SMORE: Synteny Modulator of Repetitive Elements

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
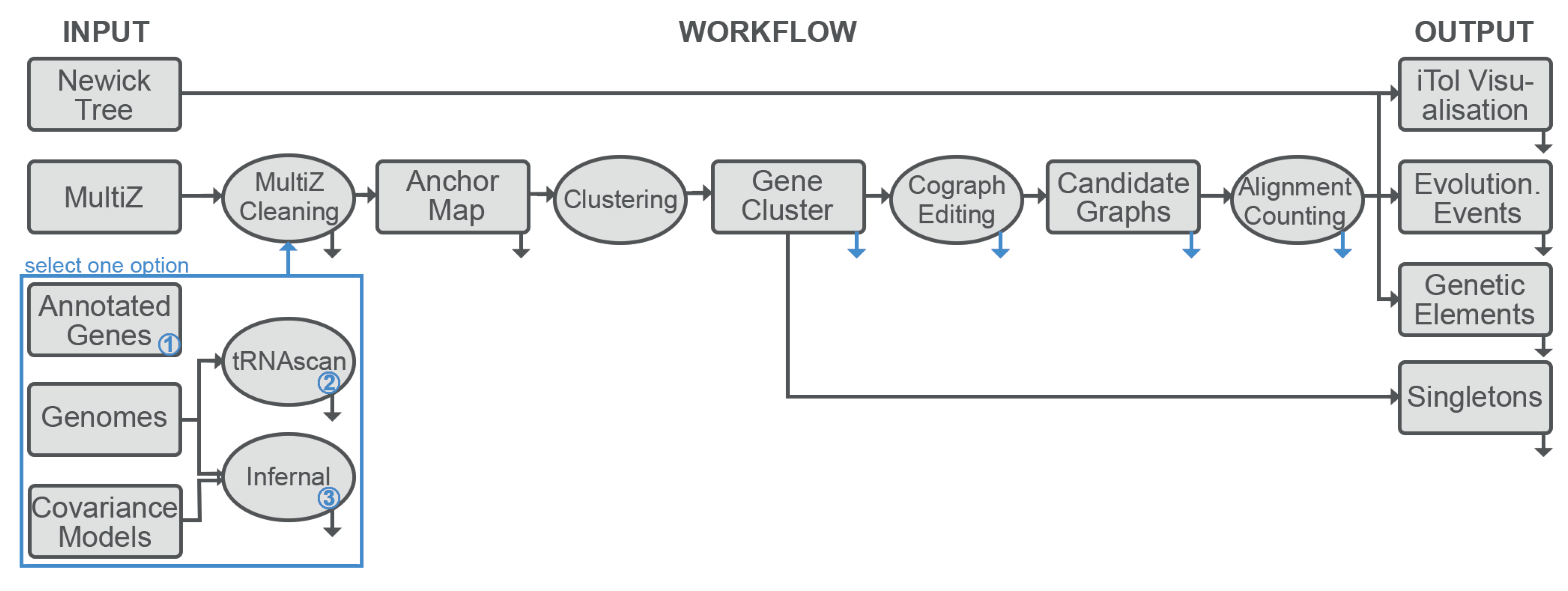
2.1. Overview
2.2. Annotation of the Loci of Interest
2.3. Genomic Anchors
- For the genetic element , we can find two flanking regions and that have orthologous counterparts and in species B on the basis of sequence similarity.
- On the basis of genomic coordinates, the order of the sequences is determined such that and .
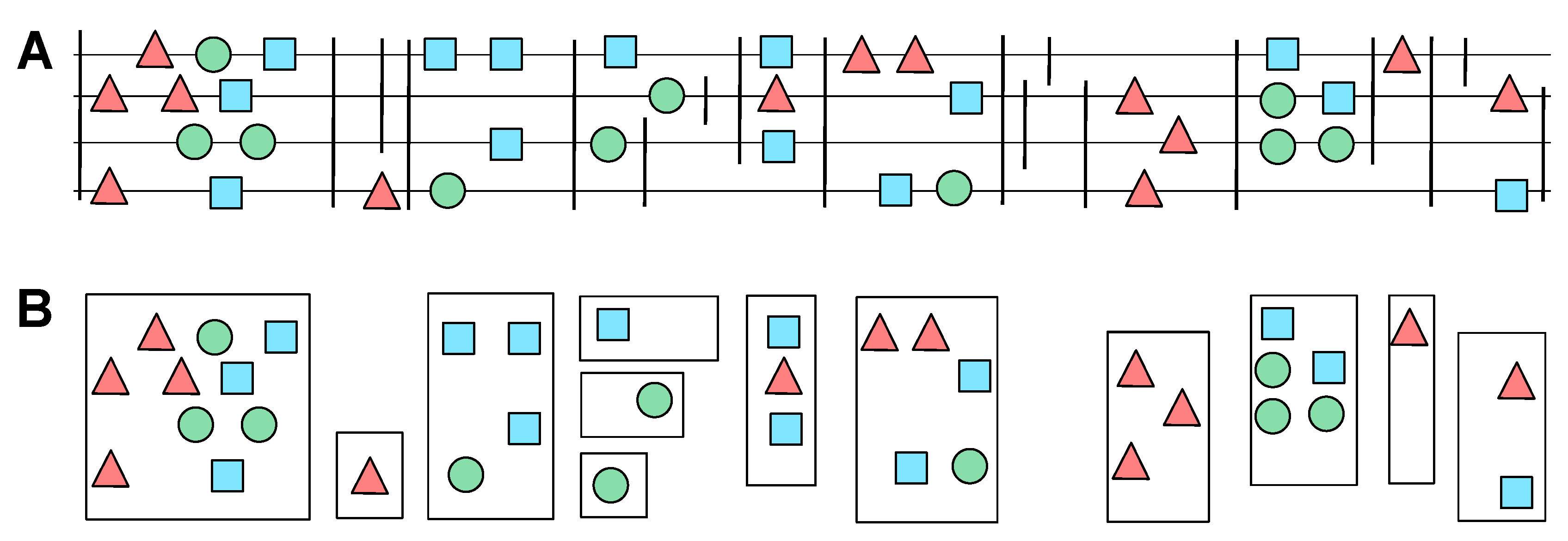
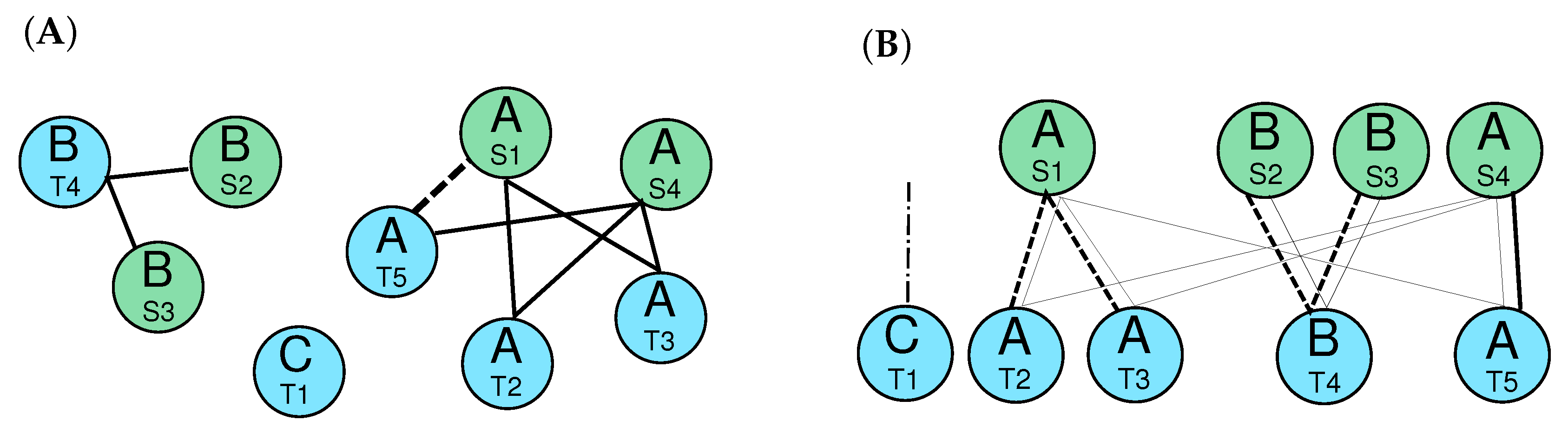
2.4. Candidate Clusters of Co-Orthologous Genes
- The relative genomic order of the elements in each cluster is the same.
- There are no elements belonging to another cluster between the the elements of and .
