
The Expansion of Animal MicroRNA Families Revisited

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. MicroRNA Detection
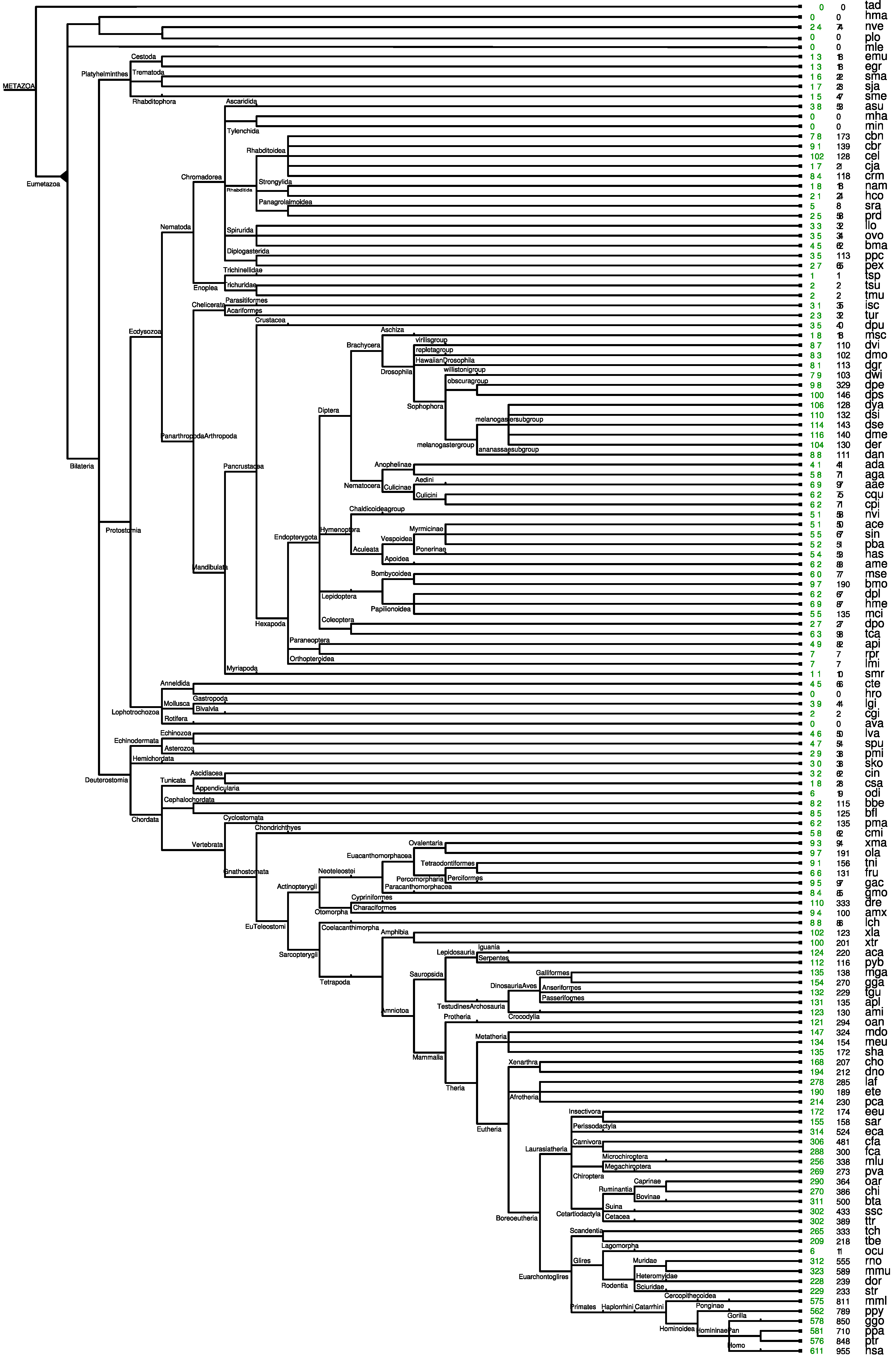
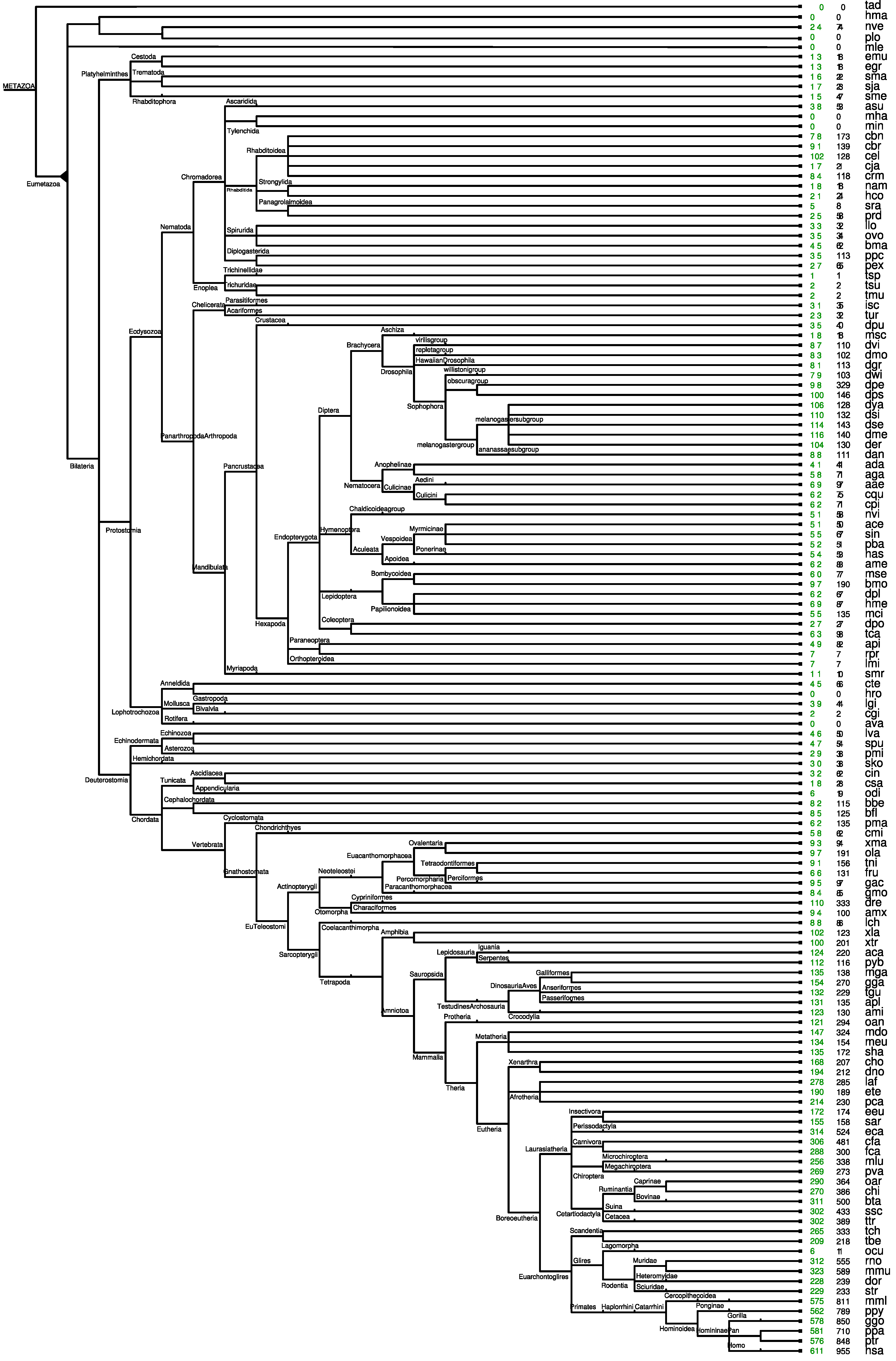
2.2. MicroRNA Age
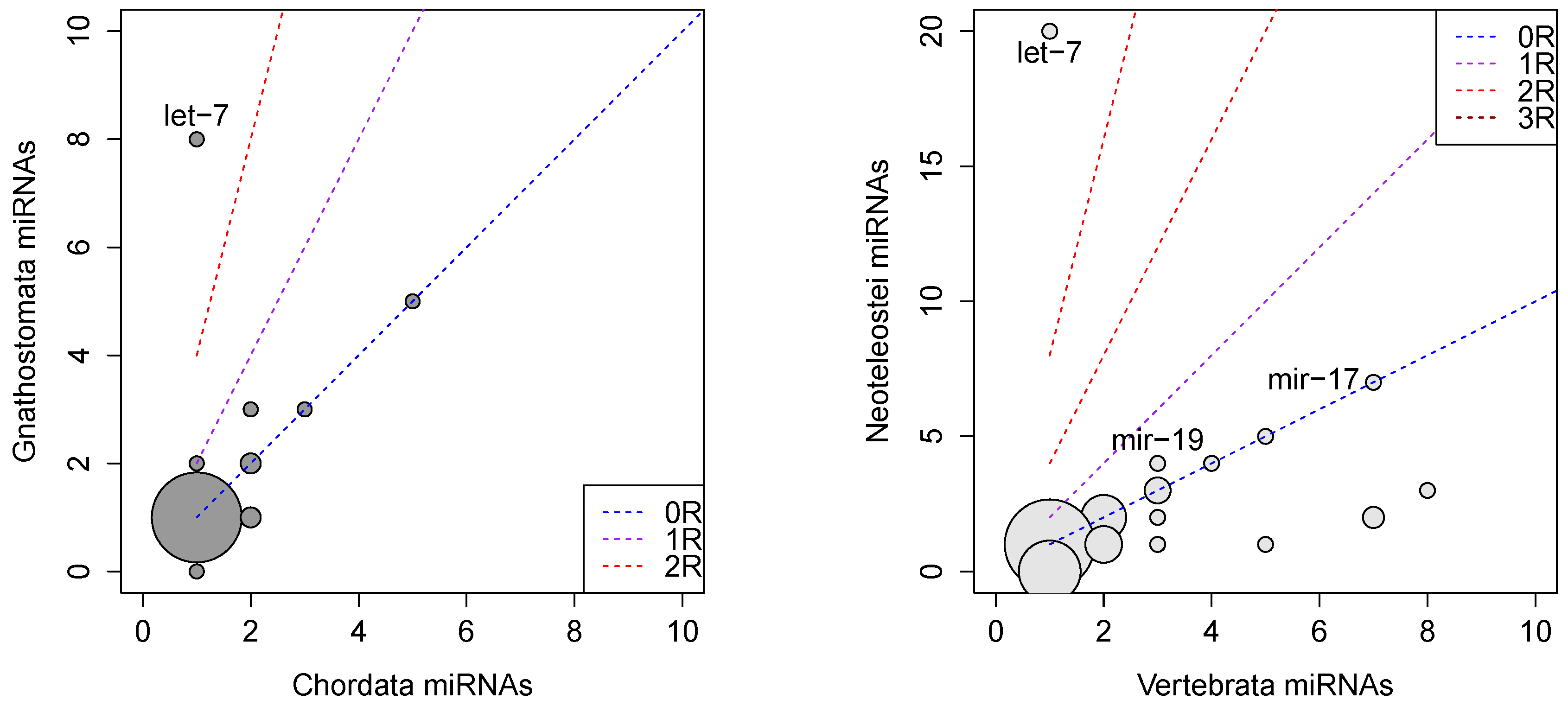
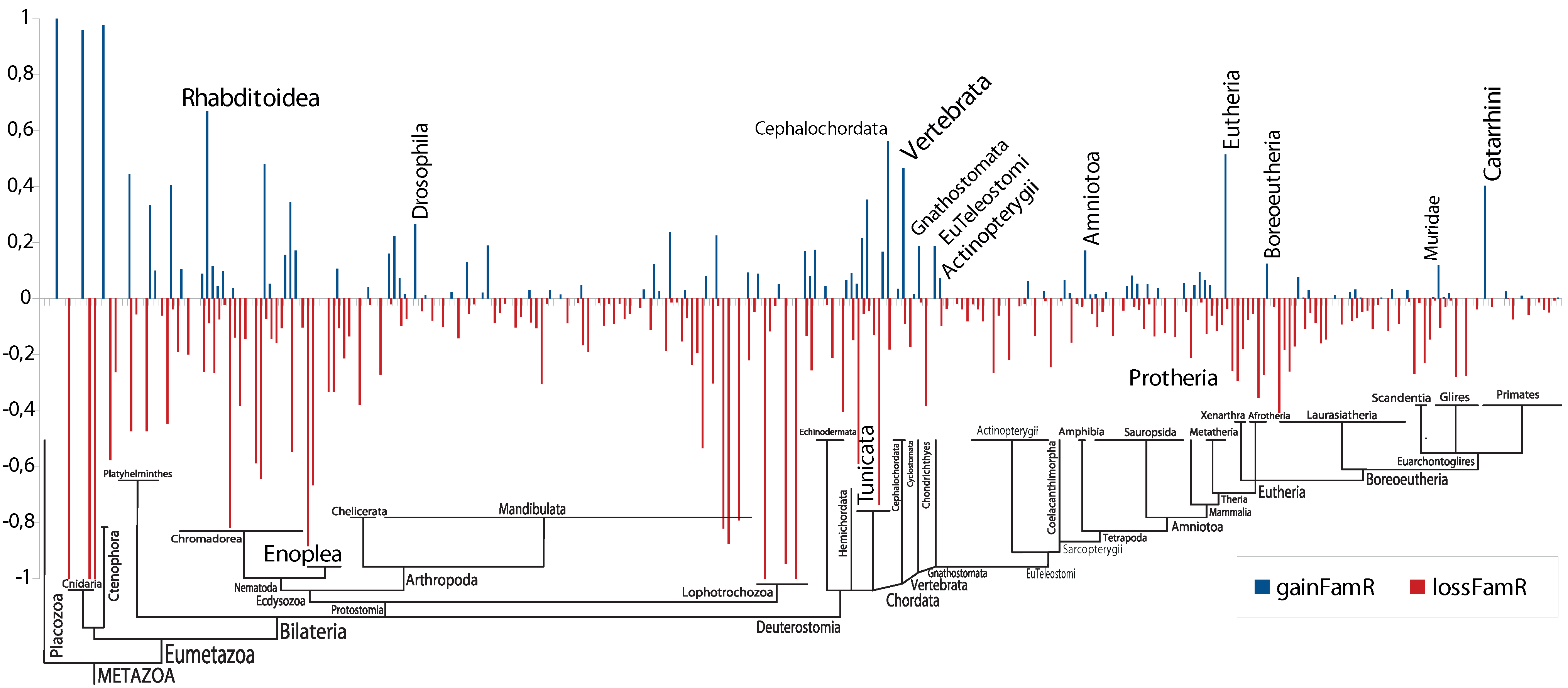
2.3. Gains and Losses of Paralogs
2.4. ePoPE: Efficient Prediction of Paralog Evolution
3. Results and Discussion


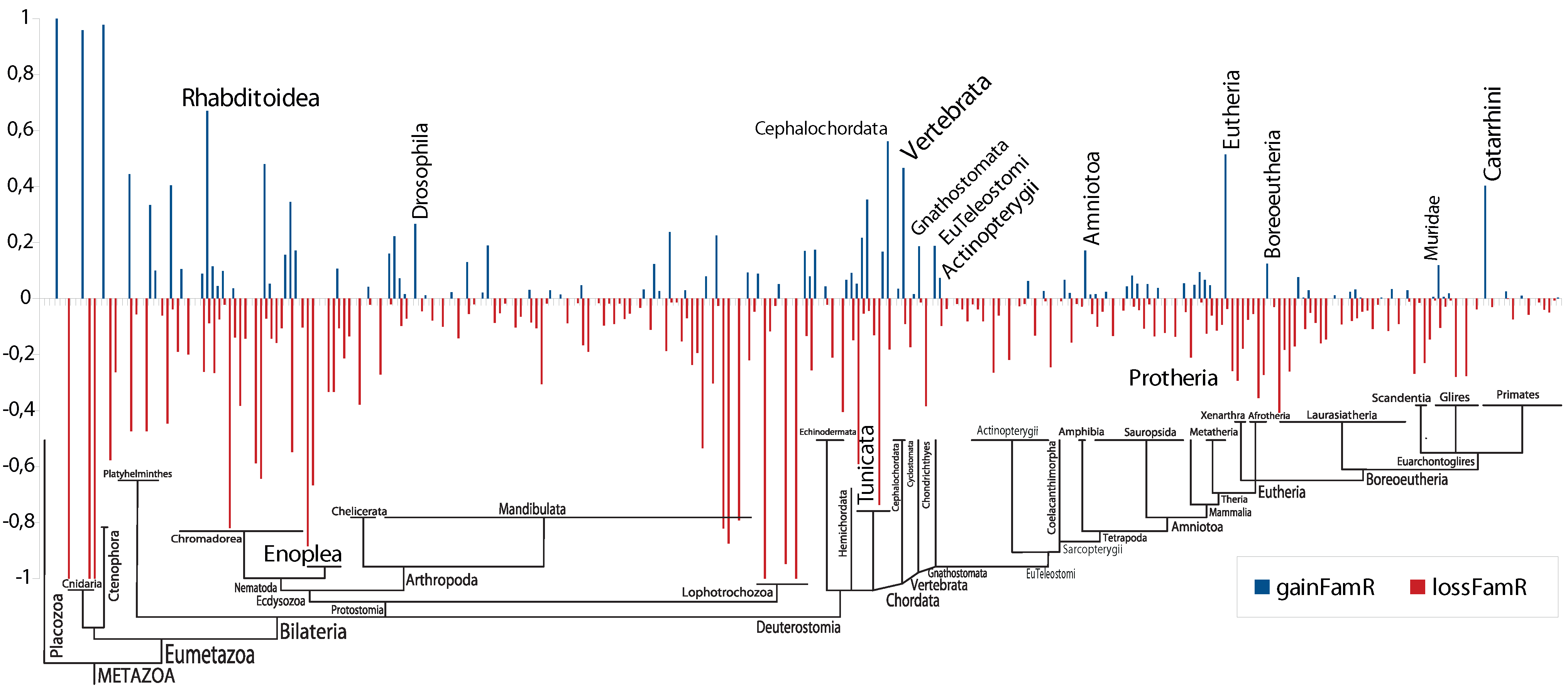
4. Conclusions
Supplementary Files
Supplementary File 1Supplementary File 2Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ameres, S.L.; Zamore, P.D. Diversifying microRNA sequence and function. Nat. Rev. Mol. Cell Biol. 2013, 14, 475–488. [Google Scholar] [PubMed]
- Cerutti, H.; Casas-Mollano, J.A. On the origin and functions of RNA-mediated silencing: From protists to man. Curr. Genet. 2006, 50, 81–99. [Google Scholar] [CrossRef] [PubMed]
- Shabalina, S.A.; Koonin, E.V. Origins and evolution of eukaryotic RNA interference. Trends Ecol. Evol. 2008, 23, 578–587. [Google Scholar] [CrossRef] [PubMed]
- Bologna, N.G.; Schapire, A.L.; Palatnik, J.F. Processing of plant microRNA precursors. Brief. Funct. Genomics 2013, 12, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Militello, K.T.; Refour, P.; Comeaux, C.A.; Duraisingh, M.T. Antisense RNA and RNAi in protozoan parasites: Working hard or hardly working? Mol. Biochem. Parasitol. 