Evolutionary Steps in the Analytics of Primordial Metabolic Evolution †




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
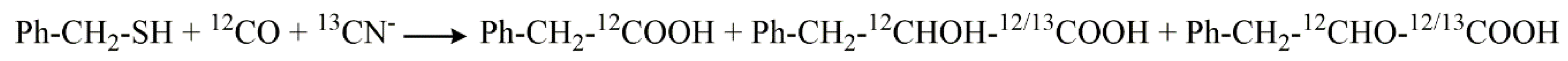{kind=link}
Abstract
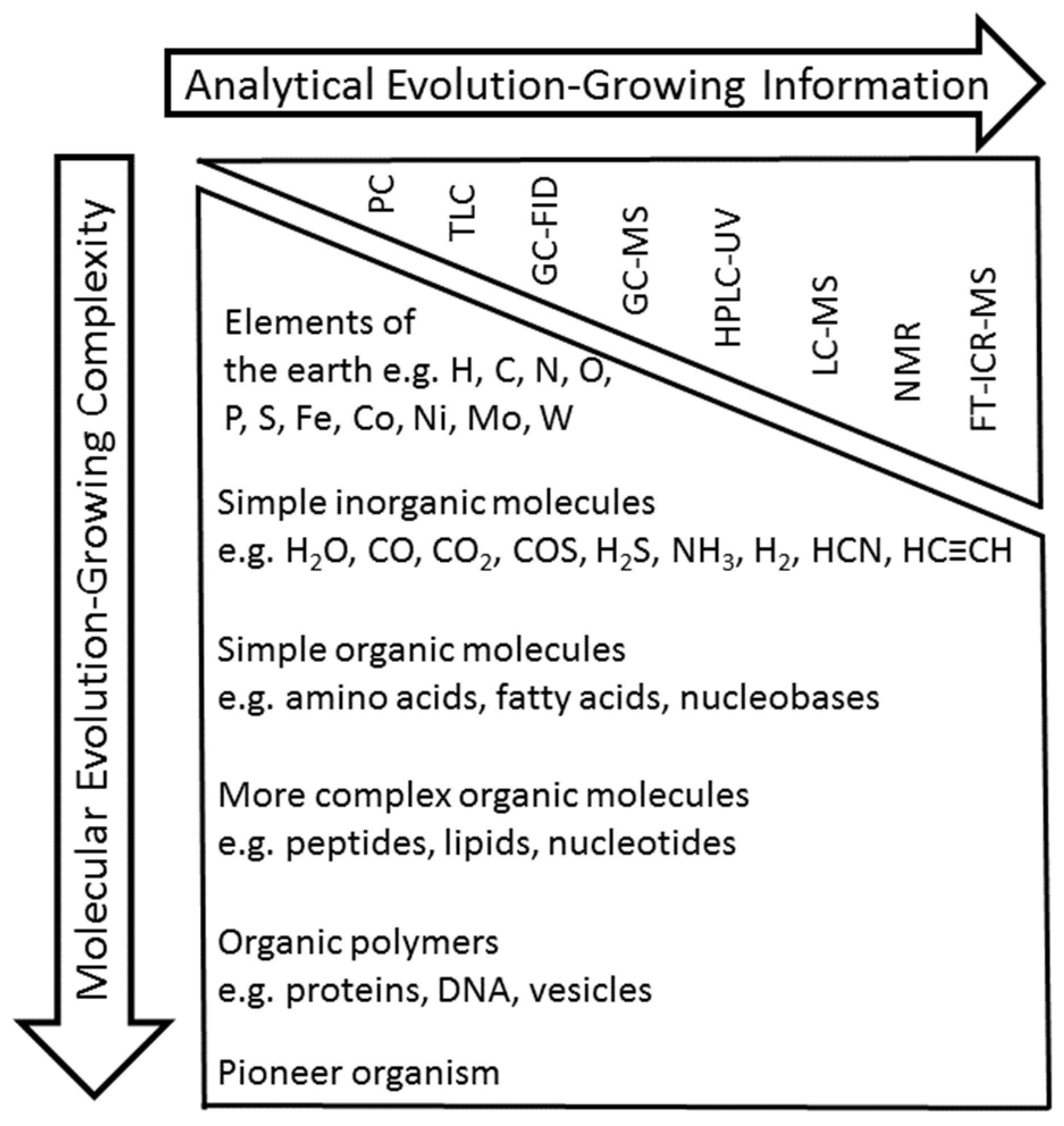
1. Introduction
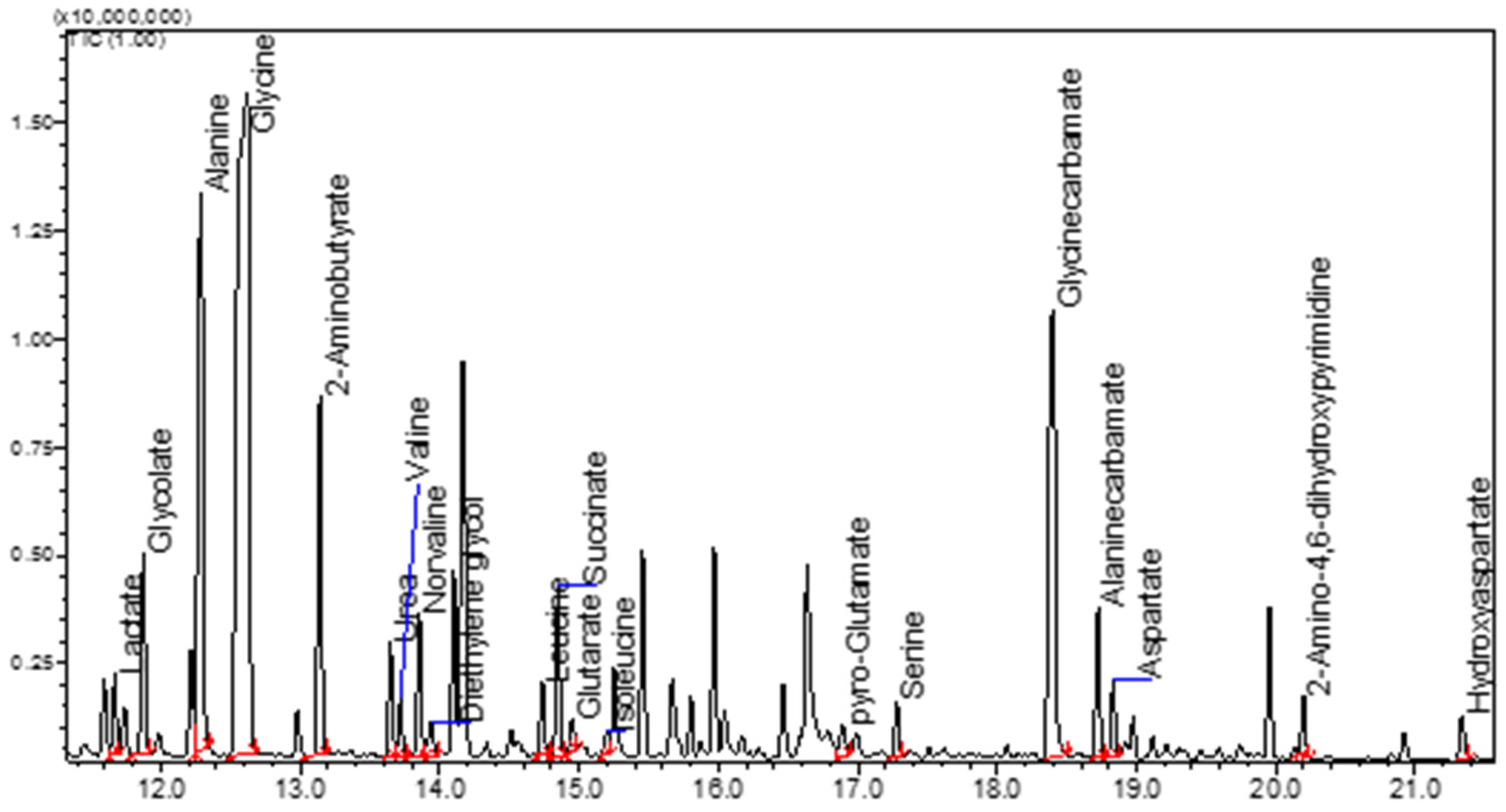
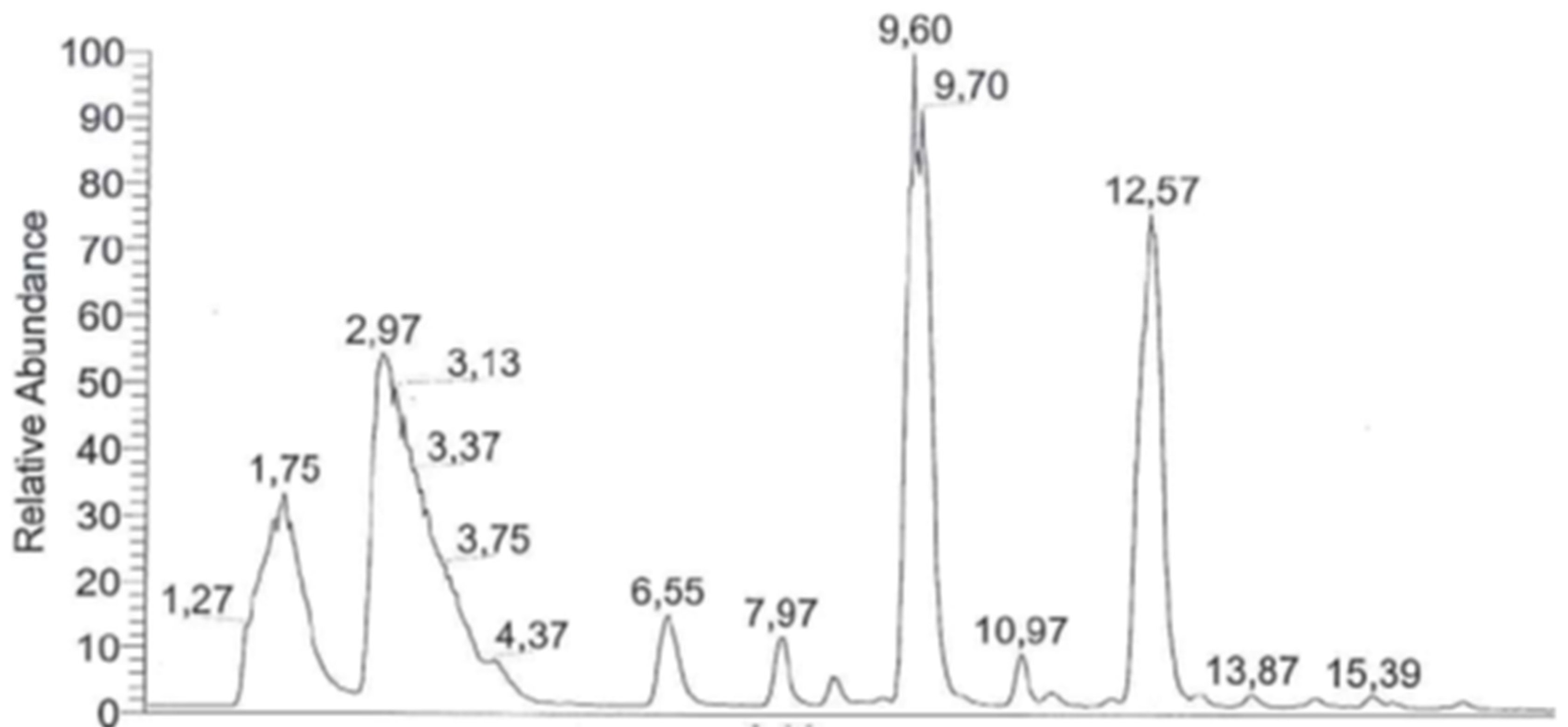
2. GC-FID and GC-MS Analysis as Robust Tools for Detecting Simple Organic Molecules Formed Under Primordial Conditions
