Making Molecules with Clay: Layered Double Hydroxides, Pentopyranose Nucleic Acids and the Origin of Life

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
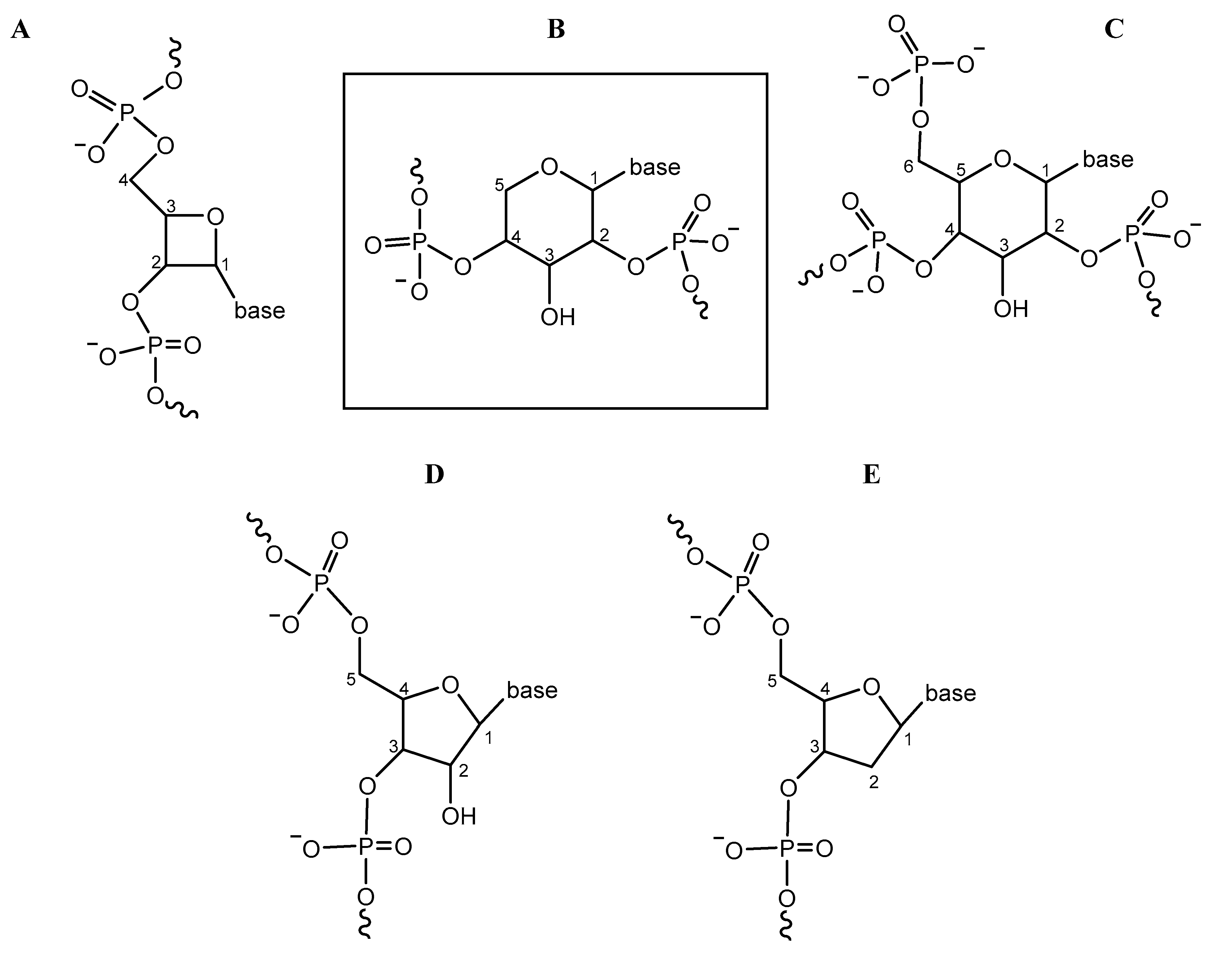
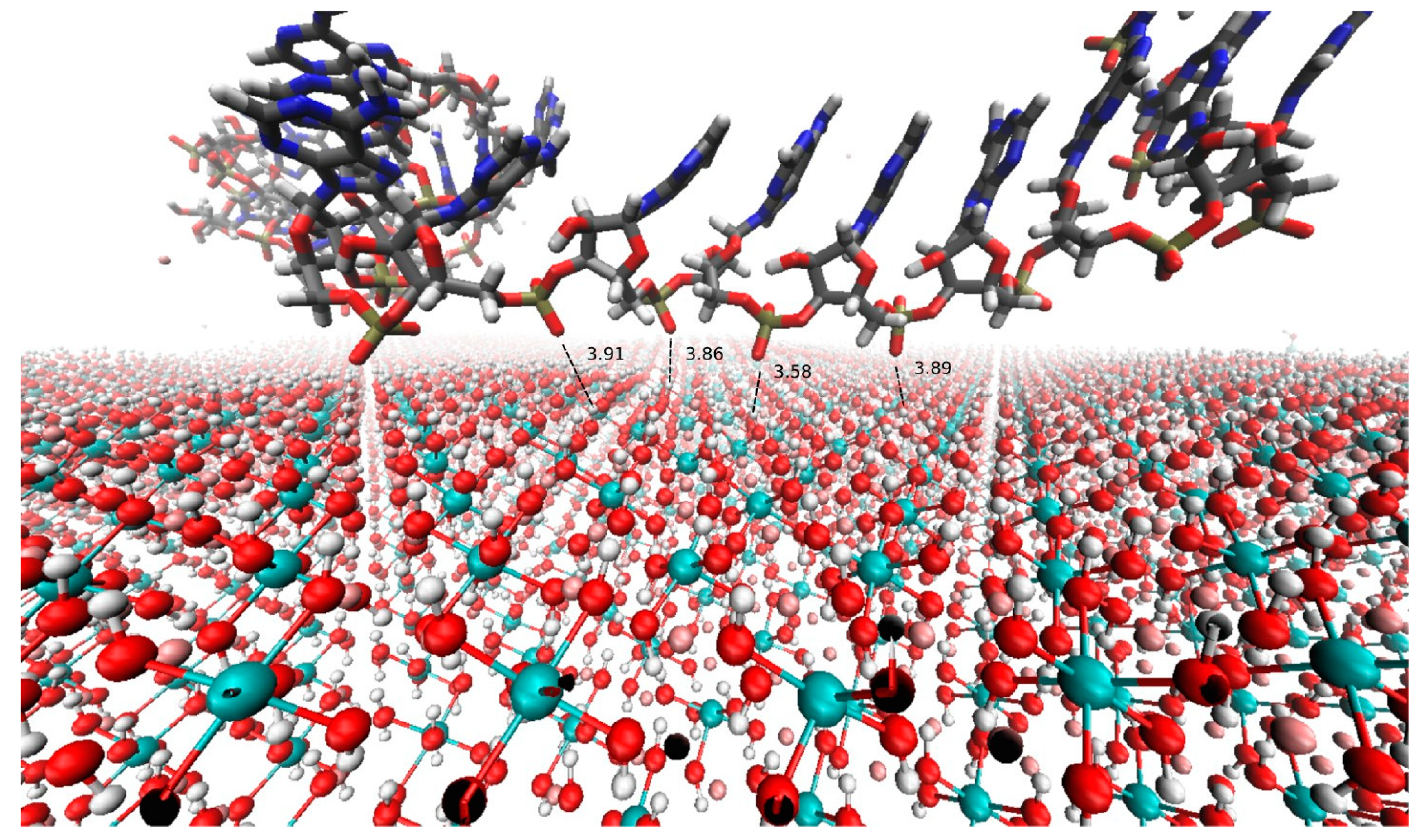