2008, 157, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Braun, L.; Cannella, D.; Ortet, P.; Barakat, M.; Sautel, C.F.; Kieffer, S.; Garin, J.; Bastien, O.; Voinnet, O.; Hakimi, M.A.; et al. A complex small RNA repertoire is generated by a plant/fungal-like machinery and effected by a Metazoan-like Argonaute in the single-cell human parasite Toxoplasma gondii. PLoS Pathog. 2010, 6, e1000920. [Google Scholar] [CrossRef] [PubMed]
- Avesson, L.; Reimegård, J.; Wagner, E.G.; Söderbom, F. MicroRNAs in Amoebozoa: Deep sequencing of the small RNA population in the social amoeba Dictyostelium discoideum reveals developmentally regulated microRNAs. RNA 2012, 18, 1771–1782. [Google Scholar] [CrossRef] [PubMed]
- Saraiya, A.A.; Wang, C.C. snoRNA, a novel precursor of microRNA in Giardia lamblia. PLoS Pathog. 2008, 4, e1000224. [Google Scholar] [CrossRef]
- Chen, X.S.; Collins, L.J.; Biggs, P.J.; Penny, D. High throughput genome-wide survey of small RNAs from the parasitic protists Giardia intestinalis and Trichomonas vaginalis. Genome Biol. Evol. 2009, 1, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Price, N.; Cartwright, R.A.; Sabath, N.; Graur, D.; Azevedo, R.B. Neutral evolution of robustness in Drosophila microRNA precursors. Mol. Biol. Evol. 2011, 28, 2115–2123. [Google Scholar] [CrossRef] [PubMed]
