Serpentinization: Connecting Geochemistry, Ancient Metabolism and Industrial Hydrogenation

 ,
,  ,
,  ,
, 
Abstract
1. Abiotic Chemical Synthesis at Hydrothermal Vents
2. The Early Earth: Magma, then Crust, then Oceans
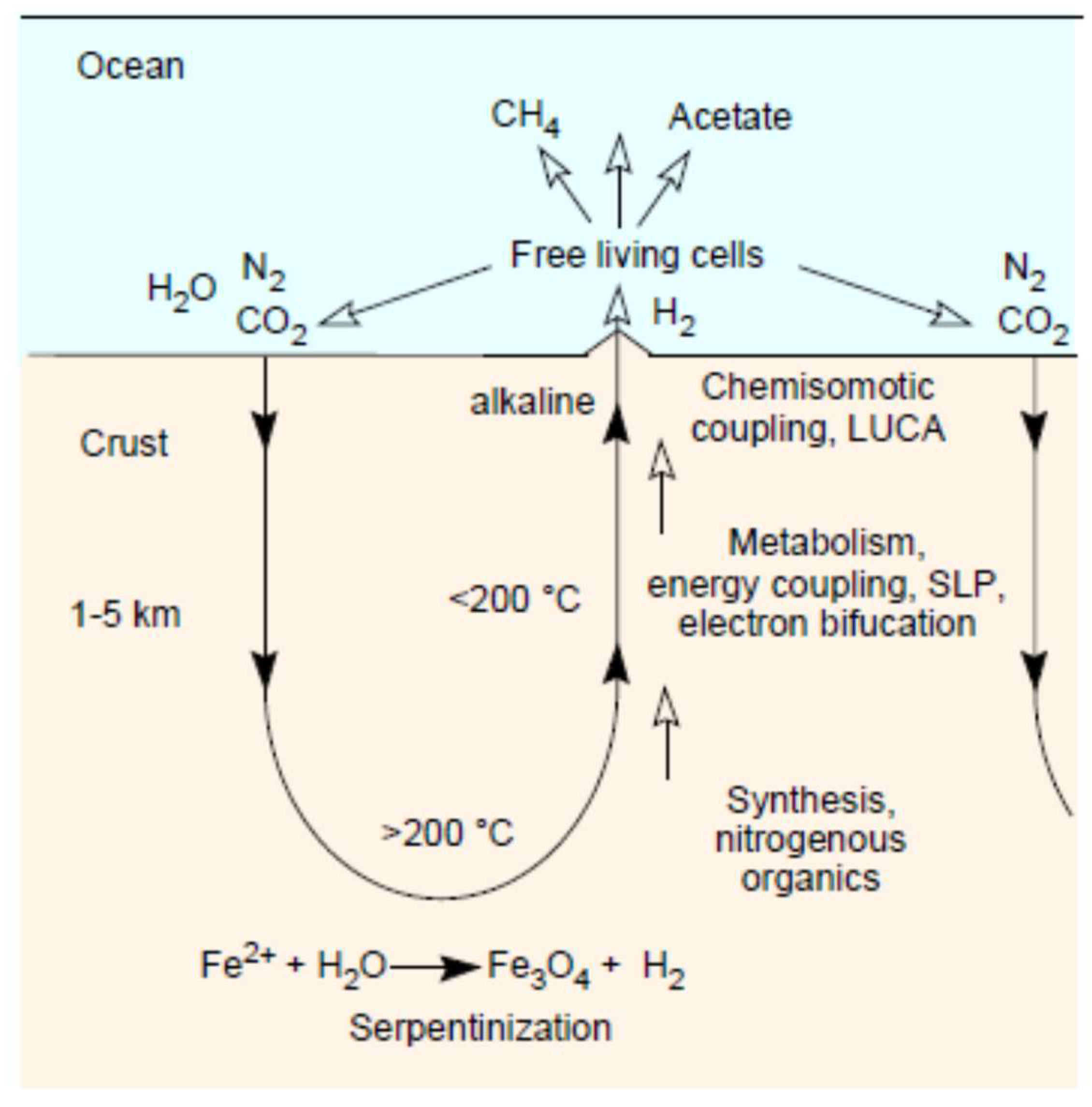
3. Serpentinization: Rock–Water Interactions
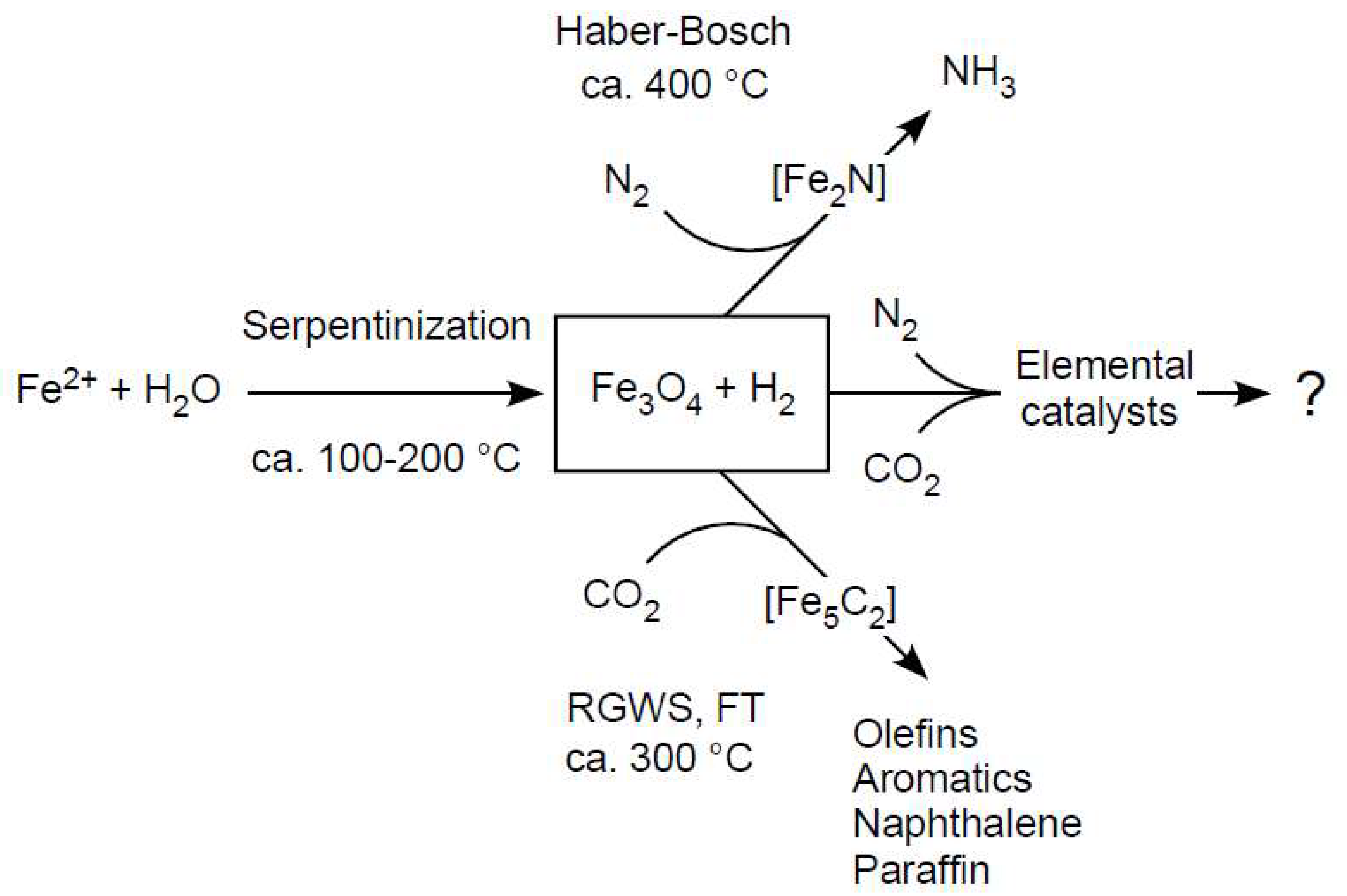
4. Serpentinization: Awaruite and Carbon
5. Serpentinization: Methane
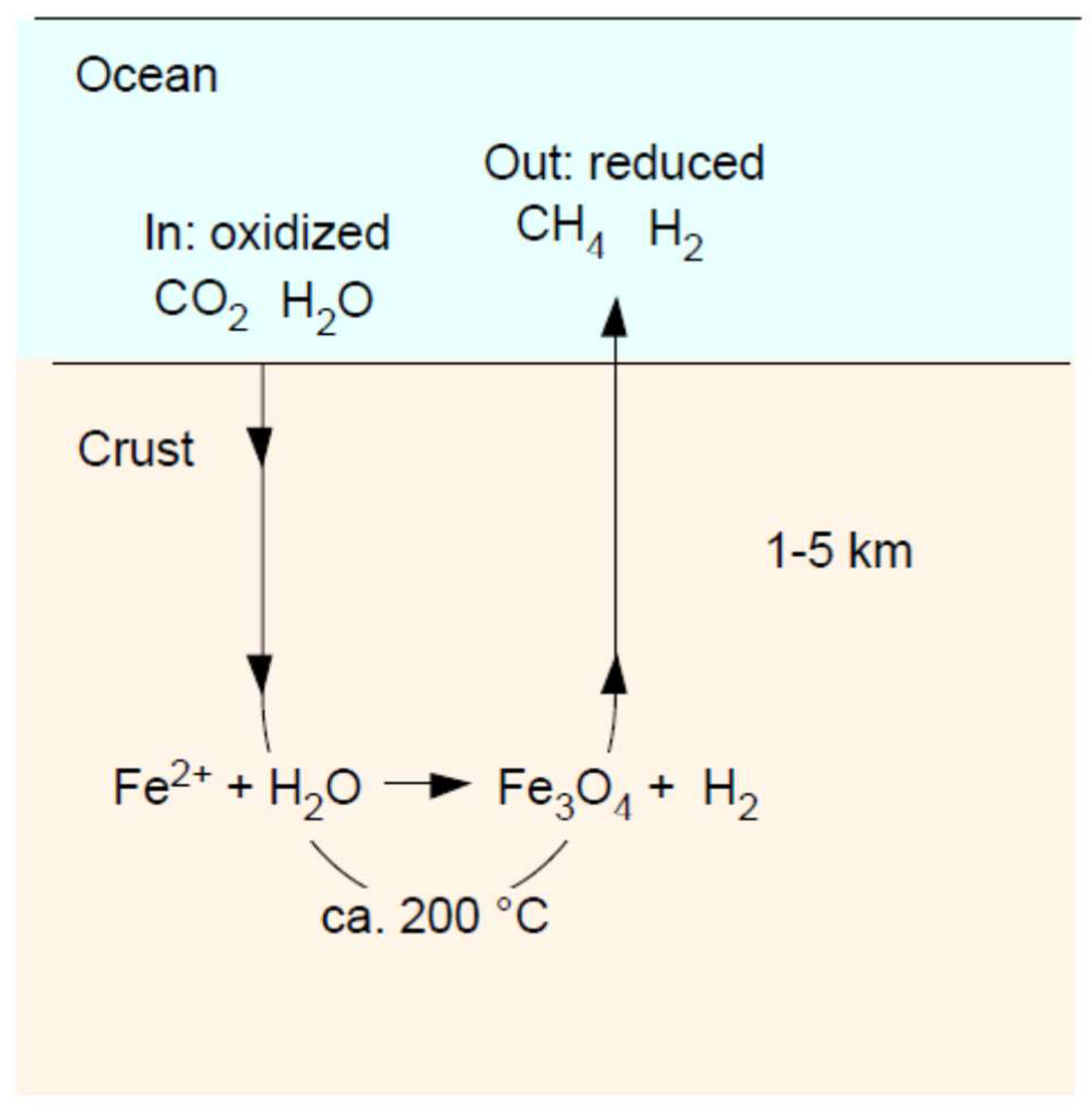
6. Serpentinization: Magnetite (Fe3O4)
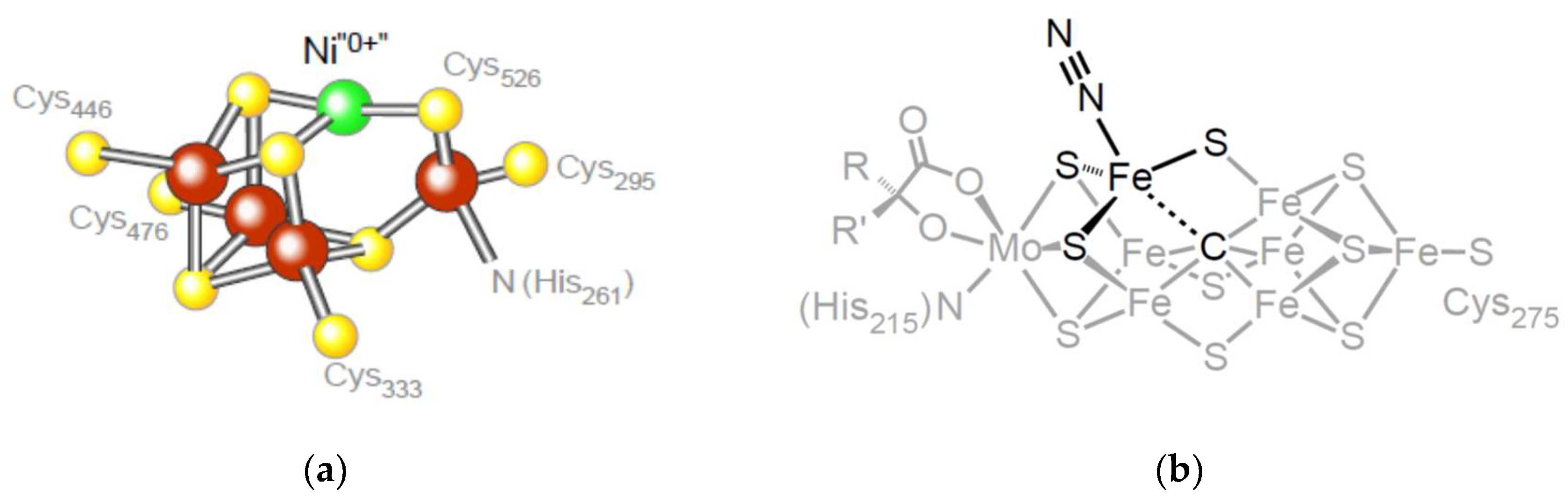
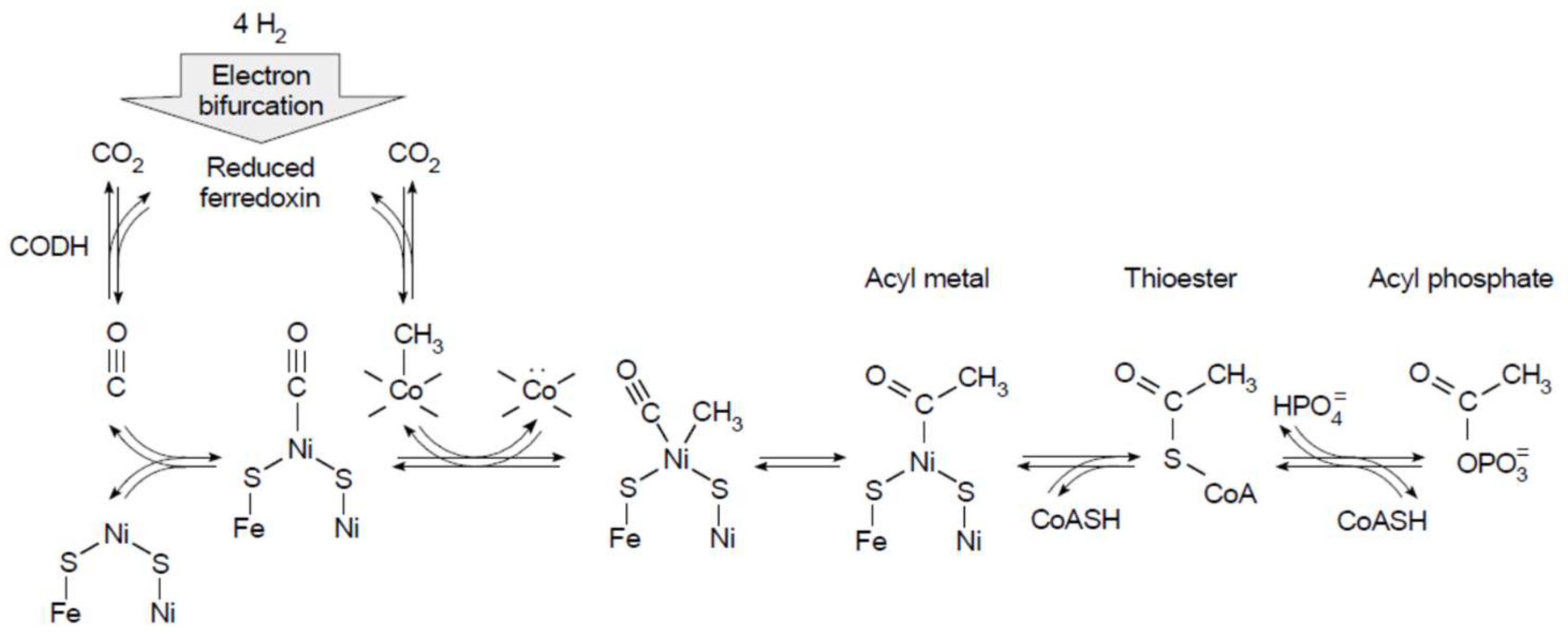
7. Conserved Relicts in Metabolism: The Ni0 in CODH
8. Conserved Relicts in Metabolism: The Carbide in Nitrogenase
9. Weighing in on Caveats
10. Do Genomes Help?
11. What Next?
Funding
Acknowledgments
Conflicts of Interest
References
- Corliss, J.B.; Baross, J.A.; Hoffman, S.E. A hypothesis concerning the relationships between submarine hot springs and the origin of life on earth. Oceanol. Acta 1981, 59–69. Available online: http://archimer.ifremer.fr/doc/00245/35661/34170.pdf (accessed on 22 September 2018).
- Baross, J.A.; Hoffman, S.E. Submarine hydrothermal vents and associated gradient environments as sites for the origin and evolution of life. Orig. Life Evol. Biosph. 1985, 15, 327–345. [Google Scholar] [CrossRef]
- Holm, N.G. Why are Hydrothermal Systems Proposed as Plausible Environments for the Origin of Life? In Marine Hydrothermal Systems and the Origin of Life; Holm, N.G., Ed.; Springer: Dordrecht, The Netherlands, 1992; pp. 5–14. [Google Scholar]
- Nisbet, E.; Sleep, N.H. The habitat and nature of early life. Nature 2001, 409, 1083–1091. [Google Scholar] [CrossRef] [PubMed]
- McCollom, T.M.; Seewald, J.S. Serpentinites, hydrogen, and life. Elements 2013, 9, 129–134. [Google Scholar] [CrossRef]
- Russell, M.J.; Hall, A.J. The emergence of life from iron monosulphide bubbles at a submarine hydrothermal redox and pH front. J. Geol. Soc. 1997, 154, 377–402. [Google Scholar] [CrossRef]
- Kelley, D.S.; Baross, J.A.; Delaney, J.R. Volcanoes, fluids, and life at mid-ocean ridge spreading centers. Annu. Rev. Earth Planet. Sci. 2002, 30, 385–491. [Google Scholar] [CrossRef]
- Freeland, S.J.; Hurst, L.D. The genetic code is one in a million. J. Mol. Evol. 1998, 47, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Tashiro, T.; Ishida, A.; Hori, M.; Igisu, M.; Koike, M.; Méjean, P.; Takahata, N.; Sano, Y.; Komiya, T. Early trace of life from 3.95 Ga sedimentary rocks in Labrador, Canada. Nature 2017, 549, 516–518. [Google Scholar] [CrossRef] [PubMed]
- Ueno, Y.; Yamada, K.; Yoshida, N.; Maruyama, S.; Isozaki, Y. Evidence from fluid inclusions for microbial methanogenesis in the early Archaean era. Nature 2006, 440, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Cleaves, H.J., II. The prebiotic geochemistry of formaldehyde. Precambrian Res. 2008, 164, 111–118. [Google Scholar] [CrossRef]
- Patel, B.H.; Percivalle, C.; Ritson, D.J.; Duffy, C.D.; Sutherland, J.D. Common origins of RNA, protein and lipid precursors in a cyanosulfidic protometabolism. Nat. Chem. 2015, 7, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Russell, M.J.; Nitschke, W.; Branscomb, E. The inevitable journey to being. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20120254. [Google Scholar] [CrossRef] [PubMed]
- Cockell, C.S. The origin and emergence of life under impact bombardment. Philos. Trans. R. Soc. B 2006, 362, 1845–1856. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y.; Sekine, T.; Oba, M.; Kakegawa, T.; Nakazawa, H. Biomolecule formation by oceanic impacts on early Earth. Nat. Geosci. 2009, 2, 62–66. [Google Scholar] [CrossRef]
- Russell, M. First Life. Am. Sci. 2006, 94, 32–39. [Google Scholar] [CrossRef]
- Jackson, J.B. Natural pH gradients in hydrothermal alkali vents were unlikely to have played a role in the origin of life. J. Mol. Evol. 2016, 83, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lane, N. Proton gradients at the origin of life. BioEssays 2017, 39, 1600217. [Google Scholar] [CrossRef] [PubMed]
- Shock, E.L.; Helgeson, H.C. Calculation of the thermodynamic and transport properties of aqueous species at high pressures and temperatures: Correlation algorithms for ionic species and equation of state predictions to 5 kb and 1000 °C. Geochim. Cosmochim. Acta 1988, 52, 2009–2036. [Google Scholar] [CrossRef]
- Shock, E.L. Geochemical constraints on the origin of organic compounds in hydrothermal systems. Orig. Life Evol. Biosph. 1990, 20, 331–367. [Google Scholar] [CrossRef]