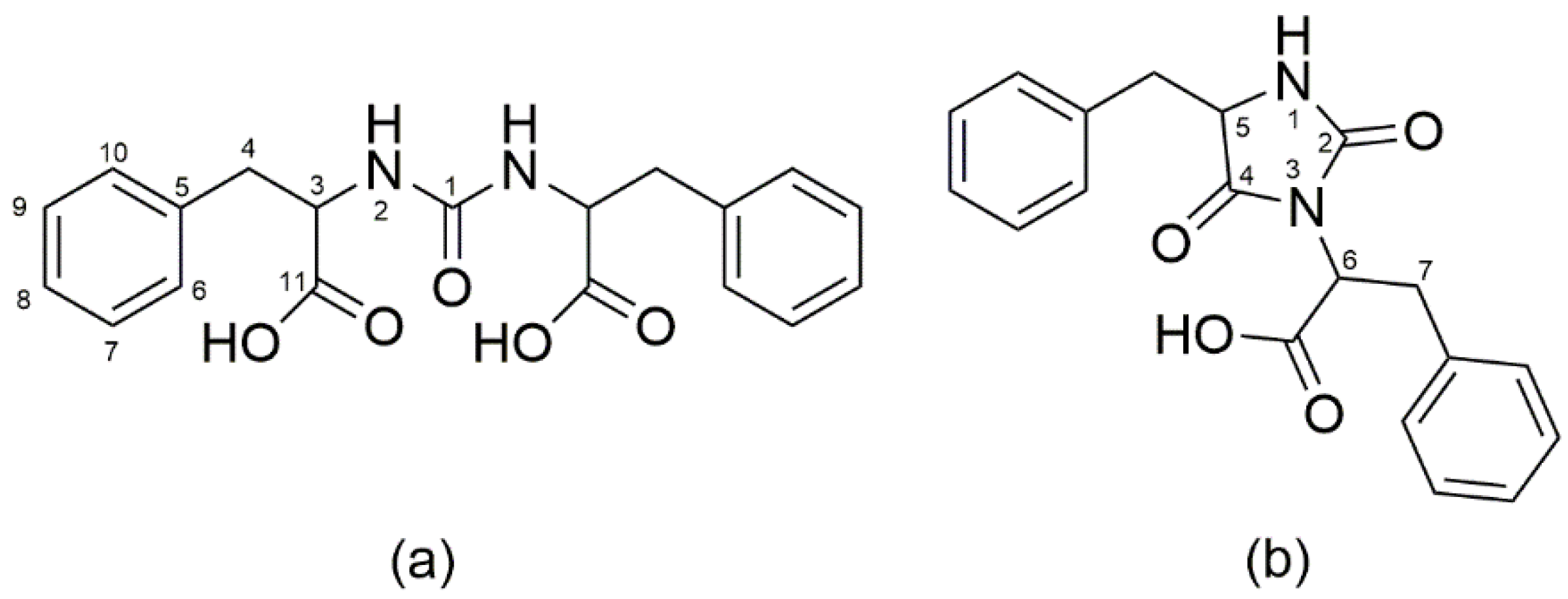
3. HPLC-UV and LC-MS for the Elucidation of the Formation of More Complex Molecules Like Peptides
4. NMR as a Powerful Tool to Determine the Structures of Complex Unknown Intermediates
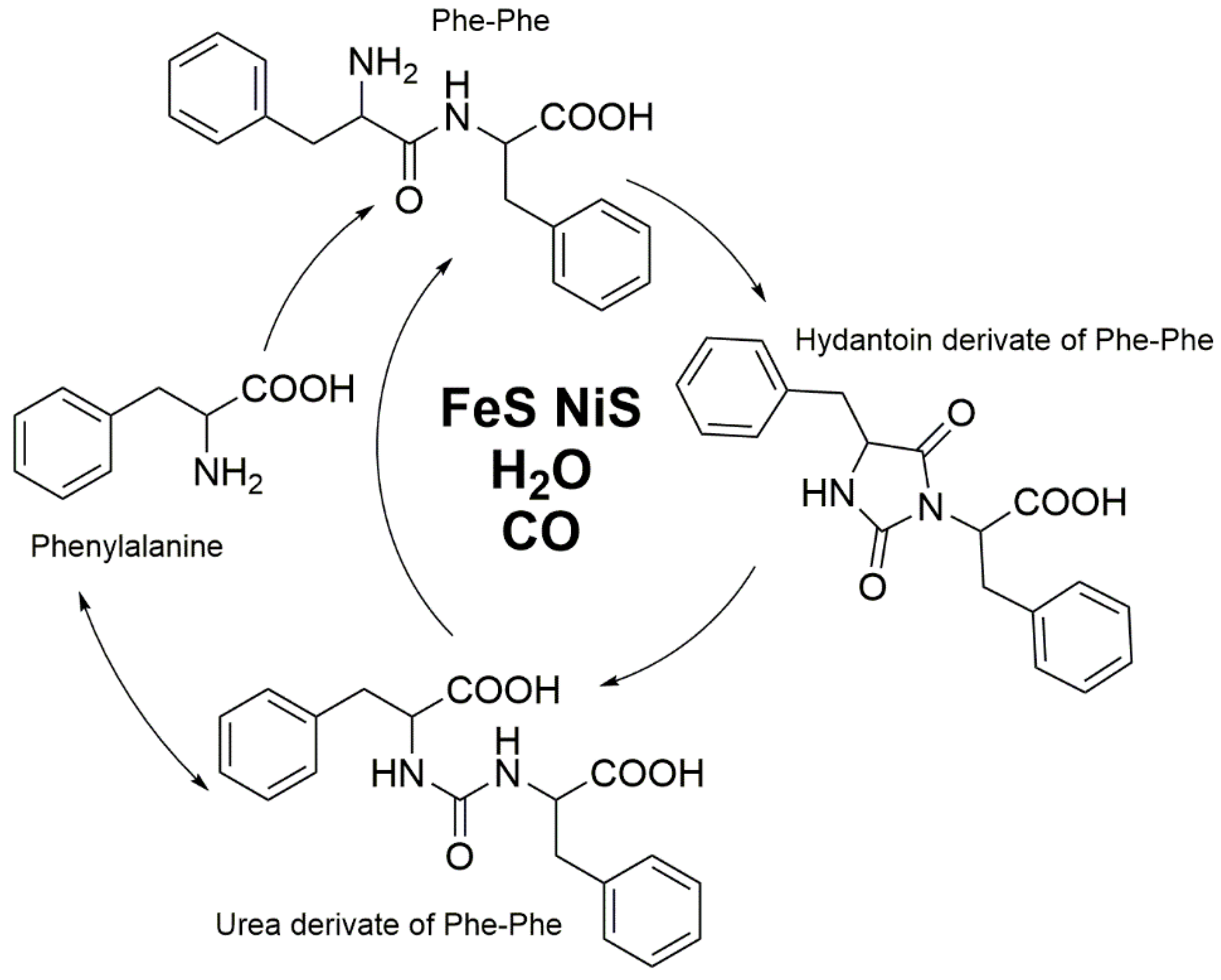
5. Stable Isotope Labeling as Advanced Tools to Verify Products and to Elucidate Mechanisms in Product Formation