- Hertel, J.; Lindemeyer, M.; Missal, K.; Fried, C.; Tanzer, A.; Flamm, C.; Hofacker, I.L.; Stadler, P.F. The Students of Bioinformatics Computer Labs 2004 and 2005. The Expansion of the Metazoan MicroRNA Repertoire. BMC Genomics 2006, 7. [Google Scholar] [CrossRef]
- Tanzer, A.; Stadler, P.F. Molecular Evolution of a MicroRNA Cluster. J. Mol. Biol. 2004, 339, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Hertel, J.; Bartschat, S.; Wintsche, A.; Otto, C.; The Students of the Bioinformatics Computer Lab 2011; Stadler, P.F. Evolution of the let-7 microRNA Family. RNA Biol. 2012, 9, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Ambros, V.; Bartel, B.; Bartel, D.P.; Burge, C.B.; Carrington, J.C.; Chen, X.; Dreyfuss, G.; Eddy, S.R.; Griffiths-Jones, S.; Marshall, M.; et al. A uniform system for microRNA annotation. RNA 2003, 9, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Sempere, L.F.; Cole, C.N.; McPeek, M.A.; Peterson, K.J. The phylogenetic distribution of Metazoan microRNAs: Insights into evolutionary complexity and constraint. J. Exp. Zool. B Mol. Dev. Evol. 2006, 306B, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Heimberg, A.M.; Sempere, L.F.; Moy, V.N.; Donoghue, P.C.J.; Peterson, K. MicroRNAs and the advent of vertebrate morphological complexity. Proc. Natl. Acad. Sci. USA 2007, 105, 2946–2950. [Google Scholar] [CrossRef]