- Konn, C.; Charlou, J.L.; Holm, N.G.; Mousis, O. The production of methane, hydrogen, and organic compounds in ultramafic-hosted hydrothermal vents of the Mid-Atlantic Ridge. Astrobiology 2015, 15, 381–399. [Google Scholar] [CrossRef] [PubMed]
- McCollom, T.M.; Sherwood Lollar, B.; Lacrampe-Couloume, G.; Seewald, J.S. The influence of carbon source on abiotic organic synthesis and carbon isotope fractionation under hydrothermal conditions. Geochim. Cosmochim. Acta 2010, 74, 2717–2740. [Google Scholar] [CrossRef]
- Schrenk, M.O.; Brazelton, W.J.; Lang, S.Q. Serpentinization, carbon, and deep life. Rev. Mineral. Geochem. 2013, 75, 575–606. [Google Scholar] [CrossRef]
- Martin, W.; Russell, M.J. On the origin of biochemistry at an alkaline hydrothermal vent. Philos. Trans. R. Soc. B Biol. Sci. 2007, 362, 1887–1925. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.; Baross, J.; Kelley, D.; Russell, M.J. Hydrothermal vents and the origin of life. Nat. Rev. Microbiol. 2008, 6, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Ragsdale, S.W. Nickel-based enzyme systems. J. Biol. Chem. 2009, 284, 18571–18575. [Google Scholar] [CrossRef] [PubMed]
- Can, M.; Armstrong, F.A.; Ragsdale, S.W. Structure, function, and mechanism of the nickel metalloenzyme, CO dehydrogenase, and acetyl-CoA synthase. Bioinorg. Enzymol. 2014, 114, 4149–4174. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, K.M.; Roemelt, M.; Ettenhuber, P.; Hu, Y.; Ribbe, M.W.; Neese, F.; Bergmann, U.; DeBeer, S. X-ray emission spectroscopy evidences a central carbon in the nitrogenase iron-molybdenum cofactor. Science 2011, 334, 974–977. [Google Scholar] [CrossRef] [PubMed]
- Spatzal, T.; Aksoyoglu, M.; Zhang, L.; Andrade, S.L.A.; Schleicher, E.; Weber, S.; Rees, D.C.; Einsle, O. Evidence for interstitial carbon in nitrogenase FeMo cofactor. Science 2011, 334, 940. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Ge, Q.; Yao, R.; Wen, Z.; Fang, C.; Guo, L.; Xu, H.; Sun, J. Directly converting CO2 into a gasoline fuel. Nat. Commun. 2017, 8, 15174. [Google Scholar] [CrossRef] [PubMed]
- Ertl, G. Primary steps in catalytic synthesis of ammonia. J. Vac. Sci. Technol. A Vac. Surf. Films 1983, 1, 1247–1253. [Google Scholar] [CrossRef]
- Kandemir, T.; Schuster, M.E.; Senyshyn, A.; Behrens, M.; Schlögl, R. The Haber–Bosch process revisited: On the real structure and stability of “ammonia iron” under working conditions. Angew. Chem. Int. Ed. 2013, 52, 12723–12726. [Google Scholar] [CrossRef] [PubMed]
- McCollom, T.M.; Seewald, J.S. Abiotic synthesis of organic compounds in deep-sea hydrothermal environments. Chem. Rev. 2007, 107, 382–401. [Google Scholar] [CrossRef] [PubMed]
- Hennet, R.J.-C.; Holm, N.G.; Engel, M.H. Abiotic synthesis of amino acids under hydrothermal conditions and the origin of life: A perpetual phenomenon? Naturwissenschaften 1992, 79, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Shock, E.; Schulte, M.D. Organic synthesis during fluid mixing in hydrothermal systems. J. Geophys. Res. 1998, 103, 28513–28527. [Google Scholar] [CrossRef]
- Amend, J.P.; Shock, E.L. Energetics of amino acid synthesis in hydrothermal ecosystems. Science 1998, 281, 1659–1662. [Google Scholar] [CrossRef] [PubMed]
- Amend, J.P.; McCollom, T.M. Energetics of biomolecule synthesis on early Earth. In Chemical Evolution II: From the Origins of Life to Modern Soc; ACS Publications: Washington, DC, USA, 2009; pp. 63–94. [Google Scholar]
- Sleep, N.H.; Bird, D.K.; Pope, E.C. Serpentinite and the dawn of life. Philos. Trans. R. Soc. B Biol. Sci. 2011, 366, 2857–2869. [Google Scholar] [CrossRef] [PubMed]
- Arndt, N.T.; Nisbet, E.G. Processes on the young Earth and the habitats of early life. Annu. Rev. Earth Planet. Sci. 2012, 40, 521–549. [Google Scholar] [CrossRef]
- Zahnle, K.; Arndt, N.; Cockell, C.; Halliday, A.; Nisbet, E.; Selsis, F.; Sleep, N.H. Emergence of a habitable planet. Space Sci. Rev. 2007, 129, 35–78. [Google Scholar] [CrossRef]
- Dasgupta, R. Ingassing, storage, and outgassing of terrestrial carbon through geologic time. Rev. Mineral. Geochem. 2013, 75, 183–229. [Google Scholar] [CrossRef]
- Etiope, G.; Schoell, M. Abiotic gas: Atypical, but not rare. Elements 2014, 10, 291–296. [Google Scholar] [CrossRef]
- Bottke, W.F.; Norman, M.D. The late heavy bombardment. Annu. Rev. Earth Planet. Sci. 2017, 45, 619–647. [Google Scholar] [CrossRef]
