Formation of Abasic Oligomers in Nonenzymatic Polymerization of Canonical Nucleotides


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Dehydration-Rehydration Cycles
2.2.2. Analysis of Deglycosylation
2.2.3. HPLC Analysis
2.2.4. Mass Analysis
3. Results and Discussion
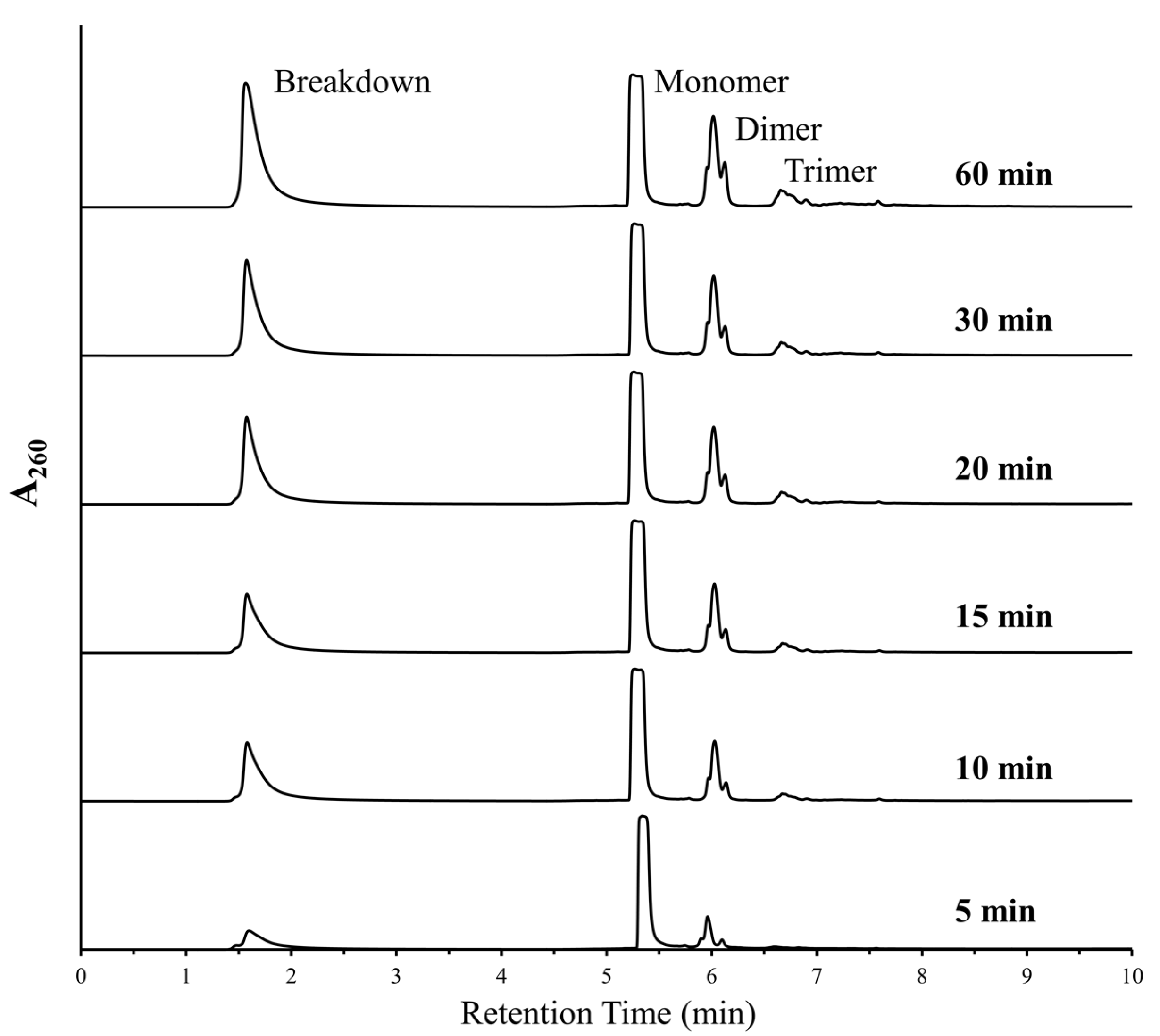
3.1. Polymerization of Canonical Nucleotides
3.2. DH-RH Reactions for Nucleotide Mixtures Capable of Hydrogen Bonding
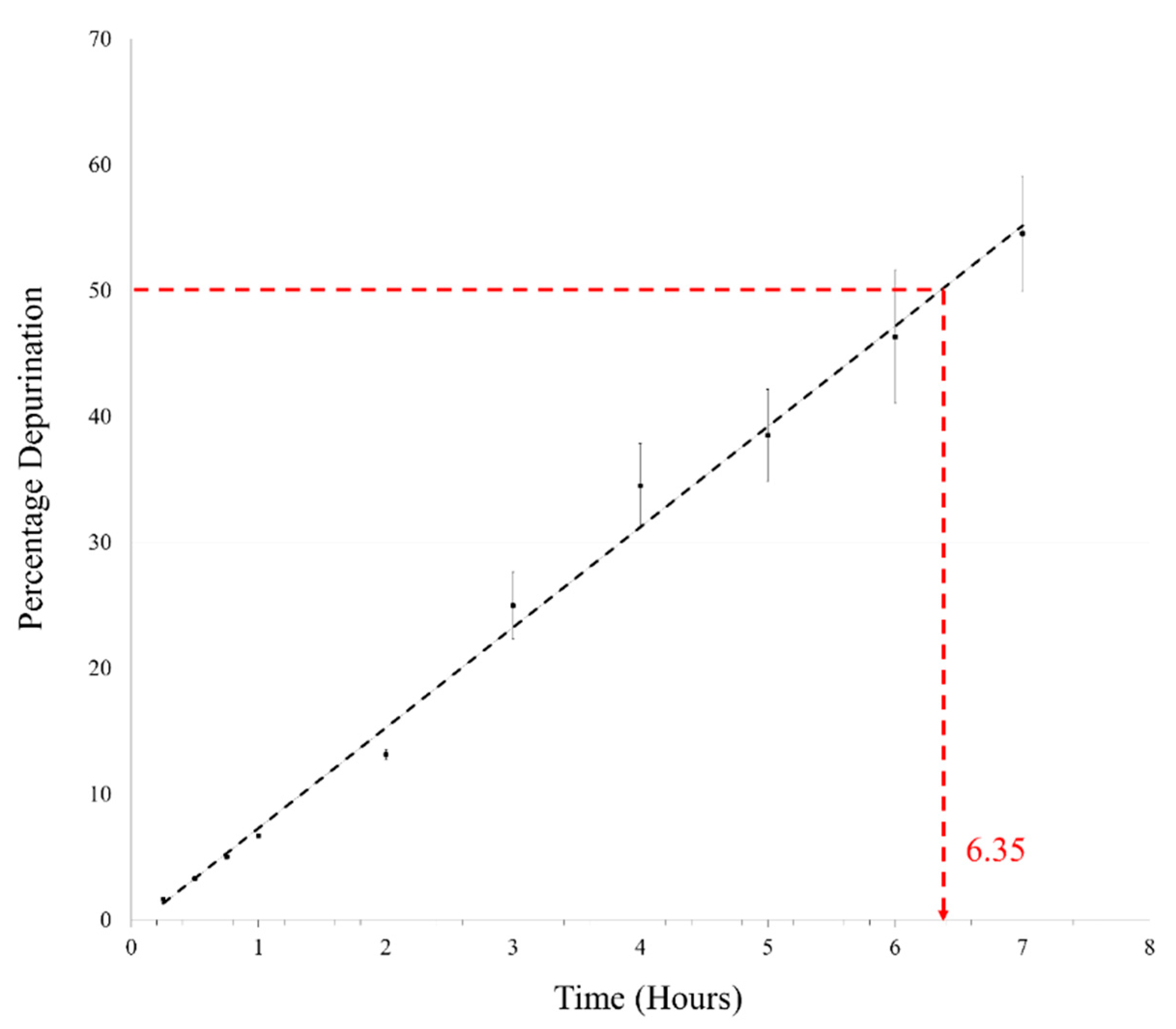
3.3. Deglycosylation Reactions during DH-RH Cycles
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Huang, W.; Ferris, J.P. One-step, regioselective synthesis of up to 50-mers of RNA oligomers by montmorillonite catalysis. J. Am. Chem. Soc. 2006, 128, 8914–8919. [Google Scholar] [CrossRef] [PubMed]
- Deamer, D. Liquid crystalline nanostructures: Organizing matrices for non-enzymatic nucleic acid polymerization. Chem. Soc. Rev. 2012, 41, 5375–5379. [Google Scholar] [CrossRef] [PubMed]
- Rajamani, S.; Vlassov, A.; Benner, S.; Coombs, A.; Olasagasti, F.; Deamer, D. Lipid-assisted synthesis of RNA-like polymers from mononucleotides. Orig. Life Evol. Biosph. 2008, 38, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Van Kranendonk, M.J.; Deamer, D.W.; Djokic, T. Life springs. Sci. Am. 2017, 317, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Djokic, T.; Van Kranendonk, M.J.; Campbell, K.A.; Walter, M.R.; Ward, C.R. Earliest signs of life on land preserved in ca. 3.5 Ga hot spring deposits. Nat. Commun. 2017, 8, 15263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forsythe, J.G.; Yu, S.; Mamajanov, I.; Grover, M.A.; Krishnamurthy, R.; Fernµndez, F.M.; Hud, N.V. Ester-Mediated Amide Bond Formation Driven by Wet–Dry Cycles: A Possible Path to Polypeptides on the Prebiotic Earth. Angew. Chem. Int. Ed. 2015, 54, 9871–9875. [Google Scholar] [CrossRef] [PubMed]