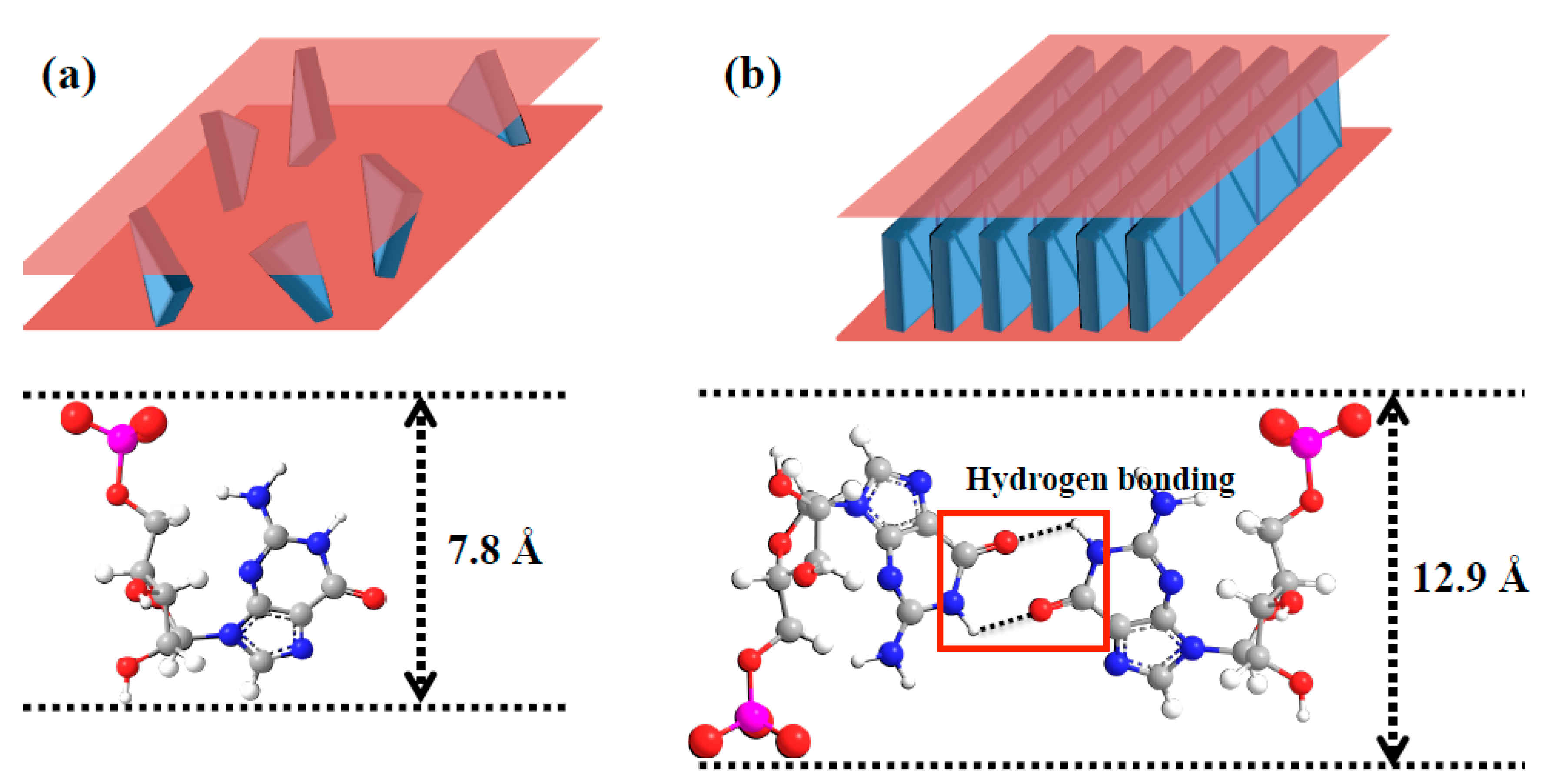
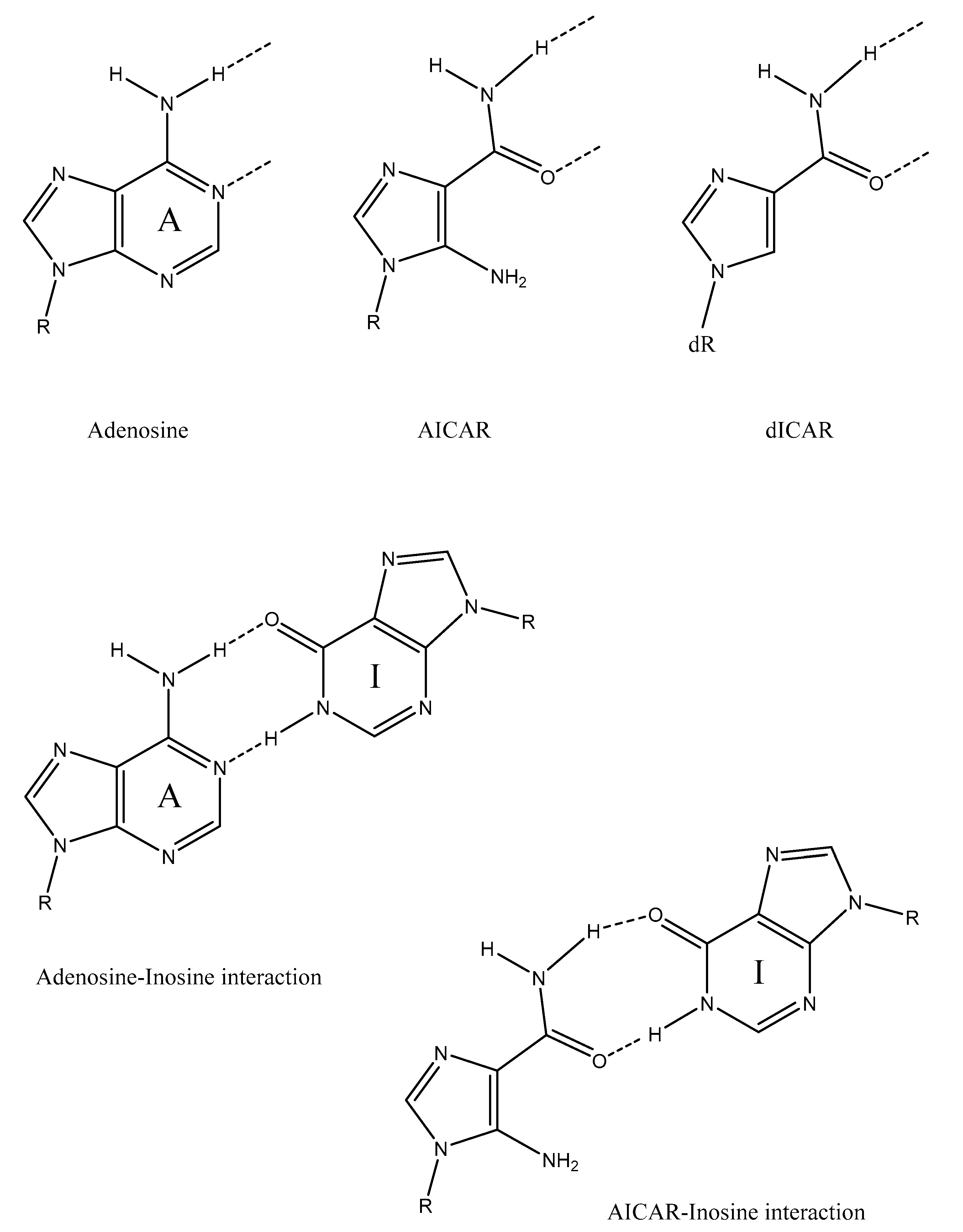
2. Hypothesis
- The demonstrated prebiotically plausible formation of the pentose–diphosphate backbone monomers; and, conversely, the absence of this for the RNA backbone subunit.