- The total extension of the merge cluster does not exceed a user-defined threshold.
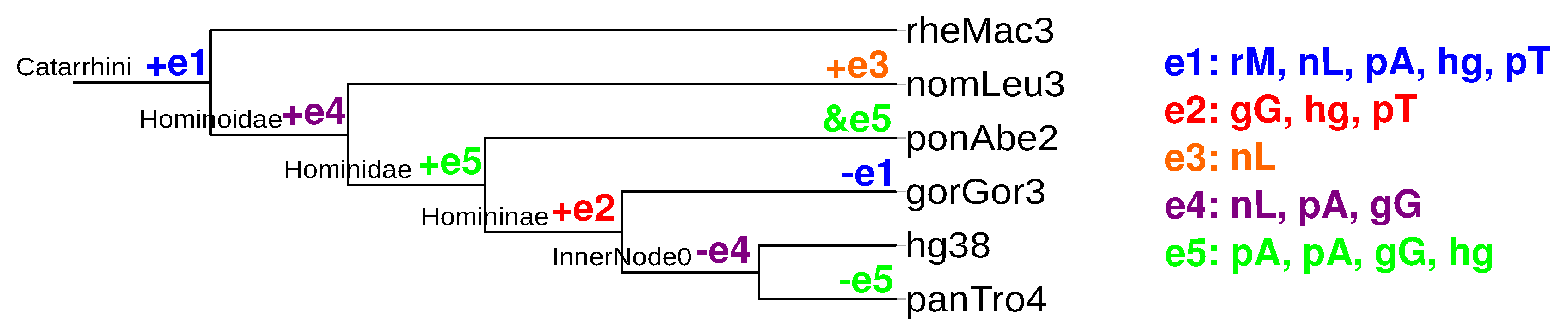
2.4.1. Counting Events Using Relaxed Adjacency Conditions
2.4.2. Orthologs
2.5. Quantitative Analysis of Evolutionary Events
2.6. Pseudogenes and Remolding Events
2.7. Implementation
- A MSA of the genomes under consideration is required to extract the synteny anchor points. Currently only Multiz format is supported.
- The corresponding genomic sequences are required for the annotation of the loci of interest. The pipeline expects fasta format. Because there is no guarantee that genome-wide MSA represents the complete genome, both MSA and genomes must be provided.