- Heimberg, A.M.; Cowper-Sal·lari, R.; Sémon, M.; Donoghue, P.C.; Peterson, K.J. MicroRNAs reveal the interrelationships of hagfish, lampreys, and gnathostomes and the nature of the ancestral vertebrate. Proc. Natl. Acad. Sci. USA 2010, 107, 19379–19383. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, B.M.; Heimberg, A.M.; Moy, V.N.; Sperling, E.A.; Holstein, T.W.; Heber, S.; Peterson, K.J. The deep evolution of Metazoan microRNAs. Evol. Dev. 2009, 11, 50–68. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Adamski, M.; Thompson, E.M. Altered miRNA Repertoire in the Simplified Chordate, Oikopleura dioica. Mol. Biol. Evol. 2008, 25, 1067–1080. [Google Scholar] [CrossRef] [PubMed]
- Thomson, R.C.; Plachetzki, D.C.; Mahler, D.L.; Moore, B.R. A critical appraisal of the use of microRNA data in phylogenetics. Proc. Natl. Acad. Sci. USA 2014, 111, E3659–E3668. [Google Scholar] [CrossRef] [PubMed]
- Bentwich, I.; Avniel, A.A.; Karov, Y.; Aharonov, R.; Gilad, S.; Barad, O.; Barzilai, A.; Einat, P.; Einav, U.; Meiri, E.; et al. Identification of hundreds of conserved and nonconserved human microRNAs. Nat. Genet. 