Microfluidic Reactors for Carbon Fixation under Ambient-Pressure Alkaline-Hydrothermal-Vent Conditions

 ,
,
Abstract
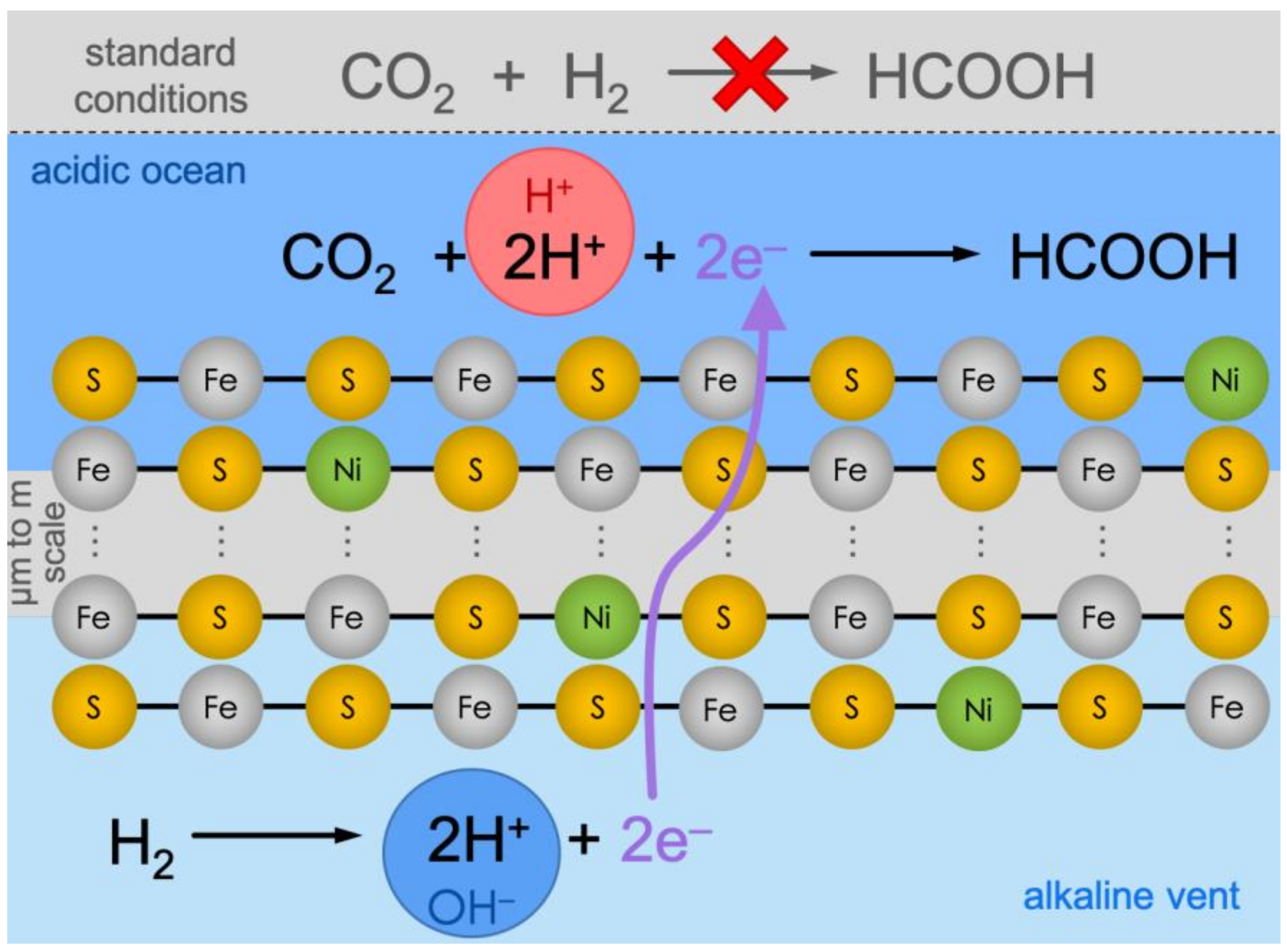
1. Introduction
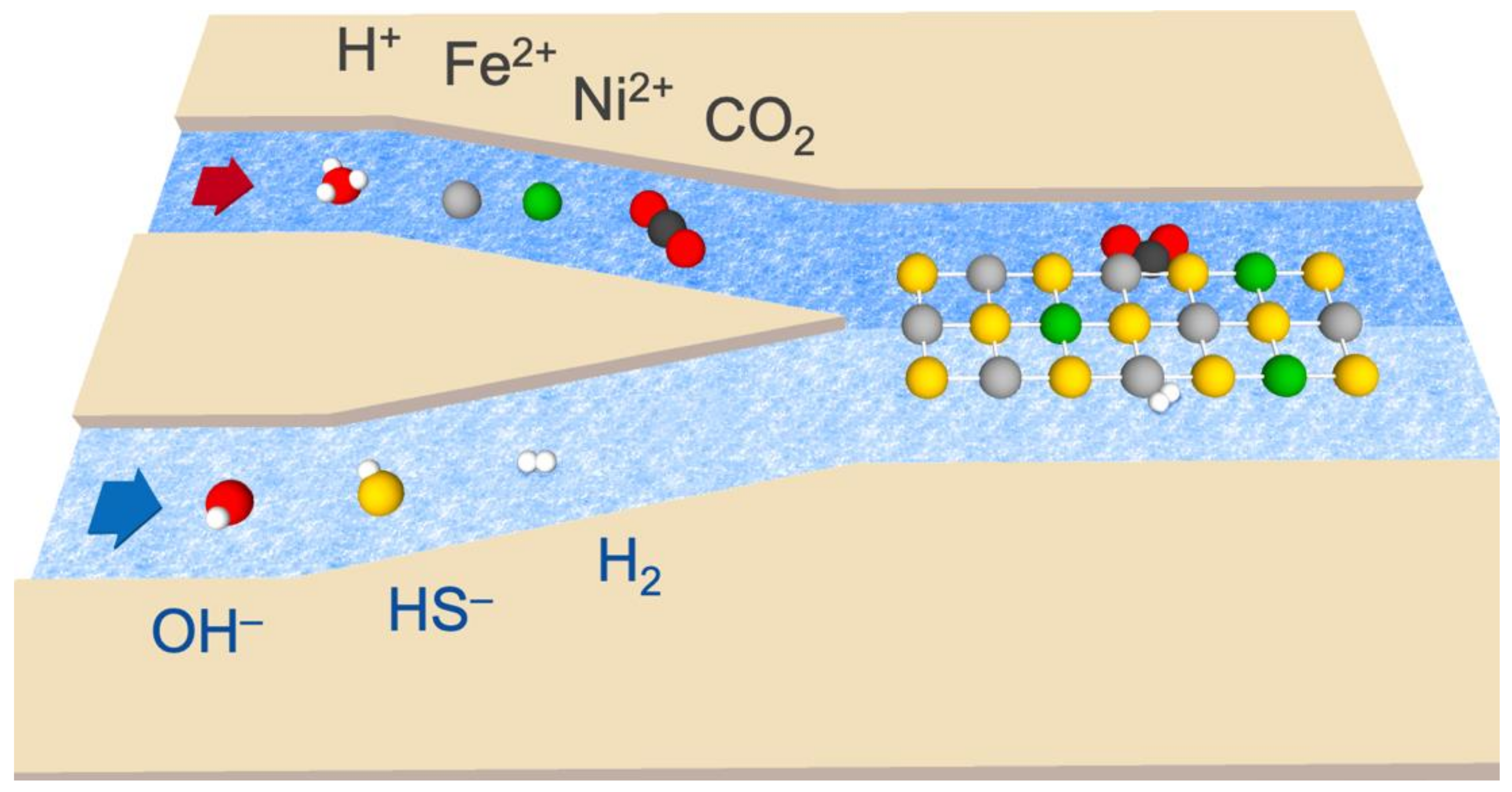
2. Methods
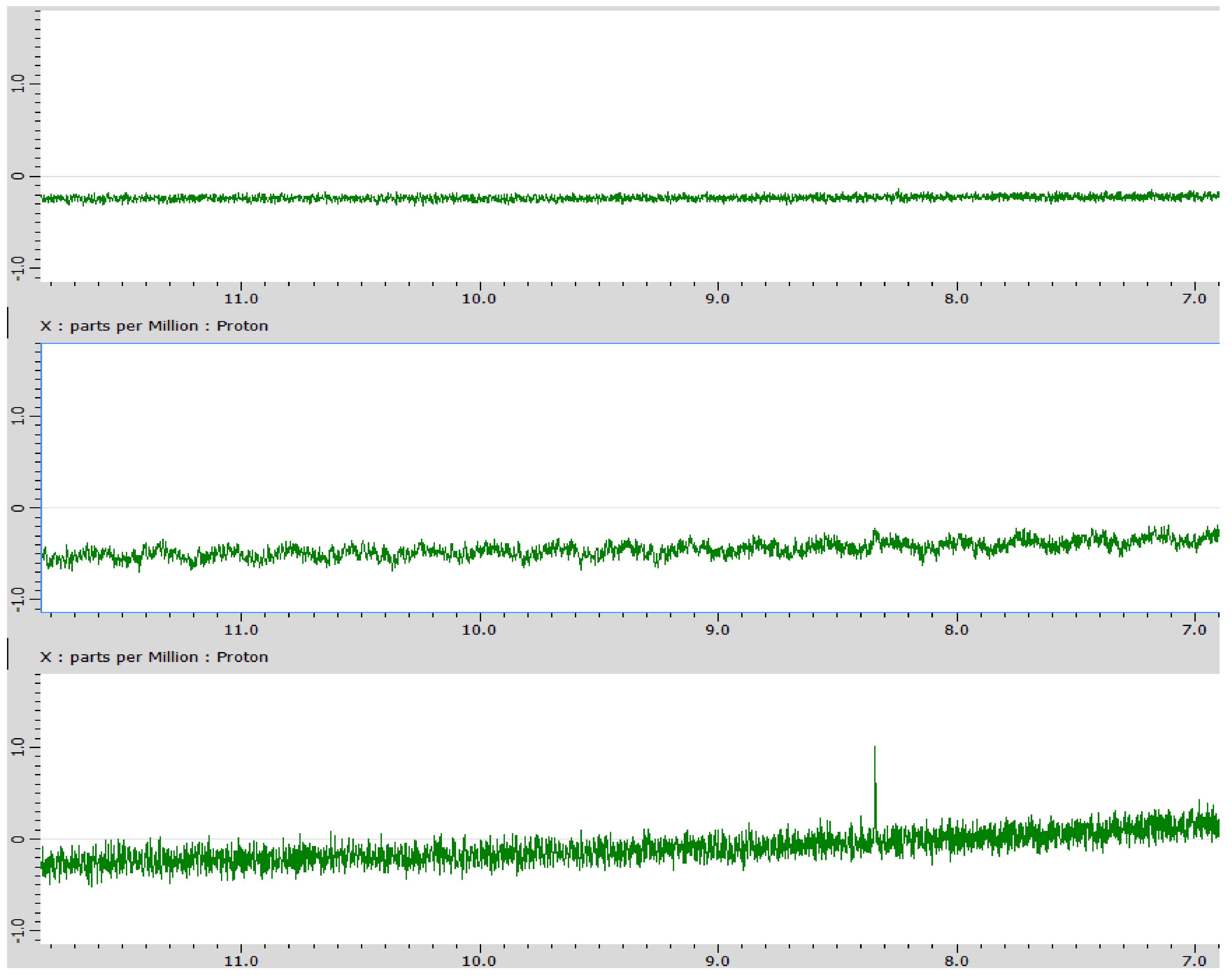
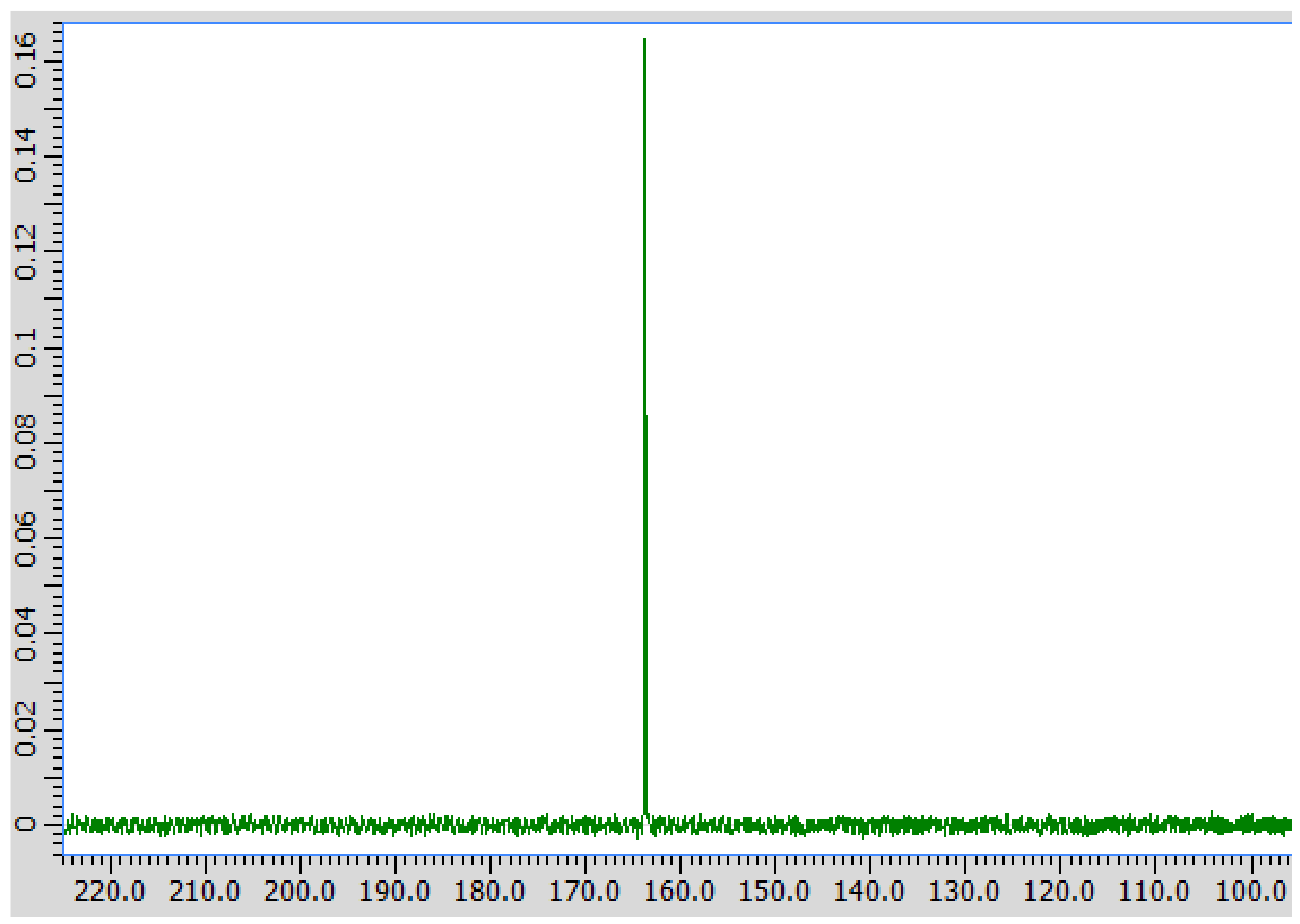
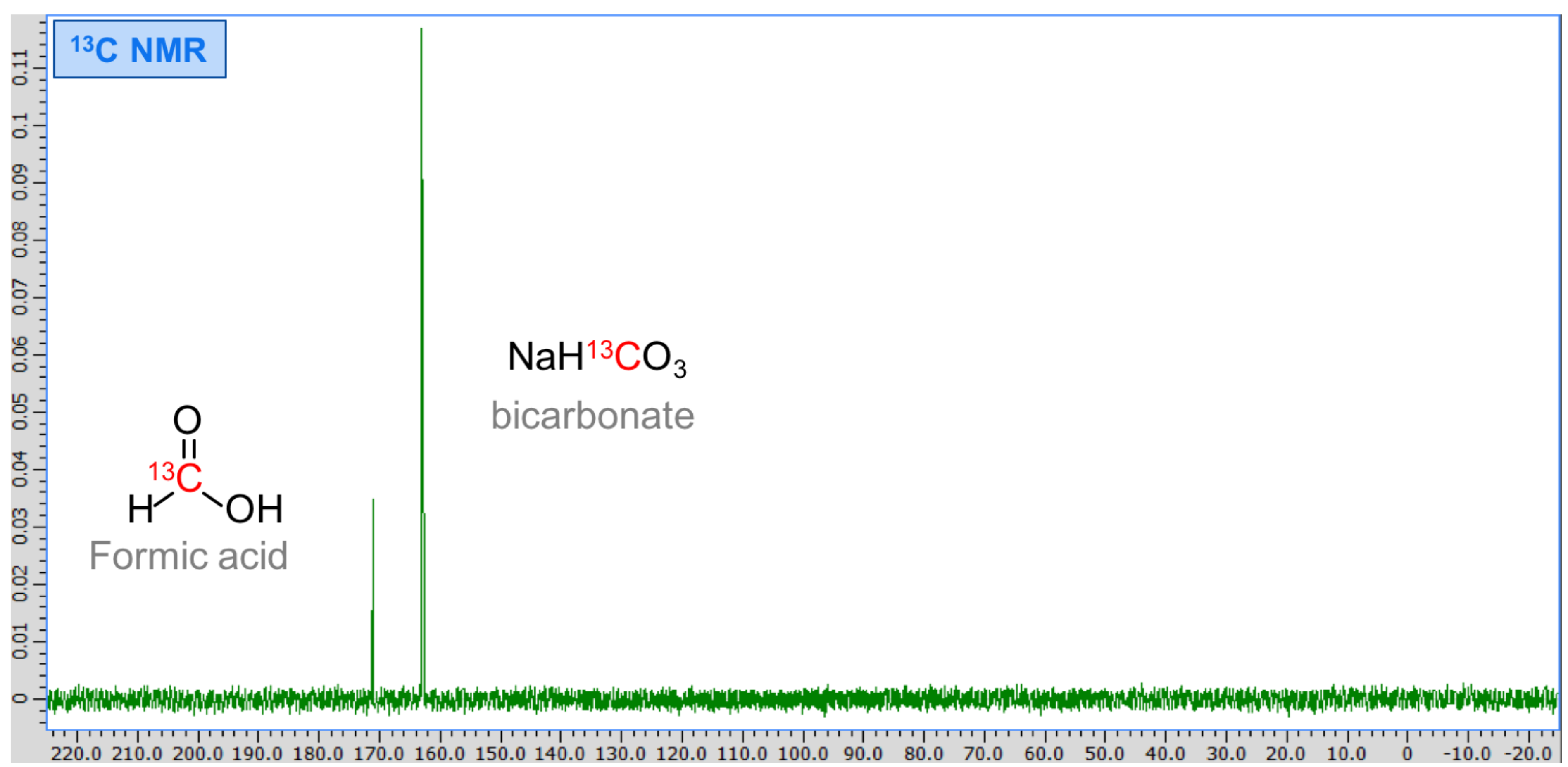
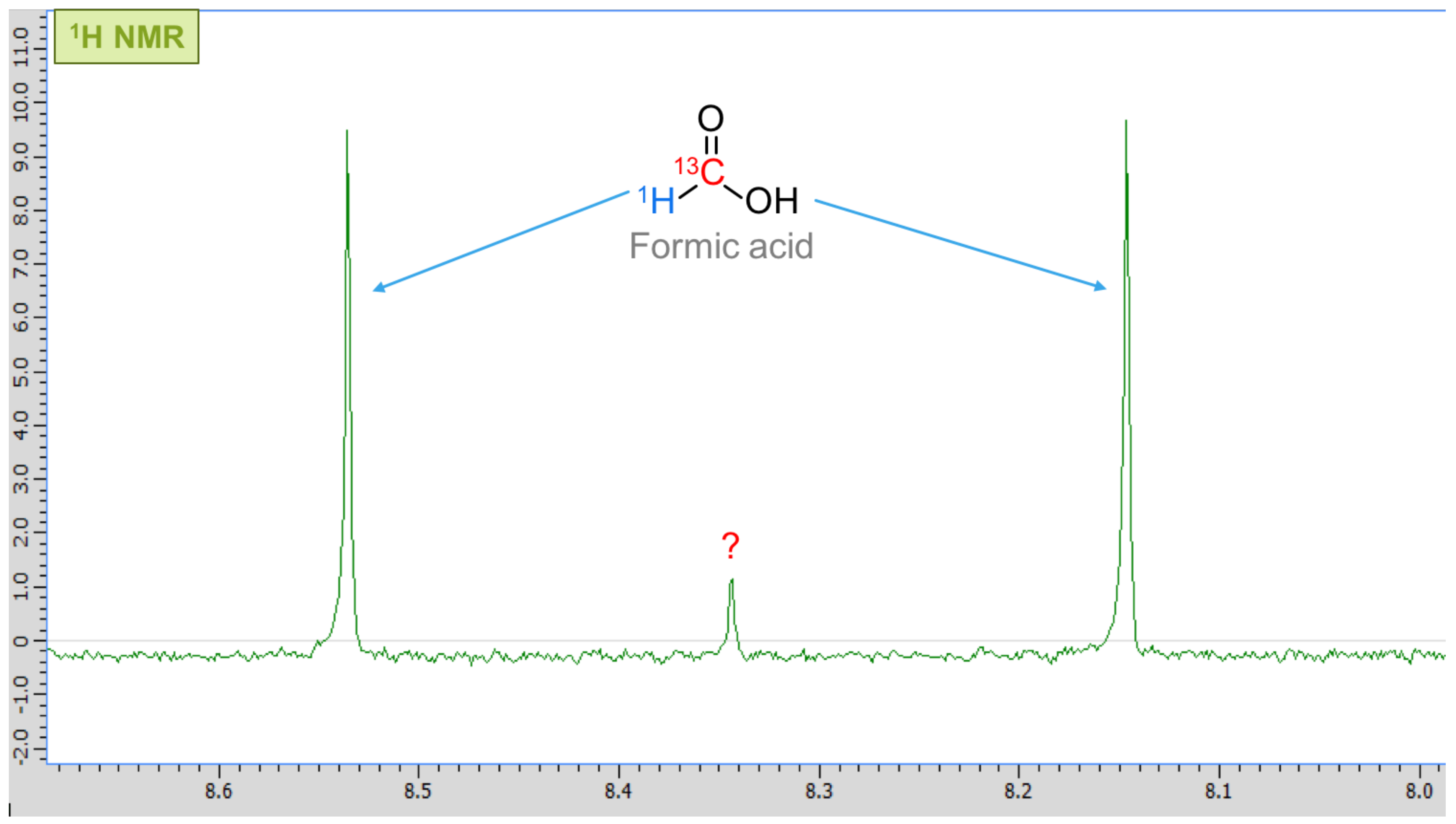
3. Results
- De-aerated water (to achieve parallel flow and take sample blanks).
- FeCl2 (50 mM) and NiCl2 (5 mM).
- De-aerated water bubbled with CO2 (at atmospheric pressure), or alternatively dissolved NaHCO3 (100 mM), acidified with HCl (1 M) to pH 6.
- De-aerated water.
- De-aerated water with Na2S (10 mM), K2HPO4 (10 mM), and Na2Si3O7 (10 mM), bubbled with H2 (at atmospheric pressure), and at a final pH of ~11.
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A




References
- Wächtershäuser, G. Pyrite formation, the first energy source for life: A hypothesis. Syst. Appl. Microbiol. 1988, 10, 207–210. [Google Scholar] [CrossRef]
- Russell, M.J.; Hall, A.J.; Turner, D. In vitro growth of iron sulphide chimneys: Possible culture chambers for origin-of-life experiments. Terra Nova 1989, 1, 238–241. [Google Scholar] [CrossRef]