- Litasov, K.D.; Shatskiy, A.F. Composition of the Earth’s core: A review. Russ. Geol. Geophys. 2016, 57, 22–46. [Google Scholar] [CrossRef]
- Kasting, J.F.; Holm, N.G. What determines the volume of the oceans? Earth Planet. Sci. Lett. 1992, 109, 507–515. [Google Scholar] [CrossRef]
- Westall, F.; Brack, A. The importance of water for life. Space Sci. Rev. 2018, 214, 1–23. [Google Scholar] [CrossRef]
- Korenaga, J.; Planavsky, N.J.; Evans, D.A.D. Global water cycle and the coevolution of the Earth’s interior and surface environment. Philos. Trans. R. Soc. A 2017, 375, 20150393. [Google Scholar] [CrossRef] [PubMed]
- McCollom, T.M.; Bach, W. Thermodynamic constraints on hydrogen generation during serpentinization of ultramafic rocks. Geochim. Cosmochim. Acta 2009, 73, 856–875. [Google Scholar] [CrossRef]
- Klein, F.; Bach, W.; McCollom, T.M. Compositional controls on hydrogen generation during serpentinization of ultramafic rocks. Lithos 2013, 178, 55–69. [Google Scholar] [CrossRef]
- Holm, N.G. Hydrothermal activity and the volume of the oceans. Deep. Res. Part II Top. Stud. Oceanogr. 1996, 43, 47–52. [Google Scholar] [CrossRef]
- Russell, M.J.; Hall, A.J.; Martin, W. Serpentinization as a source of energy at the origin of life. Geobiology 2010, 8, 355–371. [Google Scholar] [CrossRef] [PubMed]
- Holm, N.G.; Oze, C.; Mousis, O.; Waite, J.H.; Guilbert-Lepoutre, A. Serpentinization and the formation of H2 and CH4 on celestial bodies (planets, moons, comets). Astrobiology 2015, 15, 587–600. [Google Scholar] [CrossRef] [PubMed]
- Klein, F.; Bach, W. Fe-Ni-Co-O-S phase relations in peridotite–seawater interactions. J. Petrol. 2009, 50, 37–59. [Google Scholar] [CrossRef]
- Foustoukos, D.I.; Bizimis, M.; Frisby, C.; Shirey, S.B. Redox controls on Ni-Fe-PGE mineralization and Re/Os fractionation during serpentinization of abyssal peridotite. Geochim. Cosmochim. Acta 2015, 150, 11–25. [Google Scholar] [CrossRef]
- Sleep, N.H.; Meibom, A.; Fridriksson, T.; Coleman, R.G.; Bird, D.K. H2-rich fluids from serpentinization: Geochemical and biotic implications. Proc. Natl. Acad. Sci. USA 2004, 101, 12818–12823. [Google Scholar] [CrossRef] [PubMed]
- Lang, S.Q.; Früh-Green, G.; Bernasconi, S.M.; Brazelton, W.J.; Schrenk, M.O.; McGonigle, J.M. Deeply-sourced formate fuels sulfate reducers but not methanogens at Lost City hydrothermal field. Sci. Rep. 2018, 8, 755. [Google Scholar] [CrossRef] [PubMed]
- Janecky, D.R.; Seyfried, W.E. Hydrothermal serpentinization of peridotite within the oceanic crust: Experimental investigations of mineralogy and major element chemistry. Geochim. Cosmochim. Acta 1986, 50, 1357–1378. [Google Scholar] [CrossRef]
- Palandri, J.L.; Reed, M.H. Geochemical models of metasomatism in ultramafic systems: Serpentinization, rodingitization, and sea floor carbonate chimney precipitation. Geochim. Cosmochim. Acta 2004, 68, 1115–1133. [Google Scholar] [CrossRef]
- Charlou, J.L.; Donval, J.P.; Fouquet, Y.; Jean-Baptiste, P.; Holm, N. Geochemistry of high H2 and CH4 vent fluids issuing from ultramafic rocks at the Rainbow hydrothermal field (36°14′N,MAR). Chem. Geol. 2002, 191, 345–359. [Google Scholar] [CrossRef]
- Kelley, D.S.; Karson, J.A.; Früh-Green, G.L.; Yoerger, D.R.; Shank, T.M.; Butterfield, D.A.; Hayes, J.M.; Schrenk, M.O.; Olson, E.J.; Proskurowski, G.; et al. A serpentinite-hosted ecosystem: The Lost City Hydrothermal Field. Science 2005, 307, 1428–1434. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, W.E.; Foustoukos, D.I.; Fu, Q. Redox evolution and mass transfer during serpentinization: An experimental and theoretical study at 200 °C, 500 bar with implications for ultramafic-hosted hydrothermal systems at Mid-Ocean Ridges. Geochim. Cosmochim. Acta 2007, 71, 3872–3886. [Google Scholar] [CrossRef]
- McCollom, T.M.; Klein, F.; Robbins, M.; Moskowitz, B.; Berquó, T.S.; Jöns, N.; Bach, W.; Templeton, A. Temperature trends for reaction rates, hydrogen generation, and partitioning of iron during experimental serpentinization of olivine. Geochim. Cosmochim. Acta 2016, 181, 175–200. [Google Scholar] [CrossRef]
- Ueda, H.; Shibuya, T.; Sawaki, Y.; Saitoh, M.; Takai, K.; Maruyama, S. Reactions between komatiite and CO2-rich seawater at 250 and 350 °C, 500 bars: Implications for hydrogen generation in the Hadean seafloor hydrothermal system. Prog. Earth Planet. Sci. 2016, 3, 35. [Google Scholar] [CrossRef]
- McCollom, T.M.; Seewald, J.S. A reassessment of the potential for reduction of dissolved CO2 to hydrocarbons during serpentinization of olivine. Geochim. Cosmochim. Acta 2001, 65, 3769–3778. [Google Scholar] [CrossRef]
