1. Introduction
How living beings have been able to overcome the entropic forces to develop increasingly complex individuals which, in turn, maintain their functionality is an open question and one of the hardest problems of modern science [
1,
2,
3,
4,
5,
6,
7,
8,
9,
10,
11,
12]. The exhaustive analysis of the energy flows in real living entities collides with the extreme complexity of even the simplest bacteria. Therefore, one must first set what are the
physical defining properties of living beings and, then, try to attack the problem by cutting it into pieces. Each piece should incorporate a key or several key features, simple enough to accept a rigorous analysis, but complex enough to shed light to certain facets of the problem. The later integration of all pieces, however, will likely be much more than building a puzzle, for it is clear that the cross dependencies between all the building blocks will introduce an additional layer of complexity.
Following this philosophy, we will focus here on two crucial properties of living beings, according to accepted definitions of life discussed among scholars [
13,
14,
15,
16,
17]. Specifically, we will concentrate on systems able to:
Capture material resources and turn them into building blocks by the use of externally provided free energy—and eventually undergo a duplication cycle and
Keep its components together and distinguish itself from the environment. It is assumed that the compartment contains the metabolic and information system—if any. Our simplified system, thus, will lack two crucial features of living beings, namely,
To process and transmit inheritable information to progeny and
To undergo Darwinian evolution through variation of the copied inheritable information and a successive selection of the better progeny. We will thus focus on the thermodynamic properties of the
duplication process, and we will skip all the complexity arising from other phenomena. It is worth to recall here that this kind of approach, where the essential physics of the duplication problem is addressed has a long history, dating back to the late thirties of the 20th century, with the highly influential works of N. Rashevsky [
1].
In contrast to the usual top-down approaches followed in biology, we will address this problem using a bottom-up approach. In such kind of approaches to life-related phenomena, physical building blocks and chemical processes are externally assembled and triggered, creating artificial, synthetic entities that mimic some of the crucial properties of living beings. Consistently, this approach has been named
Artificial Life [
16,
18,
19,
20]. Artificial cells, or,
protocells are usually composed by emulsions [
21] made of mixtures of lipids, precursors, and water [
15,
16,
19,
20,
22,
23,
24,
25,
26,
27,
28]. The foundation of this approach is based on three main starting points: First, it provides a framework where energy imbalances trigger the emergence of cell like aggregates [
21], second, it is possible to externally drive simplified metabolic reactions [
15,
16,
26,
28], and, third, it uses the same type of building blocks—mainly lipids—that compose an important part of the structure of most of the living organisms [
29]. Crucial to our aims, it is worth to remark a couple of recent results: First, numerical approaches have shown that duplication dynamics as a consequence of energy imbalances due to geometrical frustration is expected in those systems, if properly driven out of equilibrium [
30]. Second, recent experiments succeeded
in duplicating real artificial protocells through a specific oil-in-water droplet system with replicating information templates [
31]. This result is certainly remarkable, but our approach does exclude the role of any information/replication dynamics. In doing so, we explore how far can we go by just taking into account general stability properties and energy imbalances to explain and characterize the duplication process. The work presented here runs in parallel to an interesting complementary approach taken in [
32], where the kinetics involved in the duplication events of synthetic systems was studied in detail.
In this paper we will work with a generic emulsion system [
21]. We will make use of the well understood free energy landscape of such systems, where the contributions coming from aggregate geometry and size have been long studied [
21,
33,
34], as well as the non-trivial contributions of the entropic terms [
35,
36]. The impact of a changing energy landscape—which eventually can favour a duplication event—will be studied from a generic non-equilibrium situation making use of modern methods arising from the emerging field of
Stochastic Thermodynamics [
37,
38,
39,
40,
41,
42]. Within this framework, the evolution of the system can be studied following the individual trajectories in the phase space and, importantly, exact relations between energy and work can be obtained, even in out of equilibrium cases. In addition, relations between energy, entropy and information arise naturally [
43,
44].