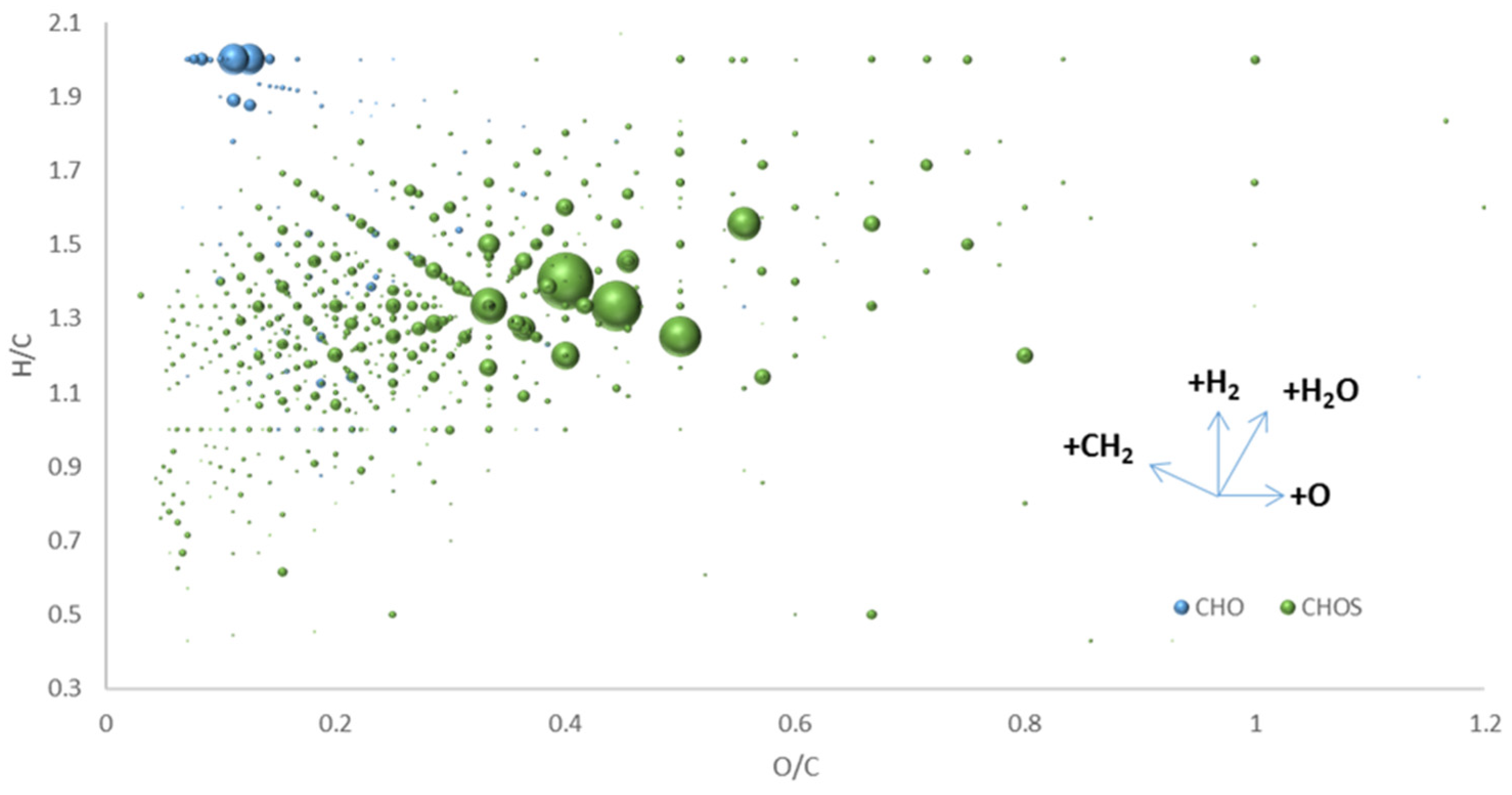
6. Extending the “Chemical Space” in “Origin-of-Life” Research by FT-ICR-MS
7. Discussion
8. Working Protocols
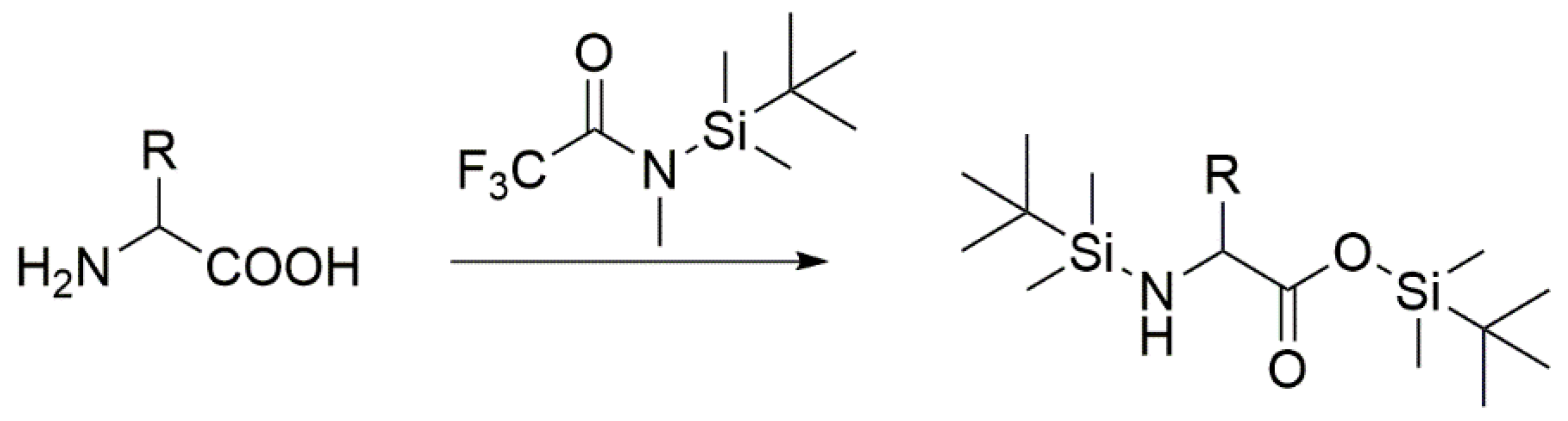
8.1. Protocol for GC-MS Analysis
- Take 1.5 mL reaction mix from reaction vessel; serum flask: syringe/needle; autoclave: pipette
- Centrifuge at 10,000 rpm for 10 min, removing solids from liquid sample
- Fill supernatant in 4 mL vial
- Freeze sample completely at −20 °C
- Freeze dry sample for minimum 24 h to remove all water from sample (derivatization agent and GC/MS column is sensitive to water)
- Dissolve sample in 250 µL Acetonitril (ACN) water free
- Add 250 µL MTBSTFA (N-tert-Butyldimetylsilyl-N-methyltrifluoroacetamide with 1% tert-Butyldimethylchlorosilane)
- Derivatize at 70 °C for 30 min
- Transfer 100 µL to an autosampler vial with 200 µL inlet.