- Früh-Green, G.L.; Connolly, J.A.D.; Plas, A.; Kelley, D.S.; Grobety, B. Serpentinization of oceanic peridotites: Implications for geochemical cycles and biological activity. In The Subseafloor Biosphere at Mid-Ocean Ridges Geophysical Monograph Series 144; Wilcock, W.S.D., Delong, E.F., Kelley, D.S., Baross, J.A., Craig, C.S., Eds.; American Geophysical Union: Washington, DC, USA, 2004; pp. 119–136. [Google Scholar]
- Frost, R. On the stability of sulfides, oxides, and native metals in serpentinite. J. Petrol. 1985, 26, 31–63. [Google Scholar] [CrossRef]
- Alt, J.C.; Shanks, W.C. Sulfur in serpentinized oceanic peridotites: Serpentinization processes and microbial sulfate reduction. J. Geophys. Res. Solid Earth 1998, 103, 9917–9929. [Google Scholar] [CrossRef]
- McCollom, T.M. Abiotic methane formation during experimental serpentinization of olivine. Proc. Natl. Acad. Sci. USA 2016, 113, 13965–13970. [Google Scholar] [CrossRef] [PubMed]
- Früh-Green, G.L.; Kelley, D.S.; Bernasconi, S.M.; Karson, J.A.; Ludwig, K.A.; Butterfield, D.A.; Boschi, C.; Proskurowski, G. 30,000 Years of hydrothermal activity at the Lost City vent field. Science 2003, 301, 495–498. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, K.A.; Shen, C.C.; Kelley, D.S.; Cheng, H.; Edwards, R.L. U-Th systematics and 230th ages of carbonate chimneys at the Lost City Hydrothermal Field. Geochim. Cosmochim. Acta 2011, 75, 1869–1888. [Google Scholar] [CrossRef]
- Proskurowski, G.; Lilley, M.D.; Seewald, J.S.; Früh-Green, G.L.; Olson, E.J.; Lupton, J.E.; Sylva, S.P.; Kelley, D.S. Abiogenic hydrocarbon production at Lost City hydrothermal field. Science 2008, 319, 319–607. [Google Scholar] [CrossRef] [PubMed]
- Lang, S.Q.; Butterfield, D.A.; Schulte, M.; Kelley, D.S.; Lilley, M.D. Elevated concentrations of formate, acetate and dissolved organic carbon found at the Lost City hydrothermal field. Geochim. Cosmochim. Acta 2010, 74, 941–952. [Google Scholar] [CrossRef]
- Holm, N.G.; Charlou, J.L. Initial indications of abiotic formation of hydrocarbons in the Rainbow ultramafic hydrothermal system, Mid-Atlantic Ridge. Earth Planet. Sci. Lett. 2001, 191, 1–8. [Google Scholar] [CrossRef]
- McDermott, J.M.; Seewald, J.S.; German, C.R.; Sylva, S.P. Pathways for abiotic organic synthesis at submarine hydrothermal fields. Proc. Natl. Acad. Sci. USA 2015, 112, 7668–7672. [Google Scholar] [CrossRef] [PubMed]
- Etiope, G.; Sherwood Lollar, B. Abiotic methane on Earth. Rev. Geophys. 2013, 51, 276–299. [Google Scholar] [CrossRef]
- Horita, J.; Berndt, M.E. Abiogenic methane formation and isotopic fractionation under hydrothermal conditions. Sci. Rep. 1999, 285, 1055–1057. [Google Scholar] [CrossRef]
- Heinen, W.; Lauwers, A. The iron-sulfur world and the origins of life: Abiotic thiol synthesis from metallic iron, H2S and CO2; a comparison of the thiol generating FeS/HCl(H2S)/CO2-system and its Fe0/H2S/CO2-counterpart. Proc. K. Ned. Akad. Van Wet. 1997, 100, 11–25. [Google Scholar]
- Guan, G.; Kida, T.; Ma, T.; Kimura, K.; Abe, E.; Yoshida, A. Reduction of aqueous CO2 at ambient temperature using zero-valent iron-based composites. Green Chem. 2003, 5, 630. [Google Scholar] [CrossRef]
- He, C.; Tian, G.; Liu, Z.; Feng, S. A mild hydrothermal route to fix carbon dioxide to simple carboxylic acids. Org. Lett. 2010, 12, 649–651. [Google Scholar] [CrossRef] [PubMed]
- Varma, S.J.; Muchowska, K.B.; Chatelain, P.; Moran, J. Native iron reduces CO2 to intermediates and endproducts of the acetyl-CoA pathway. Nat. Ecol. Evol. 2018, 2, 1019–1024. [Google Scholar] [CrossRef] [PubMed]
- Thauer, R.K.; Kaster, A.K.; Seedorf, H.; Buckel, W.; Hedderich, R. Methanogenic archaea: Ecologically relevant differences in energy conservation. Nat. Rev. Microbiol. 2008, 6, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Müller, V. Energy conservation in acetogenic bacteria. Appl. Environ. Microbiol. 2003, 69, 6345–6353. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, G. Alternative pathways of carbon dioxide fixation: Insights into the early evolution of life? Annu. Rev. Microbiol. 2011, 65, 631–658. [Google Scholar] [CrossRef] [PubMed]
- Berg, I.A.; Kockelkorn, D.; Buckel, W.; Fuchs, G. A 3-hydroxypropionate/4-hydroxybutyrate autotrophic carbon dioxide assimilation pathway in archaea. Science 2007, 318, 1782–1786. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, G. CO2 fixation in acetogenic bacteria: Variations on a theme. FEMS Microbiol. Lett. 1986, 39, 181–213. [Google Scholar] [CrossRef]