2005, 37, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Berezikov, E.; Thuemmler, F.; van Laake, L.W.; Kondova, I.; Bontrop, R.; Cuppen, E.; Plasterk, R.H. Diversity of microRNAs in human and chimpanzee brain. Nat. Genet. 2006, 38, 1375–1377. [Google Scholar] [CrossRef]
- Lu, J.; Shen, Y.; Wu, Q.; Kumar, S.; He, B.; Shi, S.; Carthew, R.W.; Wang, S.M.; Wu, C.I. The birth and death of microRNA genes in Drosophila. Nat. Genet. 2008, 40, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Campo-Paysaa, F.; Sémon, M.; Cameron, R.A.; Peterson, K.J.; Schubert, M. MicroRNA complements in deuterostomes: Origin and evolution of microRNAs. Evol. Dev. 2011, 13, 15–27. [Google Scholar] [CrossRef]
- Marco, A.; Ninova, M.; Ronshaugen, M.; Griffiths-Jones, S. Clusters of microRNAs emerge by new hairpins in existing transcripts. Nucleic Acids Res. 2013, 41, 7745–7752. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Li, W. Lowly expressed human microRNA genes evolve rapidly. Mol. Biol. Evol. 2009, 26, 1195–1198. [Google Scholar] [CrossRef] [PubMed]
- Meunier, J.; Lemoine, F.; Soumillon, M.; Liechti, A.; Weier, M.; Guschanski, K.; Hu, H.; Khaitovich, P.; Kaessmann, H. Birth and expression evolution of mammalian microRNA genes. Genome Res. 2012, 23, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.T.; Risom, T.; Strauss, W.M. Evolutionary