- The structural features of pentopyranose systems, for example the large angle of inclination between base pairs and backbone, that make them structurally suited for nonenzymatic replication using plausibly prebiotic 2′,3′-cyclic phosphate-activated monomers.
- The greater pairing strength of pentopyranose systems—important to counterbalance the initial weaker interactions between purine nucleobase precursors—to allow stable duplex formation and nonenzymatic replication.
- The fact that a pentopyranose system provides a—structural and possibly also catalytic—stepping stone to RNA, removing some of the difficulties in the RNA backbone arising de novo.
Acknowledgments
Conflicts of Interest
Abbreviations
| AICAR | 5-Aminoimidazole-4-carboxamide riboside |
| dICAR | 1-(2-Deoxy-β-d-ribofuranosyl)-imidazole-4-carboxamide |
| G2P | Glyceraldehyde-2-phosphate |
| GAP | Glycolaldehyde phosphate |
| GMP | Guanosine-5-monophosphate |
| LDH | Layered double hydroxide |
References
- Islam, S.; Powner, M.W. Prebiotic systems chemistry: Complexity overcoming clutter. Chem 2017, 2, 470–501. [Google Scholar] [CrossRef]
- Coggins, A.J.; Powner, M.W. Prebiotic synthesis of phosphoenol pyruvate by alpha-phosphorylation-controlled triose glycolysis. Nat. Chem. 2017, 9, 310–317. [Google Scholar] [CrossRef]
- Islam, S.; Bucar, D-K.; Powner, M.W. Prebiotic selection and assembly of proteinogenic amino acids and natural nucleotides from complex mixtures. Nat. Chem. 2017, 9, 584–589. [Google Scholar] [CrossRef]
- Krishnamurthy, R.; Pitsch, S.; Arrhenius, G. Mineral induced formation of pentose-2,4-bisphosphates. Orig. Life Evol. Biosph. 1999, 29, 139–152. [Google Scholar] [CrossRef]
- Pitsch, S.; Wendeborn, S.; Krishnamurthy, R.; Holzner, A.; Minton, M.; Bolli, M.; Miculca, C.; Windhab, N.; Micura, R.; Stanek, M.; et al. Pentopyranosyl oligonucleotide systems. 9th Communication. The β-D-ribopyranosyl-(2′→4’) oligonucleotide system (‘pyranosyl-RNA’): Synthesis and resumé of base-pairing properties. Helv. Chim. Acta 2003, 86, 4270–4363. [Google Scholar] [CrossRef]
- Jungmann, O.; Beier, M.; Luther, A.; Huynh, H.K.; Ebert, M.O.; Jaun, B.; Krishnamurthy, R.; Eschenmoser, A. Pentopyranosyl oligonucleotide systems. The α-L-arabinopyranosyl-(4’→2’)-oligonucleotide system: Synthesis and pairing properties. Helv. Chim. Acta 2003, 86, 1259–1308. [Google Scholar] [CrossRef]
- Eschenmoser, A. Etiology of potentially primordial biomolecular structures: From vitamin B12 to the nucleic acids and an inquiry into the chemistry of life’s origin: A retrospective. Angew. Chem. Int. Ed. 2011, 50, 12412–12472. [Google Scholar] [CrossRef] [PubMed]
- Mishra, G.; Dash, B.; Pandey, S. Layered double hydroxides: A brief review from fundamentals to application as evolving biomaterials. Appl. Clay Sci. 2018, 153, 172–186. [Google Scholar] [CrossRef]
- Kuma, K.; Paplawsky, W.; Gedulin, B.; Arrhenius, G. Mixed-valence hydroxides as bioorganic host minerals. Orig. Life Evol. Biosph. 1989, 19, 573–602. [Google Scholar] [CrossRef]