- Wächtershäuser, G. Groundworks for an evolutionary biochemistry: The iron-sulphur world. Prog. Biophys. Mol. Biol. 1992, 58, 85–201. [Google Scholar] [CrossRef]
- Maden, B. No soup for starters? Autotrophy and the origins of metabolism. Trends Biochem. Sci. 1995, 20, 337–341. [Google Scholar] [CrossRef]
- Russell, M.J.; Martin, W. The rocky roots of the acetyl-CoA pathway. Trends Biochem. Sci. 2004, 29, 358–63. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.; Russell, M.J. On the origin of biochemistry at an alkaline hydrothermal vent. Phil. Trans. R. Soc. London B 2007, 362, 1887–1925. [Google Scholar] [CrossRef] [PubMed]
- Lane, N.; Martin, W.F. The origin of membrane bioenergetics. Cell 2012, 151, 1406–1416. [Google Scholar] [CrossRef]
- Sojo, V.; Herschy, B.; Whicher, A.; Camprubí, E.; Lane, N. The origin of life in alkaline hydrothermal vents. Astrobiology 2016, 16, 181–197. [Google Scholar] [CrossRef]
- Sleep, N.H.; Bird, D.K.; Pope, E.C. Serpentinite and the dawn of life. Phil. Trans. R. Soc. London B. Biol. Sci. 2011, 366, 2857–2869. [Google Scholar] [CrossRef]