- Mungi, C.V.; Rajamani, S. Characterization of RNA-Like Oligomers from Lipid-Assisted Nonenzymatic Synthesis: Implications for Origin of Informational Molecules on Early Earth. Life 2015, 5, 65–84. [Google Scholar] [CrossRef] [Green Version]
- Leumann, C.J.; Küpfer, P.A. The chemical stability of abasic RNA compared to abasic DNA. Nucleic Acids Res. 2007, 35, 58–68. [Google Scholar]
- Suzuki, T.; Ohsumi, S.; Makino, K. Mechanistic studies on depurination and apurinic site chain breakage in oligodeoxyribonucleotides. Nucleic Acids Res. 1994, 22, 4997–5003. [Google Scholar] [CrossRef]
- Lindahl, T.; Nyberg, B. Rate of depurination of native deoxyribonucleic acid. Biochemistry 1972, 11, 3610–3618. [Google Scholar] [CrossRef]
- Rios, A.C.; Yua, H.T.; Tor, Y. Hydrolytic fitness of N-glycosyl bonds: Comparing the deglycosylation kinetics of modified, alternative, and native nucleosides. J. Phys. Org. Chem. 2015, 28, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Rios, A.C.; Tor, Y. Refining the Genetic Alphabet: A Late-Period Selection Pressure? Astrobiology 2012, 12, 884–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rios, A.C.; Tor, Y. On the Origin of the Canonical Nucleobases: An Assessment of Selection Pressures across Chemical and Early Biological Evolution. Isr. J. Chem. 2013, 53, 469–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapirof, R.; Melvyn, D.J. Acidic Hydrolysis of Deoxycytidine and Deoxyuridine Derivatives. The General Mechanism of Deoxyribonucleoside Hydrolysis. Biochemistry 1972, 11, 23–29. [Google Scholar] [CrossRef] [PubMed]
- DeGuzman, V.; Vercoutere, W.; Shenasa, H.; Deamer, D. Generation of Oligonucleotides Under Hydrothermal Conditions by Non-enzymatic Polymerization. J. Mol. Evol. 2014, 78, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Ts’o, P.O.P.; Melvin, I.S.; Olson, A.C. Interaction and Association of Bases and Nucleosides in Aqueous Solutions. J. Am. Chem. Soc. 1963, 85, 1289–1296. [Google Scholar] [CrossRef]