- Target elements can be specified either as user-supplied annotation files or as one or more covariance models for annotation with Infernal or tRNAscan-SE. The modular organization of the pipeline makes it straightforward to add, in future releases, further means of generating annotation information, such as hidden Markov models of proteins.
- A phylogenetic tree of the species of interest is necessary as a background to which evolutionary events are mapped.
2.8. Benchmarking with Artifical Data
3. Results
3.1. Automatic Pipeline for Multicopy Elements
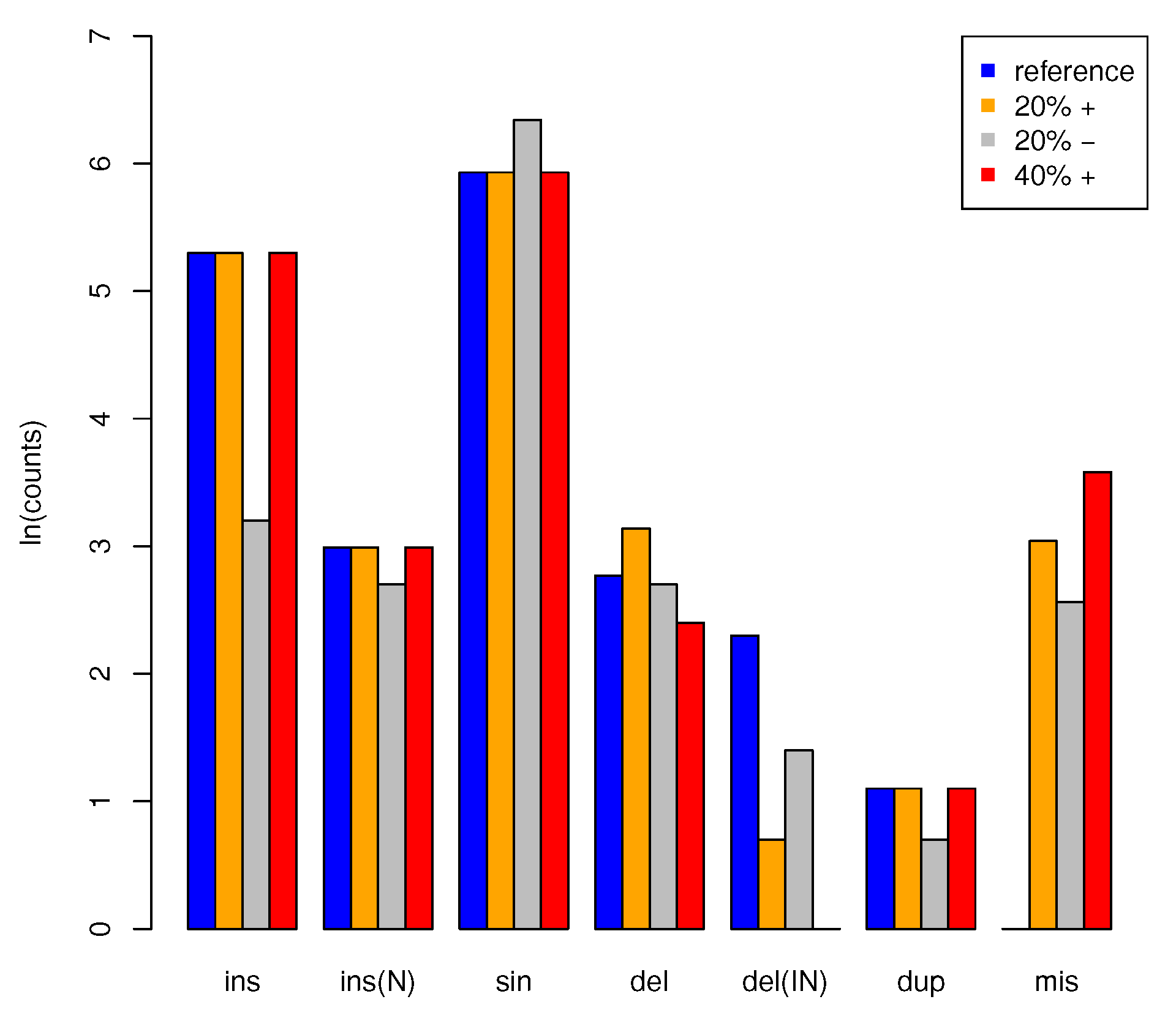
3.2. Application to Artificial Data
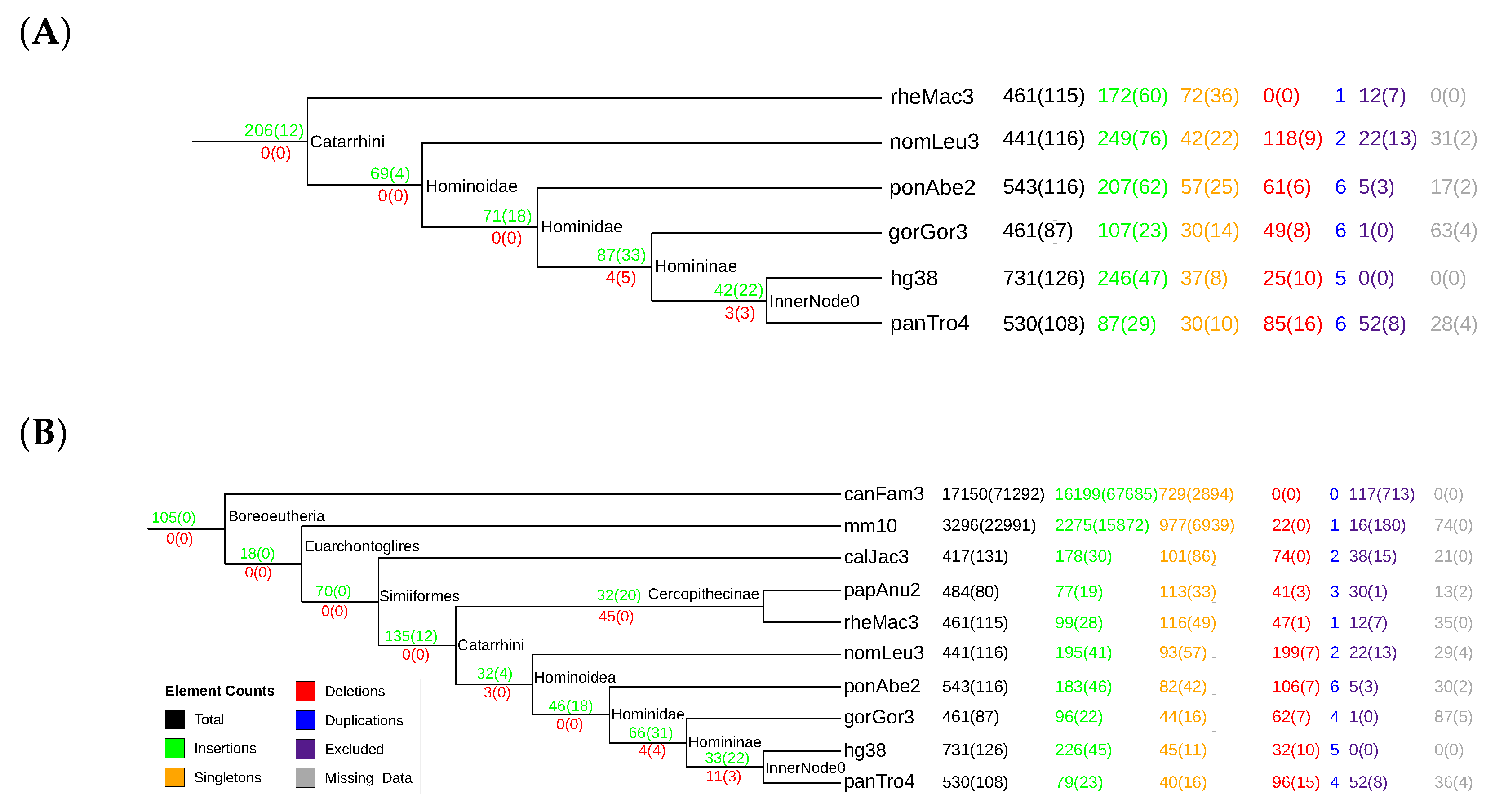
3.3. tRNAs
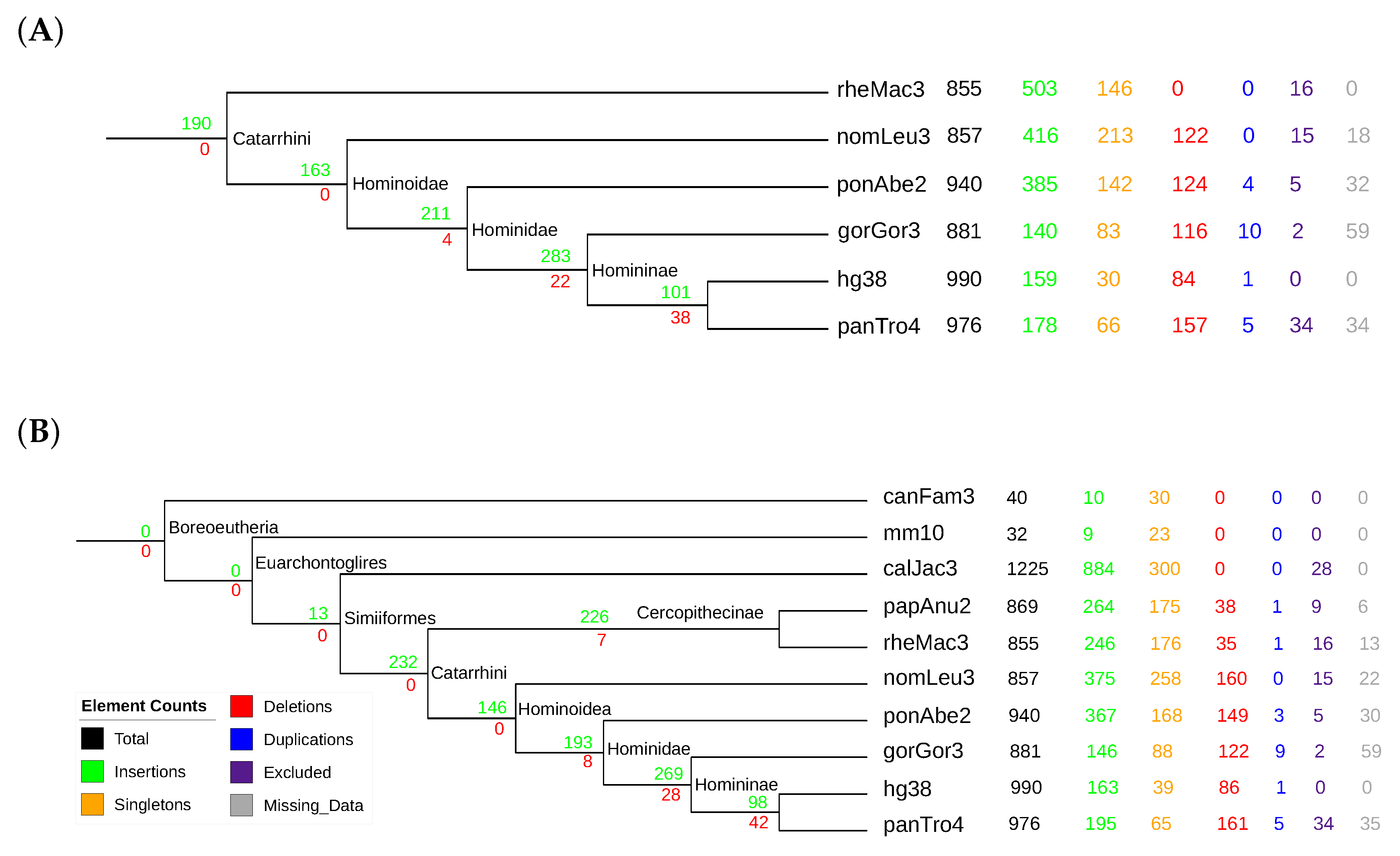
3.4. Mammalian Y RNAs
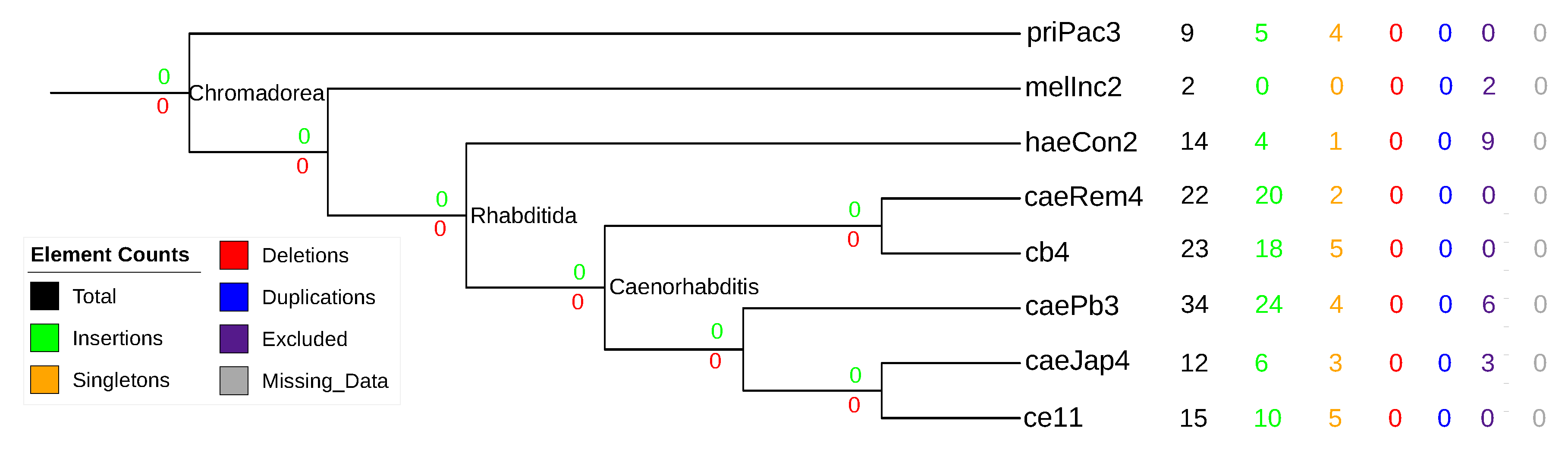
3.5. Nematode stem-bulge RNAs
4. Discussion and Concluding Remarks
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Capra, J.A.; Stolzer, M.; Durand, D.; Pollard, K.S. How old is my gene? Trends Genet. 2013, 29, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Holland, P.W. Evolution of homeobox genes. Wiley Interdiscip. Rev. Dev. Biol. 2013, 2, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Hiller, M.; Schaar, B.T.; Indjeian, V.B.; Kingsley, D.M.; Hagey, L.R.; Bejerano, G. A “forward genomics” approach links genotype to phenotype using independent phenotypic losses among related species. Cell Rep. 2012, 2, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Fitch, W.M. Distinguishing Homologous from Analogous Proteins. Syst. Biol. 1970, 19, 99–113. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Koonin, E.V.; Lipman, D.J. A genomic perspective on protein families. Science 1997, 278, 631–637. [Google Scholar]
- Lechner, M.; Findeiß, S.; Steiner, L.; Marz, M.; Stadler, P.F.; Prohaska, S.J. Proteinortho: Detection of (Co-)Orthologs in Large-Scale Analysis. BMC Bioinform. 2011, 12, 124. [Google Scholar] [CrossRef] [PubMed]
- Altenhoff, A.M.; Dessimoz, C. Phylogenetic and functional assessment of orthologs inference projects and methods. PLoS Comput. Biol. 2009, 5, e1000262. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, D.M.; Wolf, Y.I.; Mushegian, A.R.; Koonin, E.V. Computational methods for Gene Orthology inference. Brief. Bioinform. 2011, 12, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Salichos, L.; Rokas, A. Evaluating Ortholog Prediction Algorithms in a Yeast Model Clade. PLoS ONE 2011, 6, e18755. [Google Scholar] [CrossRef] [PubMed]