- Measure on GC-MS
- Take 1.5 mL reaction mix from reaction vessel
- Centrifuge at 10,000 rpm for 10 min, removing solids from liquid sample
- Separate supernatant and residue
- Extract supernatant/residue with ethyl acetate or n-hexane
- Dry solvent supernatant with sodium sulfate (Na2SO4)
- Transfer 100 µL to an auto sampler vial with 200 µL inlet
- Measure directly with GC-MS without derivatization
- Method 1: 0.2 µL Injection, Injection Temp. 260 °C, Split 1/10; 60 °C for 3 min, with 10 °C/min to 280 °C hold for 3 min; pressure 72.8 Pa; Equity TM5 30 m Film 25 µm; MS Ion source 200 °C, Interface 260 °C, EI, 40–700 m/z, scan speed 1428, column flow 1.20 mL/min; take care of overloaded signals due to derivatization agent
- Method 2: 0.5 µL Injection, Injection Temp. 260 °C, Split 1/5; 90 °C for 3 min, with 10 °C/min to 310 °C hold for 10 min; pressure 82.8 Pa; Equity TM5 30 m Film 25 µm; MS Ion source 200 °C, Interface 260 °C, EI, 40–950 m/z, scan speed 2000, column flow 1.17 mL/min; take care of overloaded signals due to derivatization agent
8.2. Protocol for LC/MS Analysis
- Take 1.5 mL of reaction mix from reaction vessel
- Centrifuge at 10,000 rpm for 10 min, removing solids from liquid sample
- Take supernatant to 4 mL vial
- Filter 200 µL through 0.45 µm PTFE membrane filter into auto sampler vial with 200 µL inlet
- Measure with HPLC or LC-MS
- HPLC Merck-Hitachi (Düsseldorf, Germany) Pump L-7100, Merck-Hitachi L-7400 UV-detector
- Nucleosil 100-5-C18 5 µm column (Bischoff Chromatography)
- A: H2O, 1‰ H3PO4, B: MeOH, 1 ‰ H3PO4; linear gradient 20–90% B; Flow rate 1 mL/min
- Injection: 10 µL
- LC-MS Hewlett-Packard (Böblingen, Germany) series 1100 (HPLC), LCQ Finnigan Mat (MS)
- Hypersil Gold AQ RP-18 5 µm column
- A: H2O, B: ACN; linear gradient 10–60% B for 24 min; Flow rate 0.7 mL/min
- Ionization: ESI positive mode
- Injection: 1 µL
8.3. Protocol for NMR Analysis
- Take 1.5 mL reaction mix from reaction vessel
- Centrifuge at 10,000 rpm for 10 min, removing solids from liquid sample
- Transfer supernatant to 4 mL vial
- Freeze sample completely at −20 °C
- Freeze dry sample for minimum 24 h to remove extant water from sample
- Dissolve dried residue in 520 µL D2O
- Add 40 µL of TSP (sodium trimethylsilyl propionate) as internal standard to calibrate 0.0 ppm
- Transfer sample to NMR tube
- Take 1.5 mL reaction mix from reaction vessel
- Centrifuge at 10,000 rpm for 10 min, removing solids from liquid sample
- Separate supernatant and residue
- Extract supernatant/residue with e.g., 520 µL of CDCl3 or d6-benzene
- Dry solvent with sodium sulfate (Na2SO4)
- Transfer solvent phase in NMR tube
- Add 40 µL of TMS (tetramethylsilane) to calibrate 0.0 ppm
8.4. Protocol for FT-ICR/MS and Data Analysis
- Take 100 µL reaction mix from reaction vessel
- Centrifuge at 15,000 rpm for 5 min, removing solids from liquid sample
- Take 6 µL supernatant
- Dilute in 994 µL Methanol
- Direct infusion at 2 µL/min
- Formula assignment following Tziotis et al. [91]
Supplementary Materials
Supplementary File 1Author Contributions
Funding
Conflicts of Interest
References
- Wachtershauser, G. Before Enzymes and Templates—Theory of Surface Metabolism. Microbiol. Rev. 1988, 52, 452–484. [Google Scholar] [PubMed]
- Wachtershauser, G. From volcanic origins of chemoautotrophic life to Bacteria, Archaea and Eukarya. Philos. Trans. R. Soc. Lond B Biol. Sci. 2006, 361, 1787–1808. [Google Scholar] [CrossRef] [PubMed]
- Wachtershauser, G. Pyrite Formation, the 1st Energy-Source for Life–A Hypothesis. Syst. Appl. Microbiol. 1988, 10, 207–210. [Google Scholar] [CrossRef]
- Wachtershauser, G. Groundworks for an evolutionary biochemistry: The iron-sulphur world. Prog. Biophys. Mol. Biol. 1992, 58, 85–201. [Google Scholar] [CrossRef]
- Wachtershauser, G. On the chemistry and evolution of the pioneer organism. Chem. Biodivers. 2007, 4, 584–602. [Google Scholar] [CrossRef]
- Miller, S.L. A production of amino acids under possible primitive earth conditions. Science 1953, 117, 528–529. [Google Scholar] [CrossRef] [PubMed]
- Consden, R.; Gordon, A.H.; Martin, A.J. Qualitative analysis of proteins: A partition chromatographic method using paper. Biochem. J. 1944, 38, 224–232. [Google Scholar] [CrossRef]
- Gordon, H.T.; Thornburg, W.; Werum, L.N. Rapid Paper Chromatography or Carbohydrates and Related Compounds. Anal. Chem. 1956, 28, 849–855. [Google Scholar] [CrossRef]
- Ferris, J.P.; Sanchez, R.A.; Orgel, L.E. Studies in prebiotic synthesis. 3. Synthesis of pyrimidines from cyanoacetylene and cyanate. J. Mol. Biol. 1968, 33, 693–704. [Google Scholar] [CrossRef]
- Dickson, H.; Kittredge, K.W.; Sarquis, A.M. Thin-Layer Chromatography: The “Eyes” of the Organic Chemist. J. Chem. Educ. 2004, 81, 1023. [Google Scholar] [CrossRef]
- Anumukonda, L.N.; Young, A.; Lynn, D.G.; Buckley, R.; Warrayat, A.; Graves, C.L.; Bean, H.D.; Hud, N.V. Adenine Synthesis in a Model Prebiotic Reaction: Connecting Origin of Life Chemistry with Biology. J. Chem. Educ. 2011, 88, 1698–1701. [Google Scholar] [CrossRef] [PubMed]
- Dolowy, M.; Pyka, A.; Pluta, M. Comparison of the Detection Limit and the Quantification Limit of Nicotinamide on Different Chromatographic Sorbents in TLC. Austin Chromatogr. 2015, 2, 1034. [Google Scholar]
- Prior, E. Determination of Adsorption Heats of Gases and Vapors by the Application of the Chromatographic Method in the Gas Phase; University of Innsbruck: Innsbruck, Austria, 1947. [Google Scholar]
- Cremer, E.; Prior, E. Application of the Chromatographic Method to the Separation of Gases and to the Determination of Adsorption Energies. Z. Elekrochem. 1951, 55, 217–220. [Google Scholar]