- Garrett, E.R.; Seydel, J.K.; Sharpen, A.J. The Acid-Catalyzed Solvolysis of Pyrimidine Nucleosides1. J. Org. Chem. 1966, 31, 2219–2227. [Google Scholar] [CrossRef]
- Gates, K.S. An overview of chemical processes that damage cellular DNA: Spontaneous hydrolysis, alkylation, and reactions with radicals. Chem. Res. Toxicol. 2009, 22, 1747–1760. [Google Scholar] [CrossRef]
- Orgel, L.E. Prebiotic chemistry and the origin of the RNA world. Crit. Rev. Biochem. Mol. Biol. 2004, 39, 99–123. [Google Scholar]
- Hud, N.V.; Cafferty, B.J.; Krishnamurthy, R.; Williams, L.D. The origin of RNA and “my grandfather’s axe”. Chem. Biol. 2013, 20, 466–474. [Google Scholar] [CrossRef]
- Kim, H.; Benner, S.A. Prebiotic Glycosylation of Uracil with Electron-Donating Substituents. Astrobiology 2015, 15, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Mungi, C.V.; Singh, S.K.; Chugh, J.; Rajamani, S. Synthesis of barbituric acid containing nucleotides and their implications for the origin of primitive informational polymers. Phys. Chem. Chem. Phys. 2016, 18, 20144–20152. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, L.; Maurel, M.-C.C.; Deamer, D. Salt-Promoted Synthesis of RNA-like Molecules in Simulated Hydrothermal Conditions. J. Mol. Evol. 2015, 80, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Himbert, S.; Chapman, M.; Deamer, D.W.; Rheinstädter, M.C. Organization of nucleotides in different environments and the formation of pre-polymers. Sci. Rep. 2016, 6, 31285. [Google Scholar] [CrossRef] [PubMed]
- Zagorevskii, D.V.; Aldersley, M.F.; Ferris, J.P. MALDI analysis of oligonucleotides directly from montmorillonite. J. Am. Soc. Mass Spectrom. 2006, 17, 1265–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burcar, B.T.; Cassidy, L.M.; Moriarty, E.M.; Joshi, P.C.; Coari, K.M.; McGown, L.B. Potential Pitfalls in MALDI-TOF MS Analysis of Abiotically Synthesized RNA Oligonucleotides. Orig. Life Evol. Biosph. 2013, 43, 247–261. [Google Scholar] [CrossRef]