- Dalquen, D.A.; Altenhoff, A.M.; Gonnet, G.H.; Dessimoz, C. The Impact of Gene Duplication, Insertion, Deletion, Lateral Gene Transfer and Sequencing Error on Orthology Inference: A Simulation Study. PLoS ONE 2013, 8, e56925. [Google Scholar] [CrossRef] [PubMed]
- Ward, N.; Moreno-Hagelsieb, G. Quickly Finding Orthologs as Reciprocal Best Hits with BLAT, LAST, and UBLAST: How Much Do We Miss? PLoS ONE 2014, 9, e101850. [Google Scholar] [CrossRef] [PubMed]
- Liao, D. Concerted Evolution: Molecular Mechanisms and Biological Implications. Am. J. Hum. Genet. 1999, 64, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Nei, M.; Rooney, A.P. Concerted and Birth-and-Death Evolution of Multigene Families. Annu. Rev. Genet. 2005, 39, 121–152. [Google Scholar] [CrossRef] [PubMed]
- Liao, D.; Pavelitz, T.; Kidd, J.R.; Kidd, K.K.; Weiner, A.M. Concerted evolution of the tandemly repeated genes encoding human U2 snRNA (the RNU2 locus) involves rapid intrachromosomal homogenization and rare interchromosomal gene conversion. EMBO J. 1997, 16, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Amstutz, H.; Munz, P.; Heyer, W.D.; Leupoid, U.; Kohli, J. Concerted evolution of tRNA genes: Intergenic conversion among three unlinked serine tRNA genes in S. pombe. Cell 1985, 40, 879–886. [Google Scholar] [CrossRef]
- Naidoo, K.; Steenkamp, E.; Coetzee, M.P.; Wingfield, M.J.; Wingfield, B.D. Concerted evolution in the ribosomal RNA cistron. PLoS ONE 2013, 8, e59355. [Google Scholar] [CrossRef] [PubMed]
- Scienski, K.; Fay, J.C.F.; Conant, G.C. Patterns of Gene Conversion in Duplicated Yeast Histones Suggest Strong Selection on a Coadapted Macromolecular Complex. Genome Biol. Evol. 2015, 7, 3249–3258. [Google Scholar] [CrossRef] [PubMed]
- Teshima, K.M.; Innan, H. The Effect of Gene Conversion on the Divergence Between Duplicated Genes. Genetics 2004, 166, 1553–1560. [Google Scholar] [CrossRef] [PubMed]
- Bermúdez-Santana, C.; Stephan-Otto Attolini, C.; Kirsten, T.; Engelhardt, J.; Prohaska, S.J.; Steigele, S.; Stadler, P.F. Genomic Organization of Eukaryotic tRNAs. BMC Genom. 2010, 11, 270. [Google Scholar] [CrossRef] [PubMed]