conservation of microRNA regulatory circuits: An examination of microRNA gene complexity and conserved microRNA-target interactions through Metazoan phylogeny. DNA Cell Biol. 2007, 26, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Niwa, R.; Slack, F.J. The evolution of animal microRNA function. Curr. Opin. Genet. Dev. 2007, 17, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Prochnik, S.E.; Rokhsar, D.S.; Aboobaker, A.A. Evidence for a microRNA expansion in the bilaterian ancestor. Dev. Genes Evol. 2007, 217, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Berezikov, E. Evolution of microRNA diversity and regulation in animals. Nat. Rev. Genet. 2011, 12, 846–860. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Chen, Z.; Ye, H.; Zhou, L.; Cao, L.; Wang, Y.; Peng, S.; Chen, L. Characterization of microRNAs in cephalochordates reveals a correlation between microRNA repertoire homology and morphological similarity in chordate evolution. Evol. Dev. 2009, 11, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Tanzer, A.; Riester, M.; Hertel, J.; Bermudez-Santana, C.I.; Gorodkin, J.; Hofacker, I.L.; Stadler, P.F. Evolutionary Genomics of MicroRNAs and Their Relatives. In Evolutionary Genomics and Systems Biology; Caetano-Anolles, G., Ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2010; pp. 295–327. [Google Scholar]