- Herschy, B.; Whicher, A.; Camprubi, E.; Watson, C.; Dartnell, L.; Ward, J.; Evans, J.R.G.; Lane, N. An origin-of-life reactor to simulate alkaline hydrothermal vents. J. Mol. Evol. 2014, 79, 213–227. [Google Scholar] [CrossRef]
- Sleep, N.H.; Meibom, A.; Fridriksson, T.; Coleman, R.G.; Bird, D.K. H2-rich fluids from serpentinization: Geochemical and biotic implications. Proc. Natl. Acad. Sci. 2004, 101, 12818–12823. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.S.; Baross, J.A.; Delaney, J.R. Volcanoes, fluids, and life at mid-ocean ridge spreading centers. Annu. Rev. Earth Planet. Sci. 2002, 30, 385–491. [Google Scholar] [CrossRef]
- Kelley, D.S.; Karson, J.A.; Früh-Green, G.L.; Yoerger, D.R.; Shank, T.M.; Butterfield, D.A.; Hayes, J.M.; Schrenk, M.O.; Olson, E.J.; Proskurowski, G.; et al. A serpentinite-hosted ecosystem: The Lost City hydrothermal field. Science 2005, 307, 1428–1434. [Google Scholar] [CrossRef] [PubMed]
- Wood, H.G. Life with CO or CO2 and H2 as a source of carbon and energy. FASEB J. 1991, 5, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Nitschke, W.; Russell, M. Beating the acetyl coenzyme A-pathway to the origin of life. Phil. Trans. R. Soc. B. 2013, 368, 20120258. [Google Scholar] [CrossRef] [PubMed]