- Bobleter, O. Exhibition of the first gas chromatographic work of Erika Cremer and Fritz Prior. Chromatographia 1996, 43, 444–446. [Google Scholar] [CrossRef]
- James, A.T.; Martin, A.J.P. Gas-liquid partition chromatography; the separation and micro-estimation of volatile fatty acids from formic acid to dodecanoic acid. Biochem. J. 1952, 50, 679–690. [Google Scholar] [CrossRef]
- Bartle, K.D.; Myers, P. History of gas chromatography. Trac Trends Anal. Chem. 2002, 21, 547–557. [Google Scholar] [CrossRef]
- Foley, J.P.; Dorsey, J.G. Clarification of the Limit of Detection in Chromatography. Chromatographia 1984, 18, 503–511. [Google Scholar] [CrossRef]
- Harley, J.; Nel, W.; Pretorius, V. Flame Ionization Detector for Gas Chromatography. Nature 1958, 181, 177–178. [Google Scholar] [CrossRef]
- McWilliam, I.G.; Dewar, R.A. Flame Ionization Detector for Gas Chromatography. Nature 1958, 181, 760. [Google Scholar] [CrossRef]
- Gohlke, R.S. Time-of-Flight Mass Spectrometry and Gas-Liquid Partition Chromatography. Anal. Chem. 1959, 31, 535–541. [Google Scholar] [CrossRef]
- Horvath, C.G.; Lipsky, S.R. Use of Liquid Ion Exchange Chromatography for the Separation of Organic Compounds. Nature 1966, 211, 748–749. [Google Scholar] [CrossRef] [PubMed]
- Horvath, C.G.; Preiss, B.A.; Lipsky, S.R. Fast liquid chromatography. Investigation of operating parameters and the separation of nucleotides on pellicular ion exchangers. Anal. Chem. 1967, 39, 1422–1428. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, M.A.; McLafferty, F.W. Liquid chromatography-mass spectrometry interface–I: The direct introduction of liquid solutions into a chemical ionization mass spectrometer. Org. Mass Spectrom. 1973, 7, 1111–1112. [Google Scholar] [CrossRef]
- Blakley, C.R.; McAdams, M.J.; Vestal, M.L. Crossed-beam liquid chromatoraph—Mass spectrometer combination. J. Chromatogr. A 1978, 158, 261–276. [Google Scholar] [CrossRef]
- Smith, D.F.; Podgorski, D.C.; Rodgers, R.P.; Blakney, G.T.; Hendrickson, C.L. 21 Tesla FT-ICR Mass Spectrometer for Ultrahigh-Resolution Analysis of Complex Organic Mixtures. Anal. Chem. 2018, 90, 2041–2047. [Google Scholar] [CrossRef]
- Herzsprung, P.; Hertkorn, N.; von Tümpling, W.; Harir, M.; Friese, K.; Schmitt-Kopplin, P. Molecular formula assignment for dissolved organic matter (DOM) using high-field FT-ICR-MS: Chemical perspective and validation of sulphur-rich organic components (CHOS) in pit lake samples. Anal. Bioanal. Chem. 2016, 408, 2461–2469. [Google Scholar] [CrossRef] [PubMed]
- Schmitt-Kopplin, P.; Gabelica, Z.; Gougeon, R.D.; Fekete, A.; Kanawati, B.; Harir, M.; Gebefuegi, I.; Eckel, G.; Hertkorn, N. High molecular diversity of extraterrestrial organic matter in Murchison meteorite revealed 40 years after its fall. Proc. Natl. Acad. Sci. USA 2010, 107, 2763–2768. [Google Scholar] [CrossRef]
- Halcrow, M.A.; Christou, G. Biomimetic Chemistry of Nickel. Chem. Rev. 1994, 94, 2421–2481. [Google Scholar] [CrossRef]
- Ragsdale, S.W. CO Dehydrogenase and the Central Role of This Enzyme in the Fixation of Carbon Dioxide by Anaerobic Bacteria. In Acetogenesis; Drake, H.L., Ed.; Springer: Boston, MA, USA, 1994; pp. 88–126. [Google Scholar] [CrossRef]
- Ferry, J.G. CO DEHYDROGENASE. Annu. Rev. Microbiol. 1995, 49, 305–333. [Google Scholar] [CrossRef]
- Menon, S.; Ragsdale, S.W. Unleashing Hydrogenase Activity in Carbon Monoxide Dehydrogenase/Acetyl-CoA Synthase and Pyruvate:Ferredoxin Oxidoreductase. Biochemistry 1996, 35, 15814–15821. [Google Scholar] [CrossRef]
- Isidorov, V.A.; Zenkevich, I.G.; Karpov, G.A. Volatile organic compounds in steam-gas discharge from some volcanoes and hydrothermal systems of Kamtchatka. Vulkanol Scismol 1991, 3, 19–25. [Google Scholar]
- Heinen, W.; Lauwers, A.M. Organic sulfur compounds resulting from the interaction of iron sulfide, hydrogen sulfide and carbon dioxide in an anaerobic aqueous environment. Orig. Life Evol. Biosph. 1996, 26, 131–150. [Google Scholar] [CrossRef] [PubMed]
- Holleman, F.A.; Wiberg, E. Lehrbuch der Anorganischen Chemie, 91th ed.; de Gruyter: Berlin, Germany, 1985; pp. 1126–1152. [Google Scholar]
- Gmelin, L. Gmelins, Handbuch der Anorganischen Chemie; Verlag Chimie: Berlin, Germany, 1929–1933; Volume Syst.Nr.59, pp. 62–63, 120.
- Huber, C.; Wächtershäuser, G. Activated acetic acid by carbon fixation on (Fe,Ni)S under primordial conditions. Science 1997, 276, 245–247. [Google Scholar] [CrossRef] [PubMed]
- Mukhin, L.E.V. Evolution of organic compounds in volcanic regions. Nature 1974, 251, 50–51. [Google Scholar] [CrossRef]
- Huber, C.; Wächtershäuser, G. alpha-Hydroxy and alpha-amino acids under possible Hadean, volcanic origin-of-life conditions. Science 2006, 314, 630–632. [Google Scholar] [CrossRef] [PubMed]
- Mawhinney, T.P.; Robinett, R.S.; Atalay, A.; Madson, M.A. Analysis of amino acids as their tert.-butyldimethylsilyl derivatives by gas-liquid chromatography and mass spectrometry. J. Chromatogr. 1986, 358, 231–242. [Google Scholar] [CrossRef]
- Huber, C.; Eisenreich, W.; Wächtershäuser, G. Synthesis of α-amino and α-hydroxy acids under volcanic conditions: Implications for the origin of life. Tetrahedron Lett. 2010, 51, 1069–1071. [Google Scholar] [CrossRef]
- Huber, C.; Kraus, F.; Hanzlik, M.; Eisenreich, W.; Wächtershäuser, G. Elements of metabolic evolution. Chemistry 2012, 18, 2063–2080. [Google Scholar] [CrossRef] [PubMed]
- Seiffert, G.B.; Ullmann, G.M.; Messerschmidt, A.; Schink, B.; Kroneck, P.M.H.; Einsle, O. Structure of the non-redox-active tungsten/[4Fe:4S] enzyme acetylene hydratase. Proc. Natl. Acad. Sci. USA 2007, 104, 3073–3077. [Google Scholar] [CrossRef]