- Rogers, H.H.; Bergman, C.M.; Griffiths-Jones, S. The evolution of tRNA genes in Drosophila. Genome Biol. Evol. 2010, 2, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.P.; Ruvinsky, I. Family size and turnover rates among several classes of small non-protein-coding RNA genes in Caenorhabditis nematodes. Genome Biol. Evol. 2012, 4, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Rogers, H.H.; Griffiths-Jones, S. tRNA anticodon shifts in eukaryotic genomes. RNA 2014, 20, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Velandia-Huerto, C.A.; Berkemer, S.J.; Hoffmann, A.; Retzlaff, N.; Romero Marroquín, L.C.; Hernández Rosales, M.; Stadler, P.F.; Bermúdez-Santana, C.I. Orthologs, turn-over, and remolding of tRNAs in primates and fruit flies. BMC Genom. 2016, 17, 617. [Google Scholar] [CrossRef] [PubMed]
- Eigen, M.; Lindemann, B.F.; Tietze, M.; Winkler-Oswatitsch, R.; Dress, A.W.M.; von Haeseler, A. How old is the genetic code? Statistical geometry of tRNA provides an answer. Science 1989, 244, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Eigen, M.; Winkler-Oswatitsch, R. Transfer-RNA, an early gene? Naturwissenschaften 1981, 68, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Florentz, C.; Jühling, F.; Pütz, J.; Sauter, C.; Stadler, P.F.; Giegé, R. Structure of transfer RNAs: A function-driven refined view. Wiley Interdiscip. Rev. RNA 2012, 3, 37–61. [Google Scholar]
- McFarlane, R.J.; Whitehall, S.K. tRNA genes in eukaryotic genome organization and reorganization. Cell Cycle 2009, 8, 3102–3106. [Google Scholar] [CrossRef] [PubMed]
- Soares, A.R.; Santos, M. Discovery and function of transfer RNA-derived fragments and their role in disease. Wiley Interdiscip. Rev. RNA 2017, 8, 5. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.J.; Fleurdépine, S.; Bousquet-Antonelli, C.; Caetano-Anollés, G.; Deragon, J.M. Common evolutionary trends for SINE RNA structures. Trends Genet. 2007, 23, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Rozhdestvensky, T.S.; Kopylov, A.M.; Brosius, J.; Hüttenhofer, A. Neuronal BC1 RNA structure: Evolutionary conversion of a tRNA(Ala) domain into an extended stem-loop structure. RNA 2001, 7, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Iacoangeli, A.; Rozhdestvensky, T.S.; Dolzhanskaya, N.; Tournier, B.; Schutt, J.; Brosius, J.; Denman, R.B.; Khandjian, E.W.; Kindler, S.; Tiedge, H. On BC1 RNA and the fragile X mental retardation protein. Proc. Natl. Acad. Sci. USA 2008, 105, 734–739. [Google Scholar] [CrossRef] [PubMed]