The remainder of the paper is organized as follows: In the next section, we describe the thermodynamics of the abstract emulsion system in detail. We derive its free energy landscape,
Section 2.1, the equilibrium distributions,
Section 2.2, and the detailed balance condition over transitions,
Section 2.3. Next, in
Section 2.4, we expose the generic protocol that drives the system towards the occurrence of a duplication event. We end the section where the system is presented by exploring the orders of magnitude involved in these kind of systems,
Section 2.5. Here we analyze the quantitative values of the thermodynamic functionals presented generically in the previous sections for a real microemulsion system. The thermodynamic analysis of the duplication thresholds is the core of section III. First, we derive a general relation for duplication probabilities,
Section 3.1. Then, in
Section 3.2, we explore the consequences of this result for a system evolving in a quasi-static fashion.
Section 3.3 generalizes the previous equilibrium approach by providing an exact equality between probabilities of duplication thresholds in a specific non-equilibrium scenario, in which the relaxation process that may eventually lead to duplication happens between two states which may not be in equilibrium. This equivalence leads us to define general duplication scenarios and derive the general conditions of duplication, as well as the amount of work invested over the system to trigger a duplication event, and the conditions for the perpetuation of the duplication cycle.
Section 3.4 refers to the free energy/entropy relations for the perpetuation of the duplication cycle in time. The final section is devoted to discuss the implications of the presented results. The whole paper is aimed to be self-contained and details of the derivations are provided in the
Appendix A to make it understandable to non-specialized audiences.
2. The System
Our system is conceived as being an abstract emulsion in a kind of reaction tank of volume connected to a heat reservoir at inverse temperature —we set . Let , where is a specific kind of lipid species populating the system and , where is a specific kind of precursor/surfactant species populating the system. Let the total amount of molecules of the different species of lipids and precursors that lie in aqueous solution inside our volume. We refer to as the boundary conditions. As we shall see, they may change in time, under the action of an external protocol.
Due to the hydrophobic/hydrophilic nature of the surfactant molecules, we assume that (part of them) tend to aggregate in spheroidal compartments. Surfactants are supposed to populate the surface of the aggregates. No assumptions are made on the specific nature of the membranes or the interior of the aggregates, leaving the discussion always in a general plane. A
state of our system is described by a 3-tuple:
where
and
are the amount of lipids and precursors forming aggregates, respectively, and
n the number of aggregates present in the volume. In general, and if no confusion can arise, we refer to a given state as
instead of
for notational simplicity. We keep the label subscript “
” accounting for the number of aggregates only for notational convenience. When we introduce time dependence, we write
. Not all molecules will be part of the aggregates. Therefore, we must account for these molecules in bulk. Consistently, given a state
occuring under the boundary conditions
, we wil have that
and
are the amount of lipids and precursors in bulk, respectively.
A
macrostate or
coarse-grained state
is defined as the 4-tuple:
where
is the probability distribution of finding
as a particular realization of this macrostate. This macrostate can be realized through any state containing
and
n protocellular aggregates following the distribution
. In case of time dependence we write
.
2.1. Gibbs Free Energy Landscape
The thermodynamic landscape of our system is given by the Gibbs free energy of the state
,
The Gibbs free energy is always defined over states of the system and depends on both the state and the boundary conditions . Therefore, the same state will have energy changes if the boundary conditions change. Each macrostate has a uniquely defined free energy functional. For notational simplicity, we drop the subscript , if no confusion arises.
The complex nature of these type of emulsions results in a free energy functional with several blocks, which we construct step by step. First, we focus on the free energy contribution of a single protocellular aggregate, containing
lipids,
, and precursors,
:
where
and
are the changes in chemical potential when moving lipids and surfactants from bulk into the
i-th aggregate, and
is a geometric term expressing shape and surface contributions to the free energy of the aggregate. This geometric term accounts for the membrane properties of the system, and is computed according to the existence of a minimum energy configuration or
perfect protocellular aggregate, which can be directly computed as the optimal packing from the knowledge to the sizes and geometries of the precursor molecules. The geometrical term thus reads:
where
is the surface tension,
the compressibility coefficient, and
the elastic bending modulus of the lipid membrane. The integral is the second order expansion of the contribution of the Helfrisch Hamiltonian to the overall free energy, being
H the curvature of the membrane—as a function of some coordinates parametrizing the membrane surface—of the current aggregate and
the curvature of the perfect aggregate. The integral is computed over the whole area of the membrane,
A [
33,
34].