- Kozomara, A.K.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Will, S.; Reiche, K.; Hofacker, I.L.; Stadler, P.F.; Backofen, R. Inferring Non-Coding RNA Families and Classes by Means of Genome-Scale Structure-Based Clustering. PLoS Comput. Biol. 2007, 3, e65. [Google Scholar] [CrossRef] [PubMed]
- Waegele, J.W.; Bartholomaeus, T.W. (Eds.) Deep Metazoan Phylogeny: The Backbone of the Tree of Life—New Insights from Analyses of Molecules, Morphology, and Theory of Data Analysis; Walter De Gruyter: Berlin, Germany, 2014.
- Smalheiser, N.R.; Torvik, V.I. Mammalian microRNAs derived from genomic repeats. Trends Genet. 2005, 21, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Piriyapongsa, J.; Jordan, I.K. A family of human microRNA genes from miniature inverted-repeat transposable elements. PLoS One 2007, 2, e203. [Google Scholar] [CrossRef] [PubMed]
- Farris, J.S. Phylogenetic analysis under Dollo’s law. Syst. Zool. 1977, 26, 77–88. [Google Scholar] [CrossRef]
- Sankoff, D. Minimal mutation trees of sequences. SIAM J. Appl. Math. 1975, 28, 35–42. [Google Scholar] [CrossRef]
- ePoPE: efficient Prediction of Paralog Evolution. Available online: http://www.bioinf.uni-leipzig.de/Software/ePoPE/ (accessed on 11 February 2015).