- Lane, N.; Allen, J.F.; Martin, W. How did LUCA make a living? Chemiosmosis in the origin of life. BioEssays 2010, 32, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Poehlein, A.; Schmidt, S.; Kaster, A.-K.; Goenrich, M.; Vollmers, J.; Thürmer, A.; Bertsch, J.; Schuchmann, K.; Voigt, B.; Hecker, M.; et al. An ancient pathway combining carbon dioxide fixation with the generation and utilization of a sodium ion gradient for ATP synthesis. PLoS ONE 2012, 7, e33439. [Google Scholar] [CrossRef] [PubMed]
- Maden, B.E. Tetrahydrofolate and tetrahydromethanopterin compared: Functionally distinct carriers in C1 metabolism. Biochem. J. 2000, 350, 609–629. [Google Scholar] [CrossRef]
- Yamaguchi, A.; Yamamoto, M.; Takai, K.; Ishii, T.; Hashimoto, K.; Nakamura, R. Electrochemical CO2 reduction by Ni-containing iron sulfides: How is CO2 electrochemically reduced at bisulfide-bearing deep-sea hydrothermal precipitates? Electrochim. Acta 2014, 141, 311–318. [Google Scholar] [CrossRef]
- Lane, N. The Vital Question: Energy, Evolution, and the Origins of Complex Life; WW Norton & Company: New York, NY, USA, 2015. [Google Scholar]
- Li, Y.; Kitadai, N.; Nakamura, R. Chemical diversity of metal sulfide minerals and its implications for the origin of life. Life 2018, 8, 1–26. [Google Scholar] [CrossRef]
- Martin, W.; Baross, J.; Kelley, D.; Russell, M.J. Hydrothermal vents and the origin of life. Nat. Rev. Microbiol. 2008, 6, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Sojo, V.; Pomiankowski, A.; Lane, N. A bioenergetic basis for membrane divergence in archaea and bacteria. PLoS Biol. 2014, 12, e1001926. [Google Scholar] [CrossRef] [PubMed]
- Lane, N. Bioenergetic constraints on the evolution of complex life. Cold Spring Harb. Perspect. Biol. 2014, 6, a015982. [Google Scholar] [CrossRef]
- Möller, F.M.F.M.; Kriegel, F.; Kieß, M.; Sojo, V.; Braun, D. Steep pH Gradients and Directed Colloid Transport in a Microfluidic Alkaline Hydrothermal Pore. Angew. Chemie Int. Ed. 2017, 56, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.M.A.; Ohno, A.; Sato, T.; Uozumi, Y. Instantaneous click chemistry by a copper-containing polymeric-membrane-installed microflow catalytic reactor. Chem. A Eur. J. 2015, 21, 17269–17273. [Google Scholar] [CrossRef]