- Swadling, J.B.; Suter, J.L.; Greenwell, H.C.; Coveney, P.V. Influence of surface chemistry and charge on mineral–RNA interactions. Langmuir 2013, 29, 1573–1583. [Google Scholar] [CrossRef]
- Xu, Z.P.; Lu, G.Q. Layered double hydroxide nanomaterials as potential cellular drug delivery agents. Pure Appl. Chem. 2006, 78, 1771–1779. [Google Scholar] [CrossRef]
- Arrhenius, G.O. Crystals and life. Helv. Chim. Acta 2003, 86, 1569–1586. [Google Scholar] [CrossRef]
- Boclair, J.W.; Braterman, P.S.; Brister, B.D.; Jiang, J.; Lou, S.; Wang, Z.; Yarberry, F. Cyanide self-addition, controlled adsorption, and other processes at layered double hydroxides. Orig. Life Evol. Biosph. 2001, 31, 53–69. [Google Scholar] [CrossRef] [PubMed]
- Gwak, G.-H.; Kocsis, I.; Legrand, Y.-M.; Barboiu, M.; Oh, J.-M. Controlled supramolecular structure of guanosine monophosphate in the interlayer space of layered double hydroxide. Beilstein J. Nanotechnol. 2016, 7, 1928–1935. [Google Scholar] [CrossRef] [PubMed]
- Hazen, R.M.; Sverjensky, D.A.; Azzolini, D.; Bish, D.L.; Elmore, S.C.; Hinnov, L.; Milliken, R.E. Clay mineral evolution. Am. Mineral 2013, 98, 2007–2029. [Google Scholar] [CrossRef]
- Hazen, R.M. Paleomineralogy of the Hadean eon: A preliminary species list. Am. J. Sci. 2013, 313, 807–843. [Google Scholar] [CrossRef]
- Arrhenius, G.; Sales, B.; Mojzsis, S.; Lee, T. Entropy and charge in molecular evolution – the case of phosphate. J. Theor. Biol. 1997, 187, 503–522. [Google Scholar] [CrossRef] [PubMed]
- Russell, M.J. Green rust: The simple organizing ’seed’ of all life? Life 2018, 8, E35. [Google Scholar] [CrossRef]
- Powner, M.W.; Gerland, B.; Sutherland, J.D. Synthesis of activated pyrimidine ribonucleotides in prebiotically plausible conditions. Nature 2009, 459, 239–242. [Google Scholar] [CrossRef]
- Powner, M.W.; Sutherland, J.D.; Szostak, J.W. Chemoselective multicomponent one-pot assembly of purine precursors in water. J. Am. Chem. Soc. 2010, 132, 16677–16688. [Google Scholar] [CrossRef]
- Kim, H.J.; Benner, S.A. Prebiotic stereoselective synthesis of purine and noncanonical pyrimidine nucleotide from nucleobases and phosphorylated carbohydrates. Proc. Natl Acad. Sci. USA 2017, 114, 11315–11320. [Google Scholar] [CrossRef] [PubMed]
- Becker, S.; Thoma, I.; Deutsch, A.; Gehrke, T.; Mayer, P.; Zipse, H.; Carell, T. A high-yielding, strictly regioselective prebiotic purine nucleoside formation pathway. Science 2016, 352, 833–836. [Google Scholar] [CrossRef] [PubMed]
- Becker, S.; Schneider, C.; Okamura, H.; Crisp, A.; Amatov, T.; Dejmek, M.; Carell, T. Wet-dry cycles enable the parallel origin of canonical and non-canonical nucleosides by continuous synthesis. Nat. Commun. 2018, 9, 163. [Google Scholar] [CrossRef] [PubMed]