- Nishihara, H.; Smit, A.F.A.; Okada, N. Functional noncoding sequences derived from SINEs in the mammalian genome. Genome Res. 2006, 16, 864–874. [Google Scholar] [CrossRef] [PubMed]
- Frenkel, F.E.; Chaley, M.B.; Korotkov, E.V.; Skryabin, K.G. Evolution of tRNA-like sequences and genome variability. Gene 2004, 335, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Hertel, J.; Stadler, P.F. The Expansion of Animal MicroRNA Families Revisited. Life 2015, 5, 905–920. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.A.; Margelot, K.; Wolin, S.L. Xenopus Ro ribonucleoproteins: Members of an evolutionarily conserved class of cytoplasmic ribonucleoproteins. Proc. Natl. Acad. Sci. USA 1993, 90, 7250–7254. [Google Scholar] [CrossRef] [PubMed]
- Lerner, M.R.; Boyle, J.A.; Hardin, J.A.; Steitz, J.A. Two novel classes of small ribonucleoproteins detected by antibodies associated with lupus erythematosus. Science 1981, 211, 400–402. [Google Scholar] [CrossRef] [PubMed]
- Hendrick, J.P.; Wolin, S.L.; Rinke, J.; Lerner, M.R.; Steitz, J.A. Ro small cytoplasmic ribonucleoproteins are a subclass of La ribonucleoproteins: Further characterization of the Ro and La small ribonucleoproteins from uninfected mammalian cells. Mol. Cell. Biol. 1981, 1, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Farris, A.D.; Koelsch, G.; Pruijn, G.J.; van Venrooij, W.J.; Harley, J.B. Conserved features of Y RNAs revealed by automated phylogenetic secondary structure analysis. Nucleic Acids Res. 1999, 27, 1070–1078. [Google Scholar] [CrossRef] [PubMed]
- Teunissen, S.W.M.; Kruithof, M.J.M.; Farris, A.D.; Harley, J.B.; van Venrooij, W.J.; Pruijn, G.J.M. Conserved features of Y RNAs: A comparison of experimentally derived secondary structures. Nucleic Acids Res. 2000, 28, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Christov, C.P.; Gardiner, T.J.; Szüts, D.; Krude, T. Functional Requirement of Noncoding Y RNAs for Human Chromosomal DNA Replication. Mol. Cell. Biol. 2006, 26, 6993–7004. [Google Scholar] [CrossRef] [PubMed]