- Batista, B.C.; Steinbock, O. Growing inorganic membranes in microfluidic devices: Chemical gardens reduced to linear walls. J. Phys. Chem. C 2015, 119, 27045–27052. [Google Scholar] [CrossRef]
- Pray, H.A.; Schweickert, C.E.; Minnich, B.H. Solubility of hydrogen, oxygen, nitrogen, and helium in water at elevated temperatures. Ind. Eng. Chem. 1952, 44, 1146–1151. [Google Scholar] [CrossRef]
- Kitadai, N.; Nakamura, R.; Yamamoto, M.; Takai, K.; Li, Y.; Yamaguchi, A.; Gilbert, A.; Ueno, Y.; Yoshida, N.; Oono, Y. Geoelectrochemical CO production: Implications for the autotrophic origin of life. Sci. Adv. 2018, 4, eaao7265. [Google Scholar] [CrossRef]
- Ooka, H.; Mcglynn, S.E.; Nakamura, R. Electrochemistry at deep-sea hydrothermal vents: Utilization of the thermodynamic driving force towards the autotrophic origin of life. ChemElectroChem 2018. [Google Scholar] [CrossRef]
- Muchowska, K.B.; Varma, S.J.; Chevallot-Beroux, E.; Lethuillier-Karl, L.; Li, G.; Moran, J. Metals promote sequences of the reverse Krebs cycle. Nat. Ecol. Evol. 2017, 1, 1716–1721. [Google Scholar] [CrossRef]