- Stairs, S.; Nikmal, A.; Bučar, D.K.; Zheng, S.L.; Szostak, J.W.; Powner, M.W. Divergent prebiotic synthesis of pyrimidine and 8-oxo-purine ribonucleotides. Nat. Commun. 2017, 8, 15270. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.C.; O’Flaherty, D.K.; Zhou, L.; Lelyveld, V.S.; Szostak, J.W. Inosine, but none of the 8-oxo-purines, is a plausible component of a primordial version of RNA. Proc. Natl. Acad. Sci. USA 2018, 115, 13318–13323. [Google Scholar] [CrossRef] [PubMed]
- Bernhardt, H.S.; Sandwick, R.K. Purine biosynthetic intermediate-containing ribose-phosphate polymers as evolutionary precursors to RNA. J. Mol. Evol. 2014, 79, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Morar, M.; Ealick, S.E. Structural biology of the purine biosynthetic pathway. Cell Mol. Life Sci. 2008, 65, 3699–3724. [Google Scholar] [CrossRef] [PubMed]
- de Duve, C. Blueprint for a Cell: The Nature and Origin of Life; Neil Patterson Publishers, Carolina Biological Supply Company: Burlington, NC, USA, 1991; pp. 133–134. [Google Scholar]
- Nierlich, D.P.; Magasanik, B. Phosphoribosylglycinamide synthetase of Aerobacter aerogenes. Purification and properties, and nonenzymatic formation of its substrate 5’-phosphoribosylamine. J. Biol. Chem. 1965, 240, 366–374. [Google Scholar] [PubMed]
- Mueller, E.J.; Meyer, E.; Rudolph, J.; Davisson, V.J.; Stubbe, J. N5-Carboxyaminoimidazole ribonucleotide: Evidence for a new intermediate and two new enzymatic activities in the de novo purine biosynthetic pathway of Escherichia coli. Biochemistry 1994, 33, 2269–2278. [Google Scholar] [CrossRef]
- Crick, F.H. The origin of the genetic code. J. Mol. Biol. 1968, 38, 367–379. [Google Scholar] [CrossRef]
- Wachtershauser, G. An all-purine precursor of nucleic acids. Proc. Natl. Acad. Sci. USA 1988, 85, 1134–1135. [Google Scholar] [CrossRef]
- Chandrasekhar, K.; Malathi, R. Non-Watson Crick base pairs might stabilize RNA structural motifs in ribozymes – a comparative study of group-I intron structures. J. Biosci. 2003, 28, 547–555. [Google Scholar] [CrossRef]
- Fedor, M.J.; Williamson, J.R. The catalytic diversity of RNAs. Nat. Rev. Mol. Cell Biol. 2005, 6, 399–412. [Google Scholar] [CrossRef]
- Bernhardt, H.S.; Tate, W.P. The transition from noncoded to coded protein synthesis: did coding mRNAs arise from stability-enhancing binding partners to tRNA? Biol. Direct 2010, 5, 16. [Google Scholar] [CrossRef]
- Krishnamurthy, R. On the emergence of RNA. Isr. J. Chem. 2015, 55, 837–850. [Google Scholar] [CrossRef]
- Grégoire, B.; Greenwell, H.C.; Fraser, D.G. Peptide formation on layered mineral surfaces: The key role of brucite-like minerals on the enhanced formation of alanine dipeptides. ACS Earth Space Chem. 2018, 2, 852–862. [Google Scholar] [CrossRef]
- Pitsch, S.; Krishnamurthy, R.; Arrhenius, G. Concentration of simple aldehydes by sulfite-containing double-layer hydroxide minerals: Implications for biopoesis. Helv. Chim. Acta 2000, 83, 2398–2411. [Google Scholar] [CrossRef]
- Ferris, J.P. Montmorillonite catalysis of 30-50 mer oligonucleotides: Laboratory demonstration of potential steps in the origin of the RNA world. Orig. Life Evol. Biosph. 2002, 32, 311–332. [Google Scholar] [CrossRef]