- Sousa, F.L.; Martin, W.F. Biochemical fossils of the ancient transition from geoenergetics to bioenergetics in prokaryotic one carbon compound metabolism. Biochim. Biophys. Acta Bioenerg. 2014, 1837, 964–981. [Google Scholar] [CrossRef] [PubMed]
- Weiss, M.C.; Sousa, F.L.; Mrnjavac, N.; Neukirchen, S.; Roettger, M.; Nelson-Sathi, S.; Martin, W.F. The physiology and habitat of the last universal common ancestor. Nat. Microbiol. 2016, 1, 16116. [Google Scholar] [CrossRef] [PubMed]
- Weiss, M.C.; Preiner, M.; Xavier, J.C.; Zimorski, V.; Martin, F. The last universal common ancestor between ancient Earth chemistry and the onset of genetics. PLoS Genet. 2018, 14, e1007518. [Google Scholar] [CrossRef] [PubMed]
- Früh-Green, G.L.; Orcutt, B.N.; Green, S.L.; Cotterill, C.; Morgan, S.; Akizawa, N.; Bayrakci, G.; Behrmann, J.-H.; Boschi, C.; Brazelton, W.J.; et al. Northern sites. Proc. Int. Ocean Discov. Progr. 2017, 357. [Google Scholar] [CrossRef]
- Smirnov, A.; Hausner, D.; Laffers, R.; Strongin, D.R.; Schoonen, M.A.A. Abiotic ammonium formation in the presence of Ni-Fe metals and alloys and its implications for the Hadean nitrogen cycle. Geochem. Trans. 2008, 9, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Brandes, J.A.; Boctor, N.Z.; Cody, G.D.; Cooper, B.A.; Hazen, R.M.; Yoder, H.S. Abiotic nitrogen reduction on the early Earth. Nature 1998, 395, 365–367. [Google Scholar] [CrossRef] [PubMed]
- Dörr, M.; Käßbohrer, J.; Grunert, R.; Kreisel, G.; Brand, W.A.; Werner, R.A.; Geilmann, H.; Apfel, C.; Robl, C.; Weigand, W. A possible prebiotic formation of ammonia from dinitrogen on iron sulfide surfaces. Angew. Chem. Int. Ed. 2003, 42, 1540–1543. [Google Scholar] [CrossRef] [PubMed]
- Holm, N.G.; Neubeck, A. Reduction of nitrogen compounds in oceanic basement and its implications for HCN formation and abiotic organic synthesis. Geochem. Trans. 2009, 10, 9. [Google Scholar] [CrossRef] [PubMed]
- Bradley, A.S. The sluggish speed of making abiotic methane. Proc. Natl. Acad. Sci. USA 2016, 113, 13944–13946. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.T.; Reeves, E.P.; McDermott, J.M.; Seewald, J.S.; Ono, S. Clumped isotopologue constraints on the origin of methane at seafloor hot springs. Geochim. Cosmochim. Acta 2018, 223, 141–158. [Google Scholar] [CrossRef]
- Seewald, J.S.; Zolotov, M.Y.; McCollom, T. Experimental investigation of single carbon compounds under hydrothermal conditions. Geochim. Cosmochim. Acta 2006, 70, 446–460. [Google Scholar] [CrossRef]
- Miller, H.M.; Mayhew, L.E.; Ellison, E.T.; Kelemen, P.; Kubo, M.; Templeton, A.S. Low temperature hydrogen production during experimental hydration of partially-serpentinized dunite. Geochim. Cosmochim. Acta 2017, 209, 161–183. [Google Scholar] [CrossRef]
- Oparin, A.I. Origin of Life; Dover Publications: New York, NY, USA, 1936; ISBN 0-486-60213-3. [Google Scholar]
- Dictor, R.A.; Bell, A.T. Fischer–Tropsch synthesis over reduced and unreduced iron oxide catalysts. J. Catal. 1986, 97, 121–136. [Google Scholar] [CrossRef]
- Wu, B.; Tian, L.; Xiang, H.; Zhang, Z.; Li, Y.W. Novel precipitated iron Fischer–Tropsch catalysts with Fe3O4 coexisting with α-Fe2O3. Catal. Lett. 2005, 102, 211–218. [Google Scholar] [CrossRef]
- McCollom, T.M. Miller-Urey and beyond: What have we learned about prebiotic organic synthesis reactions in the past 60 years? Annu. Rev. Earth Planet. Sci. 2013, 41, 207–229. [Google Scholar] [CrossRef]
- Holm, N.G.; Dumont, M.; Ivarsson, M.; Konn, C. Alkaline fluid circulation in ultramafic rocks and formation of nucleotide constituents: A hypothesis. Geochem. Trans. 2006, 7, 14–16. [Google Scholar] [CrossRef] [PubMed]
- McCollom, T.M. Laboratory simulations of abiotic hydrocarbon formation in Earth’s deep subsurface. Rev. Mineral. Geochem. 2013, 75, 467–494. [Google Scholar] [CrossRef]
- Porosoff, M.D.; Kattel, S.; Li, W.; Liu, P.; Chen, J.G. Identifying trends and descriptors for selective CO2 conversion to CO over transition metal carbides. Chem. Commun. 2015, 51, 6988. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.A.; Liu, P.; Stacchiola, D.J.; Senanayake, S.D.; White, M.G.; Chen, J.G. Hydrogenation of CO2 to methanol: Importance of metal-oxide and metal-carbide interfaces in the activation of CO2. ACS Catal. 2015, 5, 6696–6706. [Google Scholar] [CrossRef]