- Kasahara, M. Impact of whole-genome duplication on vertebrate development and evolution. Semin. Cell Dev. Biol. 2013, 24, 81–82. [Google Scholar] [CrossRef] [PubMed]
- Lozada-Chávez, I.; Stadler, P.F.; Prohaska, S.J. “Hypothesis for the modern RNA world”: A pervasive non-coding RNA-based genetic regulation is a prerequisite for the emergence 2 of multicellular complexity. Orig. Life Evol. Biosph. 2011, 41, 587–607. [Google Scholar] [CrossRef] [PubMed]
- Peterson, K.J.; Dietrich, M.R.; McPeek, M.A. MicroRNAs and Metazoan macroevolution: Insights into canalization, complexity, and the Cambrian explosion. BioEssays 2009, 31, 736–747. [Google Scholar] [CrossRef] [PubMed]
- Postlethwait, J.H.; Woods, I.G.; Ngo-Hazelett, P.; Yan, Y.L.; Kelly, P.D.; Chu, F.; Huang, H.; Hill-Force, A.; Talbot, W.S. Zebrafish comparative genomics and the origins of vertebrate chromosomes. Genome Res. 2000, 10, 1890–1902. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.; Conery, J.S. The evolutionary fate and consequences of duplicate genes. Science 2000, 290, 1151–1155. [Google Scholar] [CrossRef] [PubMed]
- Pascual-Anaya, J.; D’Aniello, S.; Kuratani, S.; Garcia-Fernàndez, J. Evolution of Hox gene clusters in deuterostomes. BMC Dev. Biol. 2013, 13. [Google Scholar] [CrossRef]
- Lemaire, P. Evolutionary crossroads in developmental biology: The tunicates. Development 2011, 138, 2143–2152. [Google Scholar] [CrossRef] [PubMed]
- Schulze, J.; Schierenberg, E. Embryogenesis of Romanomermis culicivorax: An alternative way to construct a nematode. Dev. Biol. 2009, 334, 10–21. [Google Scholar] [CrossRef]
- Sperling, E.; Vinther, J.; Moy, V.; Wheeler, B.; Semon, M.; Briggs, D.; Peterson, K. MicroRNAs resolve an apparent conflict between annelid systematics and their fossil record. Proc. Biol. Sci. 2009, 276, 4315–4322. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Fang, X.; Guo, X.; Li, L.; Luo, R.; Xu, F.; Yang, P.; Zhang, L.; Wang, X.; Qi, H.; et al. The oyster genome reveals stress adaptation and complexity of shell formation. Nature 2012, 490, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Simakov, O.; Marletaz, F.; Cho, S.; Edsinger-Gonzales, E.; Havlak, P.; Hellsten, U.; Kuo, D.; Larsson, T.; Lv, J.; Arendt, D.; et al. Insights into bilaterian evolution from three spiralian genomes. Nature 2013, 493, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Flot, J.; Hespeels, B.; Li, X.; Noel, B.; Arkhipova, I.; Danchin, E.; Hejnol, A.; Henrissat, B.; Koszul, R.; Aury, J.; et al. Genomic evidence for ameiotic evolution in the bdelloid rotifer Adineta vaga. Nature 2013, 500, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Menzel, P.; Gorodkin, J.; Stadler, P.F. The Tedious Task of Finding Homologous Non-coding RNA Genes. RNA 2009, 15, 2075–2082. [Google Scholar] [CrossRef] [PubMed]
- Devor, E.J. Primate MicroRNAs miR-220 and miR-492 Lie within Processed Pseudogenes. J. Hered. 2005, 97, 186–190. [Google Scholar] [CrossRef]
- Parikesit, A.A.; Steiner, L.; Stadler, P.F.; Prohaska, S.J. Pitfalls of Ascertainment Biases in Genome Annotations—Computing Comparable Protein Domain Distributions in Eukarya. Malays. J. Fundam. Appl. Sci. 2014, 10, 65–75. [Google Scholar]
- Marco, A.; Hooks, K.; Griffiths-Jones, S. Evolution and function of the extended miR-2 microRNA family. RNA Biol. 2012, 9, 242–248. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hertel, J.; Stadler, P.F. The Expansion of Animal MicroRNA Families Revisited. Life 2015, 5, 905-920. https://doi.org/10.3390/life5010905
Hertel J, Stadler PF. The Expansion of Animal MicroRNA Families Revisited. Life. 2015; 5(1):905-920. https://doi.org/10.3390/life5010905
Chicago/Turabian StyleHertel, Jana, and Peter F. Stadler. 2015. "The Expansion of Animal MicroRNA Families Revisited" Life 5, no. 1: 905-920. https://doi.org/10.3390/life5010905
APA StyleHertel, J., & Stadler, P. F. (2015). The Expansion of Animal MicroRNA Families Revisited. Life, 5(1), 905-920. https://doi.org/10.3390/life5010905