- Kheir, E.; Krude, T. Non-coding Y RNAs associate with early replicating euchromatin in concordance with the origin recognition complex. J. Cell Sci. 2017, 130, 1239–1250. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.; Turnbull, C.; Dalmay, T. Y RNAs: Recent developments. Biomol. Concepts 2013, 4, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Rutjes, S.A.; van der Heijden, A.; Utz, P.H.; van Venrooij, W.J.; Pruijn, G.J. Rapid nucleolytic degradation of the small cytoplasmic Y RNAs during apoptosis. J. Biol. Chem. 1999, 274, 24799–24807. [Google Scholar] [CrossRef] [PubMed]
- Mosig, A.; Guofeng, M.; Stadler, B.M.R.; Stadler, P.F. Evolution of the Vertebrate Y RNA Cluster. Theory Biosci. 2007, 126, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Perreault, J.; Noël, J.F.; Brière, F.; Cousineau, B.; Lucier, J.F.; Perreault, J.P.; Boire, G. Retropeudogenes derived from human Ro/SS-A autoantigen-associated hY RNAs. Nucleic Acids Res. 2005, 33, 2032–2041. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A Program for Improved Detection of Transfer RNA Genes in Genomic Sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Boria, I.; Gruber, A.R.; Tanzer, A.; Bernhart, S.; Lorenze, R.; Mueller, M.M.; Hofacker, I.L.; Stadler, P.F. Nematode sbRNAs: Homologs of vertebrate Y RNAs. J. Mol. Evol. 2010, 70, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Will, S.; Joshi, T.; Hofacker, I.L.; Stadler, P.F.; Backofen, R. LocARNA-P: Accurate boundary prediction and improved detection of structural RNAs. RNA 2012, 18, 900–914. [Google Scholar] [CrossRef] [PubMed]
- Will, S.; Reiche, K.; Hofacker, I.L.; Stadler, P.F.; Backofen, R. Inferring non-coding RNA families and classes by means of genome-scale structure-based clustering. PLoS Comput. Biol. 2007, 3, e65. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.; Heyne, S.; Richter, A.S.; Will, S.; Backofen, R. Freiburg RNA Tools: A web server integrating IntaRNA, ExpaRNA and LocARNA. Nucleic Acids Res. 2010, 38, W373–W377. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef] [PubMed]
- Gärtner, F.; Höner zu Siederdissen, C.; Müller, L.; Stadler, P.F. Coordinate Systems for Supergenomes; Allen Institute for Artificial Intelligence: Seattle, WA, USA, 2017. [Google Scholar]
- UCSC Genome Browser. Multiple Alignments of 19 Mammalian (16 Primate) Genomes with Human. Available online: http://hgdownload.cse.ucsc.edu/goldenPath/hg38/multiz20way/maf/ (accessed on 14 October 2015).