- Riedel, T.; Schulz, H.; Schaub, G.; Jun, K.W.; Hwang, J.S.; Lee, K.W. Fischer–Tropsch on iron with H2/CO and H2/CO2 as synthesis gases: The episodes of formation of the Fischer–Tropsch regime and construction of the catalyst. Top. Catal. 2003, 26, 41–54. [Google Scholar] [CrossRef]
- Pichler, H. Twenty-five years of synthesis of gasoline by catalytic conversion of carbon monoxide and hydrogen. In Advances in Catalysis; Frankenburg, W., Rideal, E., Komarewsky, V., Eds.; Academic Press Inc.: New York, NY, USA, 1952; p. 271. ISBN 9780120078042. [Google Scholar]
- Dry, M.E. The Fischer–Tropsch process: 1950–2000. Catal. Today 2002, 71, 227–241. [Google Scholar] [CrossRef]
- Goodrich, C.A.; Bird, J.M. Formation of iron-carbon alloys in basaltic magma at Uivaq, Disko Island: The role of carbon in mafic magmas. J. Geol. 1985, 93, 475–492. [Google Scholar] [CrossRef]
- Shi, N.; Bai, W.; Li, G.; Xiong, M.; Fang, Q.; Yang, J.; Ma, Z.; Rong, H. Yarlongite: A new metallic carbide mineral. Acta Geol. Sin. 2008, 83, 52–56. [Google Scholar] [CrossRef]
- Horita, J.; Polyakov, V.B. Carbon-bearing iron phases and the carbon isotope composition of the deep Earth. Proc. Natl. Acad. Sci. USA 2015, 112, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.W.; Bahnemann, D.W. Reduction of carbon dioxide by magnetite: Implications for the primordial synthesis of organic molecules. J. Am. Chem. Soc. 2000, 122, 970–971. [Google Scholar] [CrossRef]
- Russell, M.J.; Martin, W. The rocky roots of the acetyl-CoA pathway. Trends Biochem. Sci. 2004, 29, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Buckel, W.; Thauer, R.K. Flavin-based electron bifurcation, ferredoxin, flavodoxin, and anaerobic respiration with protons (Ech) or NAD+(Rnf) as electron acceptors: A historical review. Front. Microbiol. 2018, 9, 401. [Google Scholar] [CrossRef] [PubMed]
- Schuchmann, K.; Müller, V. Direct and reversible hydrogenation of CO2 to formate by a bacterial carbon dioxide reductase. Science 2013, 342, 1382–1385. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, G.; Jayamani, E.; Mai, G.; Buckel, W. Energy conservation via electron-transferring flavoprotein in anaerobic bacteria. J. Bacteriol. 2008, 190, 784–791. [Google Scholar] [CrossRef] [PubMed]
- Müller, V.; Chowdhury, N.P.; Basen, M. Electron bifurcation: A long- hidden energy-coupling mechanism. Annu. Rev. Microbiol. 2018, 72, 331–353. [Google Scholar] [CrossRef] [PubMed]
- Kaster, A.-K.; Moll, J.; Parey, K.; Thauer, R.K. Coupling of ferredoxin and heterodisulfide reduction via electron bifurcation in hydrogenotrophic methanogenic archaea. Proc. Natl. Acad. Sci. USA 2011, 108, 2981–2986. [Google Scholar] [CrossRef] [PubMed]
- Mall, A.; Sobotta, J.; Huber, C.; Tschirner, C.; Kowarschik, S.; Bačnik, K.; Mergelsberg, M.; Boll, M.; Hügler, M.; Eisenreich, W.; et al. Reversibility of citrate synthase allows autotrophic growth of a thermophilic bacterium. Science 2018, 359, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Nunoura, T.; Chikaraishi, Y.; Izaki, R.; Suwa, T.; Sato, T.; Harada, T.; Mori, K.; Kato, Y.; Miyazaki, M.; Shimamura, S.; et al. A primordial and reversible TCA cycle in a facultatively chemolithoautotrophic thermophile. Science 2018, 359, 559–563. [Google Scholar] [CrossRef] [PubMed]
- 1Martin, W.F.; Thauer, R.K. Energy in ancient metabolism. Cell 2017, 168, 953–955. [Google Scholar] [CrossRef] [PubMed]
- Xavier, J.C.; Preiner, M.; Martin, W.F. Something special about CO. FEBS J. 2018, in press. [Google Scholar] [CrossRef]
- Dos Santos, P.C.; Igarashi, R.Y.; Lee, H.; Hoffman, B.M.; Seefeldt, L.C.; Dean, D.R. Substrate interactions with the nitrogenase active site. Acc. Chem. Res. 2005, 38, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Burgess, B.K.; Lowe, D.J. Mechanism of molybdenum nitrogenase. Chem. Rev. 1996, 96, 2983–3011. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Kaiser, J.T.; Meloni, G.; Yang, K.-Y.; Spatzal, T.; Andrade, S.L.A.; Einsle, O.; Howard, J.B.; Rees, D.C. The 16th Fe in the nitrogenase MoFe–protein. Angew. Chem. Int. Ed. 2013, 52, 10529–10532. [Google Scholar] [CrossRef] [PubMed]
- Buckel, W.; Hetzel, M.; Kim, J. ATP-driven electron transfer in enzymatic radical reactions. Curr. Opin. Chem. Biol. 2004, 8, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Wiig, J.A.; Hu, Y.; Ribbe, M.W. Refining the pathway of carbide insertion into the nitrogenase M-cluster. Nat. Commun. 2015, 6, 8034. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Hu, Y.; Ribbe, M.W. Insights into hydrocarbon formation by nitrogenase cofactor homologs. MBio 2015, 6, e00307-15. [Google Scholar] [CrossRef] [PubMed]