- Choy, J.-H.; Kwak, S.-Y.; Park, J.-S.; Jeong, Y.J.; Portier, J. Intercalative nanohybrids of nucleoside monophosphates and DNA in layered metal hydroxide. J. Am. Chem. Soc. 1999, 121, 1399–1400. [Google Scholar] [CrossRef]
- Aisawa, S.; Ohnuma, Y.; Hirose, K.; Takahashi, S.; Hirahara, H.; Narita, E. Intercalation of nucleotides into layered double hydroxides by ion-exchange reaction. Appl. Clay Sci. 2005, 28, 137–145. [Google Scholar] [CrossRef]
- Swadling, J.B.; Coveney, P.V.; Greenwell, H.C. Stability of free and mineral-protected nucleic acids: Implications for the RNA world. Geochim. Cosmochim. Acta 2012, 1, 360–378. [Google Scholar] [CrossRef]
- Jellicoe, T.C.; Fogg, A.M. Synthesis and characterization of layered double hydroxides intercalated with sugar phosphates. J. Phys. Chem. Solids 2012, 73, 1496–1499. [Google Scholar] [CrossRef]
- Engelhart, A.E.; Powner, M.W.; Szostak, J.W. Functional RNAs exhibit tolerance for non-heritable 2′-5′ versus 3′-5′ backbone heterogeneity. Nat. Chem. 2013, 5, 390–394. [Google Scholar] [CrossRef]
- Torres, A.G.; Piñeyro, D.; Filonava, L.; Stracker, T.H.; Batlle, E.; Ribas de Pouplana, L. A-to-I editing on tRNAs: Biochemical, biological and evolutionary implications. FEBS Lett. 2014, 588, 4279–4286. [Google Scholar] [CrossRef]
- Carter, R.J.; Baeyens, K.J.; SantaLucia, J.; Turner, D.H.; Holbrook, S.R. The crystal structure of an RNA oligomer incorporating tandem adenosine-inosine mismatches. Nucleic Acids Res. 1997, 25, 4117–4122. [Google Scholar] [CrossRef]
- Johnson, W.T.; Zhang, P.M.; Bergstrom, D.E. The synthesis and stability of oligodeoxyribonucleotides containing the deoxyadenosine mimic 1-(2′-deoxy-β-D-ribofuranosyl)imidazole-4-carboxamide. Nucleic Acids Res. 1997, 25, 559–567. [Google Scholar] [CrossRef]
- Sala, M.; Pezo, V.; Pochet, S.; Wain-Hobson, S. Ambiguous base pairing of the purine analogue 1-(2-deoxy-β-d-ribofuranosyl)-imidazole-4-carboxamide during PCR. Nucleic Acids Res. 1996, 24, 3302–3306. [Google Scholar] [CrossRef]
- Itoh, T.; Ohta, N.; Shichi, T.; Yui, T.; Takagi, K. The self-assembling properties of stearate ions in hydrotalcite clay. Langmuir 2003, 19, 9120–9126. [Google Scholar] [CrossRef]





© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernhardt, H.S. Making Molecules with Clay: Layered Double Hydroxides, Pentopyranose Nucleic Acids and the Origin of Life. Life 2019, 9, 19. https://doi.org/10.3390/life9010019
Bernhardt HS. Making Molecules with Clay: Layered Double Hydroxides, Pentopyranose Nucleic Acids and the Origin of Life. Life. 2019; 9(1):19. https://doi.org/10.3390/life9010019
Chicago/Turabian StyleBernhardt, Harold S. 2019. "Making Molecules with Clay: Layered Double Hydroxides, Pentopyranose Nucleic Acids and the Origin of Life" Life 9, no. 1: 19. https://doi.org/10.3390/life9010019
APA StyleBernhardt, H. S. (2019). Making Molecules with Clay: Layered Double Hydroxides, Pentopyranose Nucleic Acids and the Origin of Life. Life, 9(1), 19. https://doi.org/10.3390/life9010019