- Igari, S.; Maekawa, T.; Sakata, S. Light hydrocarbons in fumarolic gases: A case study in the Kakkonda geothermal area. Chikyukagau 2000, 34, 103–109. [Google Scholar]
- Scheidler, C.; Sobotta, J.; Eisenreich, W.; Wachtershauser, G.; Huber, C. Unsaturated C-3,C-5,C-7,C-9-Monocarboxylic Acids by Aqueous, One-Pot Carbon Fixation: Possible Relevance for the Origin of Life. Sci. Rep. 2016, 6, 27595. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, R.V.; Epstein, S.; Cronin, J.R.; Pizzarello, S.; Yuen, G.U. Isotopic and molecular analyses of hydrocarbons and monocarboxylic acids of the Murchison meteorite. Geochim. Et Cosmochim. Acta 1992, 56, 4045–4058. [Google Scholar] [CrossRef]
- Naraoka, H.; Shimoyama, A.; Harada, K. Molecular Distribution of Monocarboxylic Acids in Asuka Carbonaceous Chondrites from Antarctica. Orig. Life Evol. Biosph. 1999, 29, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Cole, W.J.; Kaschke, M.; Sherringham, J.A.; Curry, G.B.; Turner, D.; Russell, M.J. Can amino acids be synthesized by H2S in anoxic lakes? Mar. Chem. 1994, 45, 243–256. [Google Scholar] [CrossRef]
- Cody, G.D.; Boctor, N.Z.; Filley, T.R.; Hazen, R.M.; Scott, J.H.; Sharma, A.; Yoder, H.S.J. Primordial Carbonylated Iron-Sulfur Compounds and the Synthesis of Pyruvate. Science 2000, 289, 1337–1340. [Google Scholar] [CrossRef] [PubMed]
- Muchowska, K.B.; Varma, S.J.; Chevallot-Beroux, E.; Lethuillier-Karl, L.; Li, G.; Moran, J. Metals promote sequences of the reverse Krebs cycle. Nat. Ecol. Evol. 2017, 1, 1716–1721. [Google Scholar] [CrossRef] [PubMed]
- Muchowska, K.B.; Varma, S.J.; Moran, J. Synthesis and Breakdown of the Universal Precursors to Biological Metabolism Promoted by Ferrous Iron. Chem Rxiv 2018. [Google Scholar] [CrossRef]
- Varma, S.J.; Muchowska, K.B.; Chatelain, P.; Moran, J. Native iron reduces CO2 to intermediates and end-products of the acetyl-CoA pathway. Nat. Ecol. Evol. 2018, 2, 1019–1024. [Google Scholar] [CrossRef]
- Pallmann, S.; Steflova, J.; Haas, M.; Lamour, S.; Henß, A.; Trapp, O. Schreibersite: An effective catalyst in the formose reaction network. New J. Phys. 2018, 20, 055003. [Google Scholar] [CrossRef]
- Kvenvolden, K.; Lawless, J.; Pering, K.; Peterson, E.; Flores, J.; Ponnamperuma, C.; Kaplan, I.R.; Moore, C. Evidence for Extraterrestrial Amino-acids and Hydrocarbons in the Murchison Meteorite. Nature 1970, 228, 923–926. [Google Scholar] [CrossRef]
- Lawless, J.G.; Chadha, M.S. Mass spectral analysis of C3 and C4 aliphatic amino acid derivatives. Anal. Biochem. 1971, 44, 473–485. [Google Scholar] [CrossRef]
- Lawless, J.G. Amino acids in the Murchison meteorite. Geochim. Et Cosmochim. Acta 1973, 37, 2207–2212. [Google Scholar] [CrossRef]
- OrÓ, J.; Gibert, J.; Lichtenstein, H.; Wikstrom, S.; Flory, D.A. Amino-acids, Aliphatic and Aromatic Hydrocarbons in the Murchison Meteorite. Nature 1971, 230, 105–106. [Google Scholar] [CrossRef] [PubMed]
- Simkus, D.N.; Aponte, J.C.; Hilts, R.W.; Elsila, J.E.; Herd, C.D.K. Compound-specific carbon isotope compositions of aldehydes and ketones in the Murchison meteorite. Meteorit. Planet. Sci. 2019, 54, 142–156. [Google Scholar] [CrossRef]
- Burton, A.S.; Stern, J.C.; Elsila, J.E.; Glavin, D.P.; Dworkin, J.P. Understanding prebiotic chemistry through the analysis of extraterrestrial amino acids and nucleobases in meteorites. Chem. Soc. Rev. 2012, 41, 5459–5472. [Google Scholar] [CrossRef]
- Schreiner, E.; Nair, N.N.; Marx, D. Peptide Synthesis in Aqueous Environments: The Role of Extreme Conditions on Peptide Bond Formation and Peptide Hydrolysis. J. Am. Chem. Soc. 2009, 131, 13668–13675. [Google Scholar] [CrossRef] [PubMed]
- Huber, C.; Wachtershauser, G. Peptides by activation of amino acids with CO on (Ni,Fe)S surfaces: Implications for the origin of life. Science 1998, 281, 670–672. [Google Scholar] [CrossRef] [PubMed]
- Leman, L.J.; Orgel, L.E.; Ghadiri, M.R. Carbonyl sulfide-mediated prebiotic formation of peptides. Science 2004, 306, 283–286. [Google Scholar] [CrossRef]
- Kimoto, T.; Fujinaga, T. PB81 Non-Biotic synthesis of cellular organic polymers in the presence of hydrogen sulfide. Int. Symp. Chem. Nat. Prod. 1988, 1988, 327. [Google Scholar] [CrossRef]
- Kimoto, T.; Fujinaga, T. Non-biotic synthesis of organic polymers on H2S-rich sea-floor: A possible reaction in the origin of life. Mar. Chem. 1990, 30, 179–192. [Google Scholar] [CrossRef]
- Messner, C.B.; Driscoll, P.C.; Piedrafita, G.; De Volder, M.F.L.; Ralser, M. Nonenzymatic gluconeogenesis-like formation of fructose 1,6-bisphosphate in ice. Proc. Natl. Acad. Sci. USA 2017, 114, 7403–7407. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.A.; Turchyn, A.V.; Ralser, M. Non-enzymatic glycolysis and pentose phosphate pathway-like reactions in a plausible Archean ocean. Mol. Syst. Biol. 2014, 10, 725. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.A.; Kampjut, D.; Harrison, S.A.; Ralser, M. Sulfate radicals enable a non-enzymatic Krebs cycle precursor. Nat Ecol Evol 2017, 1, 0083. [Google Scholar] [CrossRef] [PubMed]
- Fahrenbach, A.C.; Giurgiu, C.; Tam, C.P.; Li, L.; Hongo, Y.; Aono, M.; Szostak, J.W. Common and Potentially Prebiotic Origin for Precursors of Nucleotide Synthesis and Activation. J. Am. Chem. Soc. 2017, 139, 8780–8783. [Google Scholar] [CrossRef] [PubMed]
- Huber, C.; Eisenreich, W.; Hecht, S.; Wachtershauser, G. A possible primordial peptide cycle. Science 2003, 301, 938–940. [Google Scholar] [CrossRef] [PubMed]
- Whicher, A.; Camprubi, E.; Pinna, S.; Herschy, B.; Lane, N. Acetyl Phosphate as a Primordial Energy Currency at the Origin of Life. Orig. Life Evol. Biosph. 2016, 48, 159–179. [Google Scholar] [CrossRef] [PubMed]