- Boyd, E.S.; Peters, J.W. New insights into the evolutionary history of biological nitrogen fixation. Front. Microbiol. 2013, 4, 201. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.S.; Rittle, J.; Peters, J.C. Catalytic conversion of nitrogen to ammonia by an iron model complex. Nature 2013, 501, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.F. Hydrogen, metals, bifurcating electrons, and proton gradients: The early evolution of biological energy conservation. FEBS Lett. 2012, 586, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.A.; Szöllösi, G.J.; Spang, A.; Foster, P.G.; Heaps, S.E.; Boussau, B.; Ettema, T.J.G.; Embley, T.M. Integrative modeling of gene and genome evolution roots the archaeal tree of life. Proc. Natl. Acad. Sci. USA 2017, 114, E4602–E4611. [Google Scholar] [CrossRef] [PubMed]
- Takami, H.; Noguchi, H.; Takaki, Y.; Uchiyama, I.; Toyoda, A.; Nishi, S.; Chee, G.; Arai, W.; Nunoura, T.; Itoh, T.; et al. A deeply branching thermophilic bacterium with an ancient acetyl-CoA pathway dominates a subsurface ecosystem. PLoS ONE 2012, 7, e30559. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.F.; Bryant, D.A.; Beatty, J.T. A physiological perspective on the origin and evolution of photosynthesis. FEMS Microbiol. Rev. 2018, 42, 205–231. [Google Scholar] [CrossRef] [PubMed]
- Chapelle, F.H.; Neill, K.O.; Bradley, P.M.; Methe, B.A.; Ciufo, S.A.; Knobel, L.L.; Lovley, D.R. A hydrogen-based subsurface microbial community dominated by methanogens. Nature 2002, 415, 312–315. [Google Scholar] [CrossRef] [PubMed]
- Lever, M.A.; Rouxel, O.; Alt, J.C.; Shimizu, N.; Ono, S.; Coggon, R.M.; Shanks, W.C.; Lapham, L.; Elvert, M.; Prieto-Mollar, X.; et al. Evidence for microbial carbon and sulfur cycling in deeply buried ridge flank basalt. Science 2013, 339, 1305–1308. [Google Scholar] [CrossRef] [PubMed]
- Reeves, E.P.; Mcdermott, J.M.; Seewald, J.S. The origin of methanethiol in midocean ridge hydrothermal fluids. Proc. Natl. Acad. Sci. USA 2014, 111, 5474–5479. [Google Scholar] [CrossRef] [PubMed]
- Schrenk, M.O.; Kelley, D.S.; Bolton, S.A.; Baross, J.A. Low archaeal diversity linked to subseafloor geochemical processes at the Lost City Hydrothermal Field, Mid-Atlantic Ridge. Environ. Microbiol. 2004, 6, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Brazelton, W.J.; Schrenk, M.O.; Kelley, D.S.; Baross, J.A. Methane- and sulfur-metabolizing microbial communities dominate the Lost City Hydrothermal Field ecosystem. Appl. Environ. Microbiol. 2006, 72, 6257–6270. [Google Scholar] [CrossRef] [PubMed]
- Whitman, W.B.; Coleman, D.C.; Wiebe, W.J. Prokaryotes: The unseen majority. Proc. Natl. Acad. Sci. USA 1998, 95, 6578–6583. [Google Scholar] [CrossRef] [PubMed]
- Bar-On, Y.M.; Phillips, R.; Milo, R. The biomass distribution on Earth. Proc. Natl. Acad. Sci. USA 2018, 115, 6506–6511. [Google Scholar] [CrossRef] [PubMed]
- Heberling, C.; Lowell, R.P.; Liu, L.; Fisk, M.R. Extent of the microbial biosphere in the oceanic crust. Geochem. Geophys. Geosyst. 2010, 11. [Google Scholar] [CrossRef]
- Milucka, J.; Ferdelman, T.G.; Polerecky, L.; Franzke, D.; Wegener, G.; Schmid, M.; Lieberwirth, I.; Wagner, M.; Widdel, F.; Kuypers, M.M.M. Zero-valent sulphur is a key intermediate in marine methane oxidation. Nature 2012, 491, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.L.; Bada, J.L. Submarine hot springs and the origin of life. Nature 1988, 334, 609–611. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.S.; Karson, J.A.; Blackman, D.K.; Früh-Green, G.L.; Butterfield, D.A.; Lilley, M.D.; Olson, E.J.; Schrenk, M.O.; Roe, K.K.; Lebon, G.T.; et al. An off-axis hydrothermal vent field near the Mid-Atlantic Ridge at 30 degrees N. Nature 2001, 412, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Benner, S.A.; Kim, H.-J.; Carrigan, M.A. Asphalt, water, and the prebiotic synthesis of ribose, ribonucleosides, and RNA. Acc. Chem. Res. 2012, 45, 2025–2034. [Google Scholar] [CrossRef] [PubMed]
- Ricardo, A.; Carrigan, M.A.; Olcott, A.N.; Benner, S.A. Borate minerals stabilize ribose. Science 2004, 303, 196. [Google Scholar] [CrossRef] [PubMed]
- Schönheit, P.; Buckel, W.; Martin, W.F. On the origin of heterotrophy. Trends Microbiol. 2016, 24, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Stouthamer, A.H. Energy-yielding pathways. In The Bacteria Vol 6; Gunsalus, I.C., Ed.; Academic Press: New York, NY, USA, 1978; pp. 389–462. [Google Scholar]