Once we have properly characterized the free energies of a single aggregate, we proceed to construct the free energy of the whole state
. The next task will be to compute the entropy for a system in the state
under the boundary conditions
. To compute the entropy of such state, we apply directly Boltzmann’s definition over the amount of configurations the state
can adopt,
[
45]:
Clearly,
. However, we do not write this dependence explicitly for the sake of readability, if no confusion can arise. This entropic term has two contributions, the
translational entropy and the
configurational entropy. We start with the translational contribution. We consider that the system of
n indistinguishable aggregates has
degrees of freedom and that each aggregate diffuse around within a volume
and that
is an appropriate length scale for such a diffusive process. Accordingly, one has that the amount of configurations provided by the translational term is:
We emphasize that, in the approach take here,
has been chosen as a typical volume unit whose purpose is to render the argument of the logarithm dimensionless—for a deeper discussion on the choice of the right length scale see [
35,
36]. For each configuration described above, we must account for the potential degeneracy of states, or, in other words, the amount of configurations given by the amount of molecules in bulk and forming the aggregates. For each chemical species, e.g., the
i-th lipid, this amount of configurations is
Therefore, assuming that there are no cross dependencies among the different chemical species, one has that the amount of configurations of molecules in bulk and aggregates is:
Considering these two contributions, the entropy term reads:
The overall entropy of the state
, under the boundary conditions given by
,
, is:
where we used the fact the
and the Stirling approximation for the factorial for the first term, namely
. Collecting all the above ingredients, we have that the Gibbs free energy of the system in the state
under boundary conditions
becomes:
with the standard chemical potentials
and
of lipids and precursors, respectively.
2.2. Helmholtz Free Energy
Let the system be subject to the boundary conditions
. In equilibrium, the probability that the system is in the particular state
, belonging to the macrostate
is given by the Boltzmann distribution,
[
45]:
being
the partition function, namely:
Accordingly, the Helmholtz free energy of the macrostate
,
is:
being
the average over all states of the macrostate and
the entropy of the macrostate, namely:
where
is now defined as:
We point out that we will refer to a given probability distribution associated to a macrostate
either as
or
, indistinctly. We finally recall that we assume that the equilibrium distribution macrostate
is such that:
where we emphasized the dependency on the boundary conditions
only for clarity. In words, we assume that the equilibrium distribution is defined around the absolute minimum of Gibbs free energies, and that such a minimum is unique.