- Varma, S.J.; Muchowska, K.B.; Chatelain, P.; Moran, J. Native iron reduces CO2 to intermediates and end-products of the acetyl-CoA pathway. Nat. Ecol. Evol. 2018, 2, 1019–1024. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.B. Natural pH gradients in hydrothermal alkali vents were unlikely to have played a role in the origin of life. J. Mol. Evol. 2016, 83, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wächtershäuser, G. In praise of error. J. Mol. Evol. 2016, 82, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, J.D. Studies on the origin of life—the end of the beginning. Nat. Rev. Chem. 2017, 1, 0012. [Google Scholar] [CrossRef]
- Lane, N. Proton gradients at the origin of life. BioEssays 2017, 39, 1600217. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ACIDIC-SIDE CONCENTRATIONS | ALKALINE-SIDE CONCENTRATIONS | ||
|---|---|---|---|
| [FeCl2] | 50 mM | [Na2S] | 10 and 100 mM |
| [NiCl2] | 0 and 10 mM | [K2HPO4] | 10 mM |
| [NaHCO3] | 10, 50, and 100 mM (acidified to pH ~6) | [Na2Si3O7] | 0 and 10 mM |
| [Na2MoO4] | 0 and 1 mM | ||
| CO2 | Bubbled at atmospheric pressure (final pH ~6) | [H2] | Bubbled at atmospheric pressure (final pH ~11) |