- UCSC Genome Browser. Multiple Alignments of 25 Nermatodes with C. elegans. Available online: http://hgdownload.cse.ucsc.edu/goldenPath/ce11/multiz26way/ (accessed on 3 February 2016).
- Hellmuth, M.; Hernandez-Rosales, M.; Huber, K.T.; Moulton, V.; Stadler, P.F.; Wieseke, N. Orthology Relations, Symbolic Ultrametrics, and Cographs. J. Math. Biol. 2013, 66, 399–420. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, J.; Guo, J.; Chen, J. Complexity and parameterized algorithms for Cograph Editing. Theor. Comput. Sci. 2012, 461, 45–54. [Google Scholar] [CrossRef]
- Needleman, S.B.; Wunsch, C.D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970, 48, 443–453. [Google Scholar] [CrossRef]
- Hellmuth, M.; Wieseke, N.; Lechner, M.; Lenhof, H.P.; Middendorf, M.; Stadler, P.F. Phylogenetics from Paralogs. Proc. Natl. Acad. Sci. USA 2015, 112, 2058–2063. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Rosales, M.; Hellmuth, M.; Wieseke, N.; Huber, K.T.; Moulton, V.; Stadler, P.F. From Event-Labeled Gene Trees to Species Trees. BMC Bioinform. 2012, 13, S6. [Google Scholar]
- Rawlings, T.A.; Collins, T.M.; Bieler, R. Changing identities: tRNA duplication and remolding within animal mitochondrial genomes. Proc. Natl. Acad. Sci. USA 2003, 100, 15700–15705. [Google Scholar] [CrossRef] [PubMed]
- Sahyoun, A.H.; Hölzer, M.; Jühling, F.; Höner zu Siederdissen, C.; Al-Arab, M.; Tout, K.; Marz, M.; Middendorf, M.; Stadler, P.F.; Bernt, M. Towards a Comprehensive Picture of Alloacceptor tRNA Remolding in Metazoan Mitochondrial Genomes. Nucleic Acids Res. 2015, 43, 8044–8056. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef] [PubMed]
- Perreault, J.; Perreault, J.P.; Boire, G. Ro-associated Y RNAs in metazoans: Evolution and diversification. Mol. Biol. Evol. 2007, 24, 1678–1689. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Zhu, X.; Skogerbo, G.; Zhao, Y.; Fu, Z.; Wang, Y.; He, H.; Cai, L.; Sun, H.; Liu, C.; et al. Organization of the Caenorhabditis elegans small non-coding transcriptome: genomic features, biogenesis, and expression. Genome Res. 2006, 16, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Aftab, M.N.; He, H.; Skogerbø, G.; Chen, R. Microarray analysis of ncRNA expression patterns in Caenorhabditis elegans after RNAi against snoRNA associated proteins. BMC Genom. 2008, 9, 278. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, M.P.; Baylis, H.A.; Krude, T. Non-coding stem-bulge RNAs are required for cell proliferation and embryonic development in C. elegans. J. Cell Sci. 2015, 128, 2118–2129. [Google Scholar] [CrossRef] [PubMed]
- Coghlan, A.; Wolfe, K.H. Fourfold Faster Rate of Genome Rearrangement in Nematodes Than in Drosophila. Genome Res. 2002, 12, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Lin, Y.; Tang, J. MLGO: Phylogeny reconstruction and ancestral inference from gene-order data. BMC Bioinform. 2014, 15, 354. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Merkle, D.; Rasch, K.; Fritzsch, G.; Perseke, M.; Bernhard, D.; Schlegel, M.; Stadler, P.F.; Middendorf, M. CREx: Inferring Genomic Rearrangements Based on Common Intervals. Bioinformatics 2007, 23, 2957–2958. [Google Scholar] [CrossRef] [PubMed]
- Feijão, P. Reconstruction of ancestral gene orders using intermediate genomes. BMC Bioinform. 2015, 16 (Suppl. S14), S3. [Google Scholar]
- Braga, M.D.V.; Stoye, J. Sorting Linear Genomes with Rearrangements and Indels. IEEE/ACM Trans. Comput. Biol. Bioinform. 2015, 12, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Tremblay-Savard, O.; Benzaid, B.; Lang, B.F.; El-Mabrouk, N. Evolution of tRNA Repertoires in Bacillus Inferred with OrthoAlign. Mol. Biol. Evol. 2015, 32, 1643–1656. [Google Scholar] [CrossRef] [PubMed]








© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berkemer, S.J.; Hoffmann, A.; Murray, C.R.A.; Stadler, P.F. SMORE: Synteny Modulator of Repetitive Elements. Life 2017, 7, 42. https://doi.org/10.3390/life7040042
Berkemer SJ, Hoffmann A, Murray CRA, Stadler PF. SMORE: Synteny Modulator of Repetitive Elements. Life. 2017; 7(4):42. https://doi.org/10.3390/life7040042
Chicago/Turabian StyleBerkemer, Sarah J., Anne Hoffmann, Cameron R. A. Murray, and Peter F. Stadler. 2017. "SMORE: Synteny Modulator of Repetitive Elements" Life 7, no. 4: 42. https://doi.org/10.3390/life7040042
APA StyleBerkemer, S. J., Hoffmann, A., Murray, C. R. A., & Stadler, P. F. (2017). SMORE: Synteny Modulator of Repetitive Elements. Life, 7(4), 42. https://doi.org/10.3390/life7040042