2.3. Detailed Balance Condition in Duplication
The process of duplication/fusion of aggregates is of special interest for us, since it is the basis of duplication. It is assumed to satisfy the following transition rates between states:
where the kinetic constants relate as:
where
. Detailed balance condition is also assumed for any other transition between states. Therefore, for any two states
and
, thanks to the detailed balance condition given in Equation (
8) and assumed for all transitions, one has that, between two arbitrary states
,
:
Importantly, we recall that the functional
G must be computed under the same boundary conditions
in any evaluation of the difference, i.e.,:
2.4. The Driving Protocol
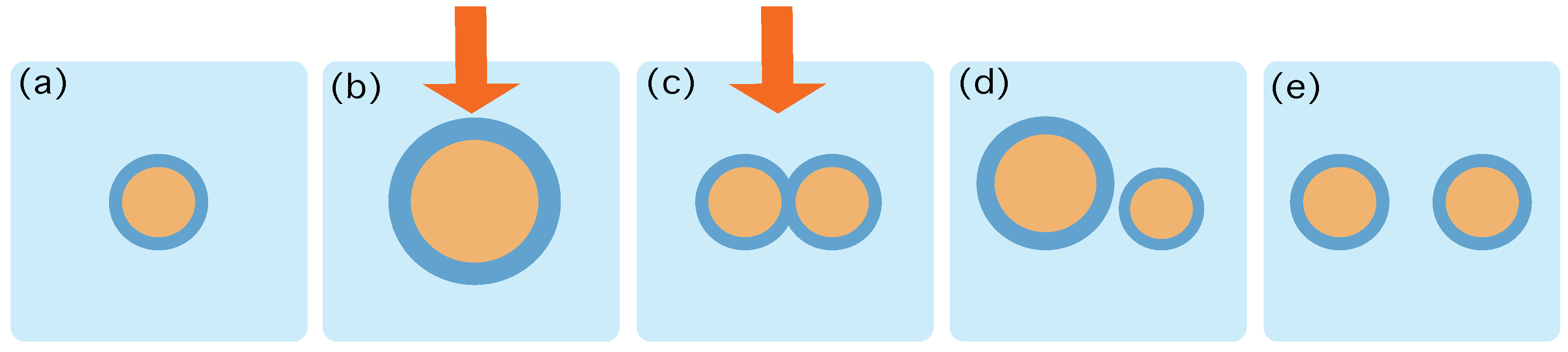
Let us assume that at time
the system is in contact to a thermal reservoir at inverse temperature
, and in an equilibrium macrostate
, that is—see
Figure 1):
From this moment on, we run a protocol that changes the energy landscape, without separating the system from the heath bath neither changing the whole system’s volume,
. This protocol runs from
to
—see
Figure 1b,c. For example, suppose that we add new lipids and that we switch on a light that triggers a metabolic reaction that transforms lipids into precursors, thereby creating new surfactants. We call this protocol
. In general, it will affect the
variables of our system. Therefore, the protocol
consists on a list of—maybe interdependent—protocols:
where the first
L elements
explicit the action of the protocol on the lipids
abundance and the last
P elements
explicit the change due to the protocol on the precursors
abundance. Let us be more specific on the action of the protocol. Assume that at time
