follow-up No changes in LV systolic or diastolic function during follow-up |
| Gehle et al. 2016 [38] | Case-control | 217 children and adults (51% female) | 339 patients referred for suspected MFS (diagnosis ruled out according to the Gh. nosology) | M-mode 2D echo Doppler TDI NT-proBNP | Increased NT-proBNP levels LV diastolic dysfunction LV dilatation No signs of LV systolic dysfunction |
| Loeper et al. 2016 [39] | Case-control | 104 adults with MFS (45% female) and 111 adults with ns-TAAD (35% female) | 148 healthy controls | 2D echo Doppler TDI | Increased aortic stiffness index in MFS and ns-TAAD Reduced LV end-systolic elastance in MFS ventricular-vascular coupling index was abnormal in MFS No difference in LV stroke work in MFS |
| Winther et al. 2019 [30] | Case-control | 69 adults (44% female) | 20 age-matched controls | 2D echo CMR | 22% had reduced LV EF (≤55%) LV systolic dysfunction |
| Author | Type of Study | Number of Patients with MFS | Prior Aortic Surgery |
|---|---|---|---|
| Kesler et al. 1994 [42] | Survey | 11 | Not stated |
| Mullen et al. 1996 [43] | Case report | 1 | Yes |
| Varghese et al. 2006 [44] | Case report | 1 | Yes |
| Botta et al. 2006 [45] | Case report | 1 | Yes |
| Knosalla et al. 2007 [46] | Case series | 10 | Yes (100%) |
| Rajagopal et al. 2009 [47] | Case report | 1 | Yes |
| Audenaert et al. 2015 [49] | Case report | 1 | Yes |
| Rao et al. 2018 [40] | Case report | 1 | Yes |
| Ogawa et al. 2019 [41] | Case report | 1 | No |
| Author | Type of Study | Number of Patients with MFS | Controls | Assessment | Findings in MFS | |
|---|---|---|---|---|---|---|
| Chen et al. 1985 [81] | Follow-up: 5.7 years * (1–22) | 24 children (63% female) | / | M-mode Resting ECG | 33% presents at least 1 PVC on resting ECG 13% had VT QTc / QTUc prolongation was associated with ventricular arrhythmias Combination of abnormal repolarization and MVP associated with ventricular arrhythmias | |
| Savolainen et al. 1997 [80] | Case-control | 45 adults (44% female) | 45 healthy age and sex-matched controls | M-mode and 2D echo Ambulatory ECG | Higher median number of PACs than controls (12/24 h vs. 6/24 h; p < 0.05) Higher median number of PVCs than controls (17/24 h vs. 1/24 h; p < 0.001) More frequently repolarization abnormalities than controls Longer PQ- and QTc-intervals compared to controls 11% had NSVT° | |
| Yetman et al. 2003 [14] | Follow-up: 6 years ^ (1.6–24.5) | 70 children and adults (51% female) | / | 2D echo Resting ECG Ambulatory ECG | 21% had ventricular ectopy (defined as >10 PVC/h) 6% had NSVT° 4% died from arrhythmias 16% had QTc prolongation and 60% had QTU prolongation Ventricular ectopy associated with LV size, MVP, and abnormalities of repolarization | |
| Hoffmann et al. 2012 [82] | Follow-up: 2.4 years ^ (2.1–2.7) | 77 adults (52% female) | / | 2D echo, Doppler and TDI Resting ECG and SAECG Ambulatory ECG | 9% reached the composite endpoint (SCD, VT, VF or AS) 7% had VT 3% had SCD | |
| Aydin et al. 2013 [82] | Follow-up: 2.6 years * | 80 children and adults (63% female) | / | M-mode and 2D echo Doppler Resting ECG Ambulatory ECG | 91% had PVCs with 35% having >10 PVC/h 11% had NSVT° 8% had ventricular tachycardia events (SCD, VT, VF or AS) 4% had SCD Ventricular tachycardia events associated with NTproBNP and mutations in exons 24–32 | |
| Schaeffer et al. 2015 [83] | Follow-up: 3.1 years * | 102 adults (56% female) | / | 2D echo Ambulatory ECG Heart rate turbulence | 12% reached the primary endpoint (SCD, survived cardiac arrest, VT/VF and AS) 9% had VT 3% had SCD | |
| Arunamata et al. 2018 [85] | Case-control | 45 children (44% female) | 37 age, BSA, sex-matched controls | M-mode and 2D echo Resting ECG | Longer QTc intervals than controls | |
| Mah et al. 2018 [79] | Cross-sectional | 274 children and adults (38% female) | / | M-mode and 2D echo Ambulatory ECG | 7% had ventricular ectopy (defined as >10 PVC/h) 5% had supraventricular ectopy (defined as >10 PAC/h) 1% had both supraventricular and ventricular ectopy None had VT or supraventricular tachycardia | |
| Muiño Mosquera et al. 2020 [78] | Follow-up Case-control | 86 children and adults (56% female) | 40 age- and sex-matched controls | 2D echo Resting ECG Ambulatory ECG NT-proBNP | Higher median number of PACs than controls (11/24 h vs. 2/24 h; p < 0.001) Higher median number of PVCs than controls (8/24 h vs. 0/24 h; p < 0.001) 23% had NSVT° Larger LVEDD and higher amount of VES were independently associated with NSVT° | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Demolder, A.; von Kodolitsch, Y.; Muiño-Mosquera, L.; De Backer, J. Myocardial Function, Heart Failure and Arrhythmia in Marfan Syndrome: A Systematic Literature Review. Diagnostics 2020, 10, 751. https://doi.org/10.3390/diagnostics10100751
Demolder A, von Kodolitsch Y, Muiño-Mosquera L, De Backer J. Myocardial Function, Heart Failure and Arrhythmia in Marfan Syndrome: A Systematic Literature Review. Diagnostics. 2020; 10(10):751. https://doi.org/10.3390/diagnostics10100751
Chicago/Turabian StyleDemolder, Anthony, Yskert von Kodolitsch, Laura Muiño-Mosquera, and Julie De Backer. 2020. "Myocardial Function, Heart Failure and Arrhythmia in Marfan Syndrome: A Systematic Literature Review" Diagnostics 10, no. 10: 751. https://doi.org/10.3390/diagnostics10100751
APA StyleDemolder, A., von Kodolitsch, Y., Muiño-Mosquera, L., & De Backer, J. (2020). Myocardial Function, Heart Failure and Arrhythmia in Marfan Syndrome: A Systematic Literature Review. Diagnostics, 10(10), 751. https://doi.org/10.3390/diagnostics10100751