- Islam, S.; Powner, M.W. Prebiotic Systems Chemistry: Complexity Overcoming Clutter. Chem 2017, 2, 470–501. [Google Scholar] [CrossRef] [Green Version]
- Cleaves, H.J. Prebiotic chemistry: Geochemical context and reaction screening. Life 2013, 3, 331–345. [Google Scholar] [CrossRef]
- Nicholas, G.; Nathaniel, V.; Kuhan, C.; Caleb, S.; Irena, M. Bulk measurements of messy chemistries are needed for a theory of the origins of life. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2017, 375, 20160347. [Google Scholar]
- Meringer, M.; Cleaves, H.J. Computational exploration of the chemical structure space of possible reverse tricarboxylic acid cycle constituents. Sci. Rep. 2017, 7, 17540. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| Chemical Species | Expected Mass | Observed Mass | ppm Error |
|---|---|---|---|
| AMP Reaction | |||
| Adenine | 136.0617 | 136.0635 | 13.2290 |
| AMP monomer | 348.0703 | 348.0691 | 3.4475 |
| Abasic Dimer | 560.0778 | 560.0764 | 2.4996 |
| Abasic Trimer | 772.0874 | 772.0899 | 3.2379 |
| GMP Reaction | |||
| Guanine | 152.0566 | 152.057 | 2.6305 |
| GMP Monomer | 364.0652 | 364.0627 | 6.8669 |
| Abasic Dimer | 576.0737 | 576.0715 | 3.8189 |
| Abasic Trimer | 788.0824 | 788.0803 | 2.6646 |
| UMP Reaction | |||
| Uracil | 113.0345 | 113.0346 | 0.8846 |
| UMP Monomer | 325.0431 | 325.0435 | 1.2306 |
| Intact Dimer | 631.0684 | 631.0682 | 0.3169 |
| Abasic Dimer | 537.0517 | 537.0533 | 2.9792 |
| CMP Reaction | |||
| Cytosine | 112.0505 | 112.0494 | 9.8170 |
| CMP Monomer | 324.0591 | 324.0596 | 1.5429 |
| Intact Dimer | 629.1004 | 629.1017 | 2.0664 |
| Abasic Dimer | 536.0677 | 536.0677 | 0.0000 |