t the boundary conditions of our system are given by
. The application of the protocol a for short time interval
to the boundary conditions, denoted by
will lead the boundary conditions to change as:
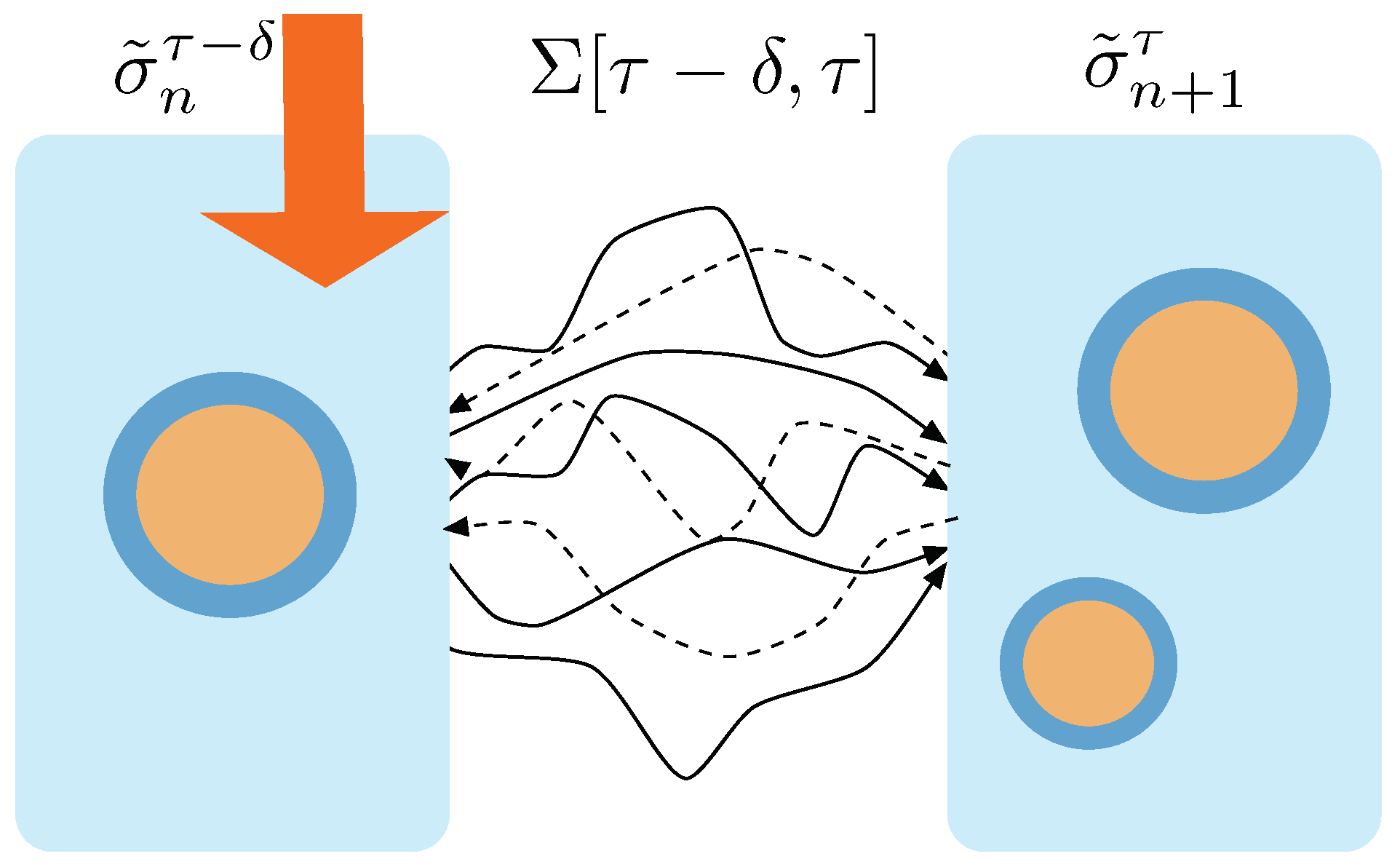
The above transformation of the boundary conditions will lead the system to change its macrostate, from
to
. This transition can be done through a set of stochastic trajectories, which will be referred to as
. At
the system will be at the macrostate
and we will stop the protocol—see
Figure 1d—letting the system relax towards an equilibrium state, achieved at time
—see
Figure 1d. The distribution of states
is assumed to obey the standard equilibrium Boltzmann statistics:
We assume that a duplication event has taken place in the time interval and that the relaxation process happening at the interval does not imply a change in the number of protocell aggregates. We remind that the whole process takes place in contact to a heat reservoir with inverse temperature and at a constant volume .
2.5. An Example: Ternary Emulsions
To grasp the orders of magnitude involved in our problem, we take a particular example of the above general system, in line to the one described in [
26,
27]. From this example, we perform a rough estimation of the orders of magnitude involved in the computation of the free energies of a single aggregate. For the sake of readability and extension, the computations provided here are not as detailed as in the other parts of the paper. We refer the interested reader to [
21,
26,
27,
46,
47,
48] for the detailed discussions on the orders of magnitude and potential experimental set ups.