- Baross, J.A.; Martin, W.F. The ribofilm as a concept for life’s origins. Cell 2015, 162, 13–15. [Google Scholar] [CrossRef] [PubMed]
- Goldford, J.E.; Hartman, H.; Smith, T.F.; Segrè, D. Remnants of an ancient metabolism without phosphate. Cell 2017, 168, 1126–1134.e9. [Google Scholar] [CrossRef] [PubMed]
- Goldford, J.E.; Segrè, D. Modern views of ancient metabolic networks. Curr. Opin. Syst. Biol. 2018, 8, 117–124. [Google Scholar] [CrossRef]
- Dayhoff, M.O.; Eck, R.V. Evolution of the structure of ferredoxin based on surviving relics of primitive amino acid sequences. Science 1966, 152, 363–366. [Google Scholar] [CrossRef]
- Wächtershäuser, G. Before enzymes and templates: Theory of surface metabolism. Microbiol. Rev. 1988, 52, 452–484. [Google Scholar] [PubMed]
- Thauer, R.K.; Kaster, A.-K.; Goenrich, M.; Schick, M.; Hiromoto, T.; Shima, S. Hydrogenases from methanogenic archaea, nickel, a novel cofactor, and H2 storage. Annu. Rev. Biochem. 2010, 79, 507–536. [Google Scholar] [CrossRef] [PubMed]
- Thauer, R.K. Hydrogenases and the global H2 cycle. Eur. J. Inorg. Chem. 2011, 2011, 919–921. [Google Scholar] [CrossRef]
- Wächtershäuser, G. Groundworks for an evolutionary biochemistry: The iron-sulphur world. Prog. Biophys. Mol. Biol. 1992, 58, 85–201. [Google Scholar] [CrossRef]
- Huber, C.; Eisenreich, W.; Wächtershäuser, G. Synthesis of α-amino and α-hydroxy acids under volcanic conditions: Implications for the origin of life. Tetrahedron Lett. 2010, 51, 1069–1071. [Google Scholar] [CrossRef]
- Huber, C.; Wächtershäuser, G. Activated acetic acid by carbon fixation on (Fe,Ni)S under primordial conditions. Science 1997, 276, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Muchowska, K.B.; Varma, S.J.; Chevallot-Beroux, E.; Lethuillier-Karl, L.; Li, G.; Moran, J. Metals promote sequences of the reverse Krebs cycle. Nat. Ecol. Evol. 2017, 1, 1716–1721. [Google Scholar] [CrossRef] [PubMed]
- Semenov, S.N.; Kraft, L.J.; Ainla, A.; Zhao, M.; Baghbanzadeh, M.; Campbell, V.E.; Kang, K.; Fox, J.M.; Whitesides, G.M. Autocatalytic, bistable, oscillatory networks of biologically relevant organic reactions. Nature 2016, 537, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Schüth, F. A vibrant science lives from within. Angew. Chem. Int. Ed. 2016, 55, 14878–14879. [Google Scholar] [CrossRef] [PubMed]
- Hickman-Lewis, K.; Cavalazzi, B.; Foucher, F.; Westall, F. Most ancient evidence for life in the Barberton greenstone belt: Microbial mats and biofabrics of the ~3.47 Ga Middle Marker horizon. Precambrian Res. 2018, 312, 45–67. [Google Scholar] [CrossRef]
- Hayatsu, R.; Anders, E. Organic compounds in meteorites and their origins. Top. Curr. Chem. 1981, 99, 1–37. [Google Scholar] [CrossRef]
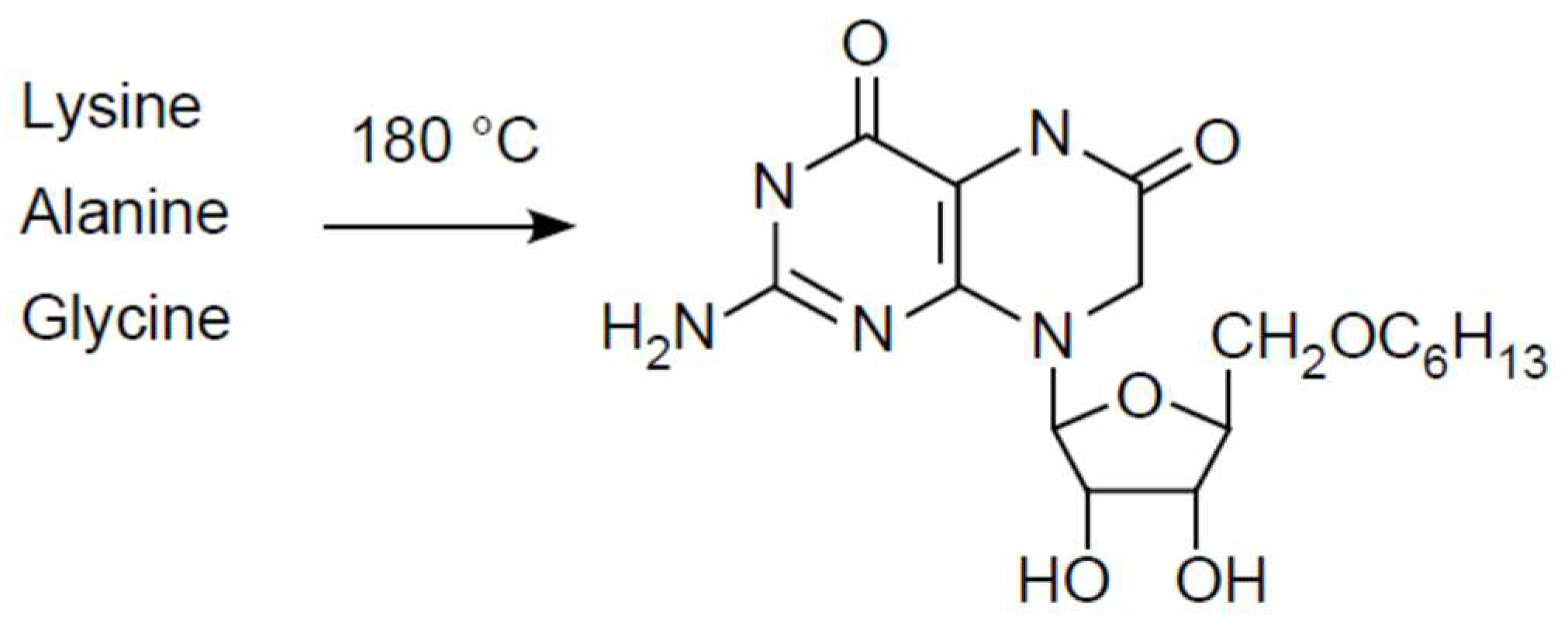
- Heinz, V.B.; Ried, W.; Dose, K. Thermische Erzeugung von Pteridinen und Flavinen aus Aminosauregemischen. Angew. Chem. 1979, 91, 510–511. [Google Scholar] [CrossRef]
- Maden, B.E.H. Tetrahydrofolate and tetrahydromethanopterin compared: Functionally distinct carriers in C1 metabolism. Biochem. J. 2000, 350, 609–629. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Preiner, M.; Xavier, J.C.; Sousa, F.L.; Zimorski, V.; Neubeck, A.; Lang, S.Q.; Greenwell, H.C.; Kleinermanns, K.; Tüysüz, H.; McCollom, T.M.; et al. Serpentinization: Connecting Geochemistry, Ancient Metabolism and Industrial Hydrogenation. Life 2018, 8, 41. https://doi.org/10.3390/life8040041
Preiner M, Xavier JC, Sousa FL, Zimorski V, Neubeck A, Lang SQ, Greenwell HC, Kleinermanns K, Tüysüz H, McCollom TM, et al. Serpentinization: Connecting Geochemistry, Ancient Metabolism and Industrial Hydrogenation. Life. 2018; 8(4):41. https://doi.org/10.3390/life8040041
Chicago/Turabian StylePreiner, Martina, Joana C. Xavier, Filipa L. Sousa, Verena Zimorski, Anna Neubeck, Susan Q. Lang, H. Chris Greenwell, Karl Kleinermanns, Harun Tüysüz, Tom M. McCollom, and et al. 2018. "Serpentinization: Connecting Geochemistry, Ancient Metabolism and Industrial Hydrogenation" Life 8, no. 4: 41. https://doi.org/10.3390/life8040041
APA StylePreiner, M., Xavier, J. C., Sousa, F. L., Zimorski, V., Neubeck, A., Lang, S. Q., Greenwell, H. C., Kleinermanns, K., Tüysüz, H., McCollom, T. M., Holm, N. G., & Martin, W. F. (2018). Serpentinization: Connecting Geochemistry, Ancient Metabolism and Industrial Hydrogenation. Life, 8(4), 41. https://doi.org/10.3390/life8040041