| Chemical Species | Expected Mass | Observed Mass | ppm Error |
|---|---|---|---|
| AMP + UMP Reaction | |||
| Adenine | 136.0617 | 136.0635 | 13.2290 |
| AMP Monomer | 348.0703 | 348.0691 | 3.4475 |
| Uracil | 113.0345 | 113.0346 | 0.8846 |
| UMP Monomer | 325.0431 | 325.0435 | 1.2306 |
| Abasic A Dimer | 560.0778 | 560.0812 | 6.0705 |
| Abasic U Dimer | 537.0517 | 537.0533 | 2.9792 |
| Abasic A Trimer | 772.0874 | 772.0899 | 3.2379 |
| Abasic U Trimer | 749.0603 | 749.0637 | 4.5390 |
| GMP + CMP Reaction | |||
| Guanine | 152.0566 | 152.057 | 2.6305 |
| GMP Monomer | 364.0652 | 364.0627 | 6.8669 |
| Cytosine | 112.0505 | 112.0494 | 9.817 |
| CMP Monomer | 324.0591 | 324.0596 | 1.5429 |
| Intact CC Dimer | 629.1004 | 629.1017 | 2.0664 |
| Intact CG Dimer | 669.1065 | 669.1042 | 3.4374 |
| Abasic G Dimer | 576.0737 | 576.0715 | 3.8189 |
| Abasic C Dimer | 536.0677 | 536.0677 | 0.0000 |
| Abasic G Trimer | 788.0824 | 788.0803 | 2.6646 |
| Abasic C Trimer | 748.0763 | 748.0787 | 3.2082 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mungi, C.V.; Bapat, N.V.; Hongo, Y.; Rajamani, S. Formation of Abasic Oligomers in Nonenzymatic Polymerization of Canonical Nucleotides. Life 2019, 9, 57. https://doi.org/10.3390/life9030057
Mungi CV, Bapat NV, Hongo Y, Rajamani S. Formation of Abasic Oligomers in Nonenzymatic Polymerization of Canonical Nucleotides. Life. 2019; 9(3):57. https://doi.org/10.3390/life9030057
Chicago/Turabian StyleMungi, Chaitanya V., Niraja V. Bapat, Yayoi Hongo, and Sudha Rajamani. 2019. "Formation of Abasic Oligomers in Nonenzymatic Polymerization of Canonical Nucleotides" Life 9, no. 3: 57. https://doi.org/10.3390/life9030057
APA StyleMungi, C. V., Bapat, N. V., Hongo, Y., & Rajamani, S. (2019). Formation of Abasic Oligomers in Nonenzymatic Polymerization of Canonical Nucleotides. Life, 9(3), 57. https://doi.org/10.3390/life9030057