Suppose that we have a Winsor type IV ternary emulsion made of a single lipid,
decanoic acid anhydride, (
), a single precursor,
decanoic acid, (
), and water. Equation (
1) now reads:
can be calculated from their partition coefficient—i.e., the fraction of lipids found in bulk solution as opposed to the aggregates. Estimations give this value to be around 14% [
46]. If
is the ratio between precursor molecules going from bulk to aggregates and precursor molecules going from aggregates to bulk, this reads:
Therefore, using Equation (
8), and setting
, one can approximate the energy gain of moving a decanoic acid molecule from bulk to aggregate,
, as
where
is the Boltzmann constant,
At
, and
being the Avogadro number, the above equation leads to:
Since the decanoic acid anhydride (
) has two hydrophobic chains, we set
which in turn evaluates to a partition coefficient of ∼2%. For the geometric term given by Equation (
2), we make the assumption that
, therefore the contribution of the Helfrisch hamiltonian will not be taken into account. The surface tension and the compressibility parameters,
can be estimated as
and
[
26]. We assume a spherical lipid core of
precursor molecules, whose individual molecular volume
. Thus, the spherical core of the aggregate has a radius,
, of:
The whole aggregate, including the surface molecules, displays a radius,
, of
where
is the length of the tail of the surfactant molecules, which is considered constant. The optimal number of surfactant molecules,
for this amount of molecules in the core of the aggregate is then computed as:
We assume that the tail length of the surfactants is around
and that their effective head area
[
21]. The typical radius of oil droplets is around
leading to a volume of
nm
, i.e., ~
femtoliter, which—assuming a typical water-to-oil ratio of 10:1—gives a system volume of
femtoliter per droplet. Therefore, a milliliter of emulsion has an order of magnitude of
oil droplets. From the ratio of precursor to droplet volume, it follows that
. With an optimal packing number of surfactants
computed from Equation (