- Ritson, D.; Sutherland, J.D. Prebiotic synthesis of simple sugars by photoredox systems chemistry. Nat. Chem. 2012, 4, 895–899. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y.; Sekine, T.; Oba, M.; Kakegawa, T.; Nakazawa, H. Biomolecule formation by oceanic impacts on early Earth. Nat. Geosci. 2008, 2, 62–66. [Google Scholar] [CrossRef]
- Weiss, M.C.; Sousa, F.L.; Mrnjavac, N.; Neukirchen, S.; Roettger, M.; Nelson-Sathi, S.; Martin, W.F. The physiology and habitat of the last universal common ancestor. Nat. Microbiol. 2016, 1, 16116. [Google Scholar] [CrossRef] [PubMed]
- Huber, H.; Gallenberger, M.; Jahn, U.; Eylert, E.; Berg, I.A.; Kockelkorn, D.; Eisenreich, W.; Fuchs, G. A dicarboxylate/4-hydroxybutyrate autotrophic carbon assimilation cycle in the hyperthermophilic Archaeum Ignicoccus hospitalis. Proc. Natl. Acad. Sci. USA 2008, 105, 7851–7856. [Google Scholar] [CrossRef] [PubMed]
- Estelmann, S.; Hugler, M.; Eisenreich, W.; Werner, K.; Berg, I.A.; Ramos-Vera, W.H.; Say, R.F.; Kockelkorn, D.; Gad’on, N.; Fuchs, G. Labeling and enzyme studies of the central carbon metabolism in Metallosphaera sedula. J. Bacteriol. 2011, 193, 1191–1200. [Google Scholar] [CrossRef] [PubMed]
- Mall, A.; Sobotta, J.; Huber, C.; Tschirner, C.; Kowarschik, S.; Bacnik, K.; Mergelsberg, M.; Boll, M.; Hugler, M.; Eisenreich, W.; et al. Reversibility of citrate synthase allows autotrophic growth of a thermophilic bacterium. Science 2018, 359, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Jahn, U.; Huber, H.; Eisenreich, W.; Hugler, M.; Fuchs, G. Insights into the autotrophic CO2 fixation pathway of the archaeon Ignicoccus hospitalis: Comprehensive analysis of the central carbon metabolism. J. Bacteriol. 2007, 189, 4108–4119. [Google Scholar] [CrossRef] [PubMed]
- Marshall, A.G.; Hendrickson, C.L.; Jackson, G.S. Fourier transform ion cyclotron resonance mass spectrometry: A primer. Mass Spectrom. Rev. 1998, 17, 1–35. [Google Scholar] [CrossRef]
- Marshall, A.G.; Hendrickson, C.L. Fourier transform ion cyclotron resonance detection: Principles and experimental configurations. Int. J. Mass Spectrom. 2002, 215, 59–75. [Google Scholar] [CrossRef]
- Sobotta, J. Investigations of Carbon Fixation in Model Organisms and in Cell-Free Prebiotic Transition Metal-Catalyzed Reactions. Ph.D. Dissertation, Technial University Munich, Munich, Germany, 2018. [Google Scholar]
- Van Krevelen, D.W. Graphical-Statistical Method for the Study of Structure and Reaction Processes of Coal. Fuel 1950, 29, 228–269. [Google Scholar]
- Kendrick, E. A Mass Scale Based on CH2 = 14.0000 for High Resolution Mass Spectrometry of Organic Compounds. Anal. Chem. 1963, 35, 2146–2154. [Google Scholar] [CrossRef]
- Hemmler, D.; Roullier-Gall, C.; Marshall, J.W.; Rychlik, M.; Taylor, A.J.; Schmitt-Kopplin, P. Evolution of Complex Maillard Chemical Reactions, Resolved in Time. Sci. Rep. 2017, 7, 3227. [Google Scholar] [CrossRef]
- Forsythe, J.G.; Yu, S.-S.; Mamajanov, I.; Grover, M.A.; Krishnamurthy, R.; Fernández, F.M.; Hud, N.V. Ester-Mediated Amide Bond Formation Driven by Wet–Dry Cycles: A Possible Path to Polypeptides on the Prebiotic Earth. Angew. Chem. Int. Ed. 2015, 54, 9871–9875. [Google Scholar] [CrossRef]
- Sandford, S.A. The AstroBiology Explorer (ABE) MIDEX Mission Concept: Using Infrared Spectroscopy to Identify Organic Molecules in Space. In Proceedings of the NASA Laboratory Astrophysics Workshop, Moffett Field, CA, USA, 1–3 May 2002; p. 153. [Google Scholar]
- Mutschler, H.; Wochner, A.; Holliger, P. Freeze–thaw cycles as drivers of complex ribozyme assembly. Nat. Chem. 2015, 7, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Ross, D.S.; Deamer, D. Dry/Wet Cycling and the Thermodynamics and Kinetics of Prebiotic Polymer Synthesis. Life 2016, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.A.; Driscoll, P.C.; Messner, C.B.; Ralser, M. 1H-NMR as implemented in several origin of life studies artificially implies the absence of metabolism-like non-enzymatic reactions by being signal-suppressed. Wellcome Open Res. 2018. [Google Scholar] [CrossRef]
- Buttery, R.G.; Teranishi, R. Gas-Liquid Chromatography of Aroma of Vegetables and Fruit. Direct Injection of Aqueous Vapors. Anal. Chem. 1961, 33, 1439–1441. [Google Scholar] [CrossRef]
- Zhang, Z.; Pawliszyn, J. Headspace solid-phase microextraction. Anal. Chem. 1993, 65, 1843–1852. [Google Scholar] [CrossRef]
- Tziotis, D.; Hertkorn, N.; Schmitt-Kopplin, P. Kendrick-Analogous Network Visualisation of Ion Cyclotron Resonance Fourier Transform Mass Spectra: Improved Options for the Assignment of Elemental Compositions and the Classification of Organic Molecular Complexity. Eur. J. Mass Spectrom. 2011, 17, 415–421. [Google Scholar] [CrossRef] [PubMed]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Geisberger, T.; Diederich, P.; Steiner, T.; Eisenreich, W.; Schmitt-Kopplin, P.; Huber, C. Evolutionary Steps in the Analytics of Primordial Metabolic Evolution. Life 2019, 9, 50. https://doi.org/10.3390/life9020050
Geisberger T, Diederich P, Steiner T, Eisenreich W, Schmitt-Kopplin P, Huber C. Evolutionary Steps in the Analytics of Primordial Metabolic Evolution. Life. 2019; 9(2):50. https://doi.org/10.3390/life9020050
Chicago/Turabian StyleGeisberger, Thomas, Philippe Diederich, Thomas Steiner, Wolfgang Eisenreich, Philippe Schmitt-Kopplin, and Claudia Huber. 2019. "Evolutionary Steps in the Analytics of Primordial Metabolic Evolution" Life 9, no. 2: 50. https://doi.org/10.3390/life9020050
APA StyleGeisberger, T., Diederich, P., Steiner, T., Eisenreich, W., Schmitt-Kopplin, P., & Huber, C. (2019). Evolutionary Steps in the Analytics of Primordial Metabolic Evolution. Life, 9(2), 50. https://doi.org/10.3390/life9020050