| OTHER CONDITIONS | |||
| Reaction durations | ½, 1, 2, 5, 12, and 24 h | Temperature | ~25 (room), 40, 50, 60, and 70 °C |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sojo, V.; Ohno, A.; McGlynn, S.E.; Yamada, Y.M.A.; Nakamura, R. Microfluidic Reactors for Carbon Fixation under Ambient-Pressure Alkaline-Hydrothermal-Vent Conditions. Life 2019, 9, 16. https://doi.org/10.3390/life9010016
Sojo V, Ohno A, McGlynn SE, Yamada YMA, Nakamura R. Microfluidic Reactors for Carbon Fixation under Ambient-Pressure Alkaline-Hydrothermal-Vent Conditions. Life. 2019; 9(1):16. https://doi.org/10.3390/life9010016
Chicago/Turabian StyleSojo, Victor, Aya Ohno, Shawn E. McGlynn, Yoichi M.A. Yamada, and Ryuhei Nakamura. 2019. "Microfluidic Reactors for Carbon Fixation under Ambient-Pressure Alkaline-Hydrothermal-Vent Conditions" Life 9, no. 1: 16. https://doi.org/10.3390/life9010016
APA StyleSojo, V., Ohno, A., McGlynn, S. E., Yamada, Y. M. A., & Nakamura, R. (2019). Microfluidic Reactors for Carbon Fixation under Ambient-Pressure Alkaline-Hydrothermal-Vent Conditions. Life, 9(1), 16. https://doi.org/10.3390/life9010016