10) and a partition coefficient of 14%, one can estimate a total of
surfactant molecules. With these values, a rough estimation of the orders of magnitude of the free energy of a single aggregate whose packing is optimal,
, is given by:
This example gives us an orientation about the energy scales involved in our problem.
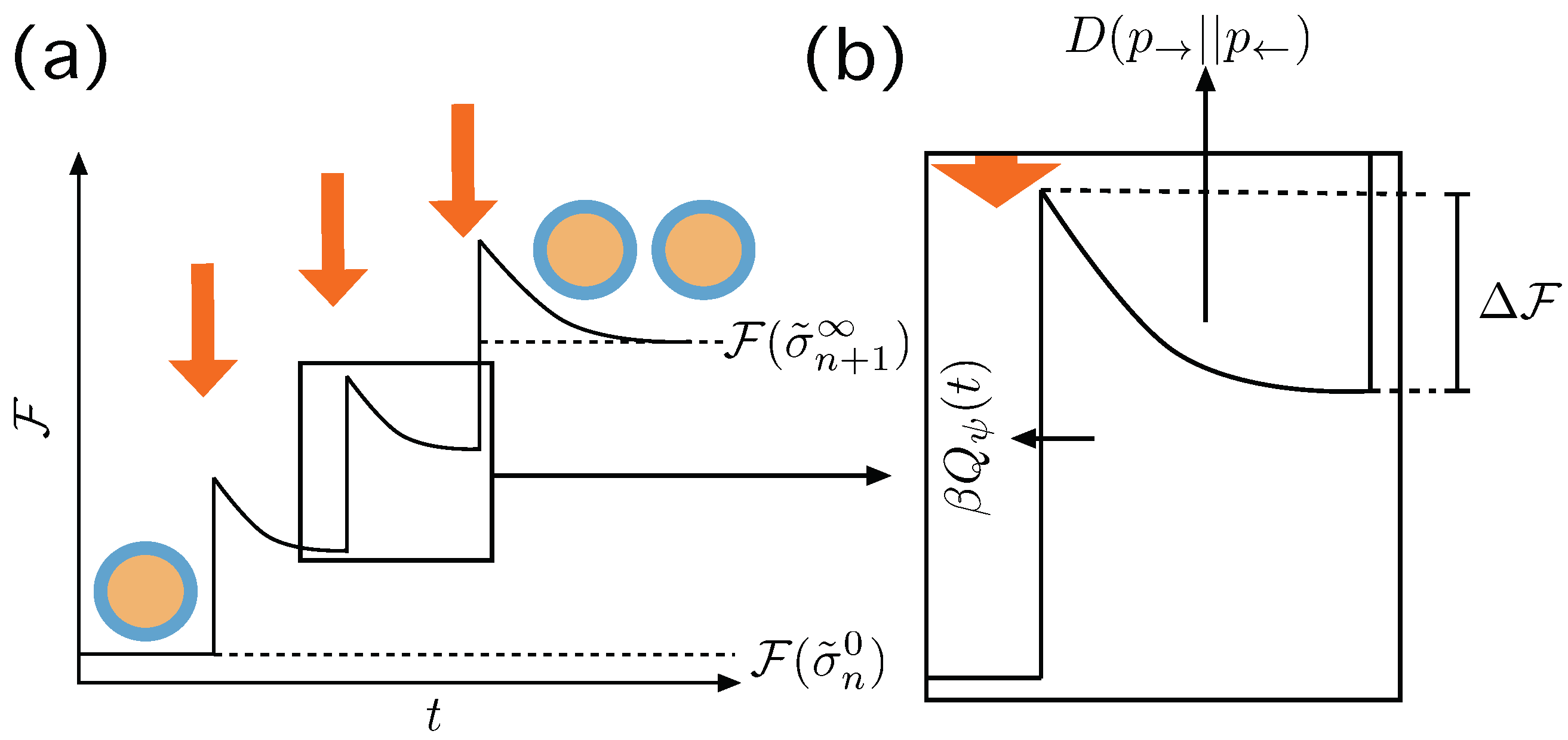
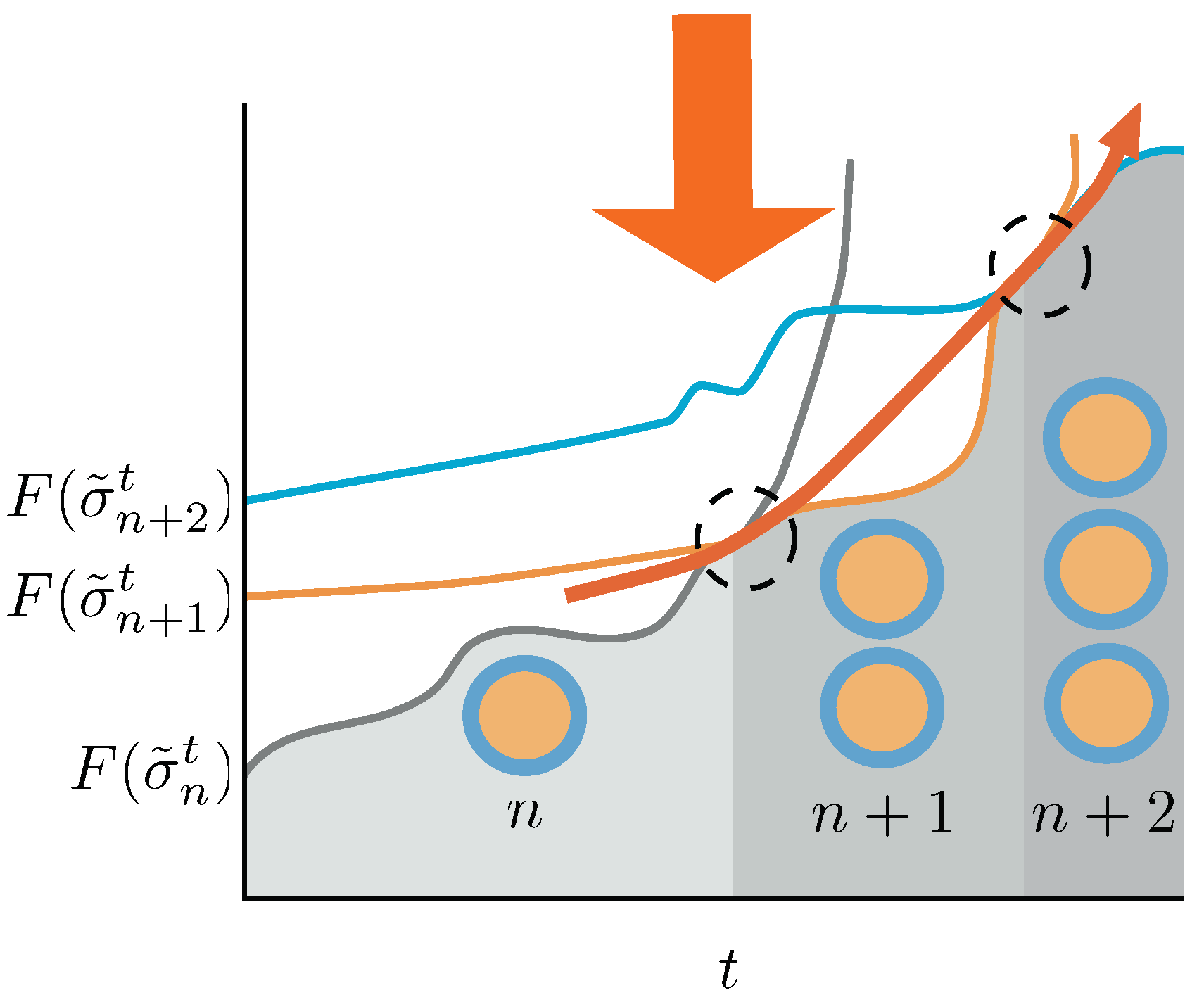
4. Discussion
In this paper we explored in depth the thermodynamics of duplication thresholds in a generic emulsion system made of an arbitrary set of lipid and precursor species. This feasible, yet artificial system enables us to overcome the tremendous complexity of the duplication process in actual living entities, such as cells. The thermodynamic landscape has been carefully constructed, accounting for the contributions due to surface tension, volume of the aggregates, entropic contributions and total amount of chemical species within the systems, all summarized in the definition of the Gibbs free energy of the state, Equation (
4). An abstract protocol is proposed, driving the system away from the equilibrium state and resulting, eventually, in a duplication event. We approached the problem from the equilibrium framework, assuming that the process is a succession of equilibrium states, and from a non-equilibrium perspective, where the visited states may not be equilibrium ones.
Fundamental relations involving free energies and duplication probabilities, Equation (
28), duplication thresholds, Equations (
29) and (
30), necessary work to be invested over the system by the protocol to trigger a duplication event, Equation (
31), dissipated work, Equation (
32) or the conditions for the perpetuation of the duplication cycle, Equations (
33) and (
34) have been derived. These relations invoke the explicit energy landscape provided by the free energies and set the abstract conditions for a duplication process to be triggered and, eventually maintained. It is worth to emphasize that they show explicitly the structure of the race between entropic forces and free energy gains to generate structure and preserve it. The synthetic approach, therefore, enabled us to convey a very detailed picture of the thermodynamical tensions involved in the process of creation and perdurability of living entities.
Further explorations should target more systematically specific systems, with quantitatively testable observables. The study of specific systems should also include the conditions of feasibility, in terms of microemulsion phases, of the aggregate duplication, avoiding transitions to non-aggregate phases, possible in emulsion systems. In the same line, a rigorous exploration of the orders of magnitude involved in the abstract relations derived above would add a necessary layer towards the quantification and, eventually, empirical test of the above predictions. Complementarily, the exploration of the constraints imposed by different protocol strategies could shed light to the potential prebiotic scenarios, where possibly circadian cycles play a crucial role in creating free energy sources driving the system towards imbalance, destabilization, and duplication. In addition, more complex free energy landscapes allowing bilayer membranes, more realistic when compared to biological structures than the single layer approach used here, could refine the triggering points for duplication events to occur. In a different direction, an in depth study of the dissipation within the trajectories themselves—assumed to observe detailed balance in the above developments—would generalize the approach, making it more realistic and providing predictions on dissipated heat which could be presumable testable. Finally, the interesting relations involving dissipation and information measures could be explored to be the seed of further developments linking information and duplication processes, in line to the results exposed in [
31], and, perhaps, clear the conditions for the emergence of inheritable information—thus the appearance of differentiated traits between elements of the system—intrinsically linked to the duplication process and, in the long term, trigger darwinian dynamics.


{kind=link}
{kind=link}
{kind=link}
{kind=link}



