
The Search for Disease Modification in Parkinson’s Disease—A Review of the Literature


 ,
,
Abstract
1. Parkinson’s Disease—Epidemiology, Aetiology, Current Treatments and the Search for Disease Modification
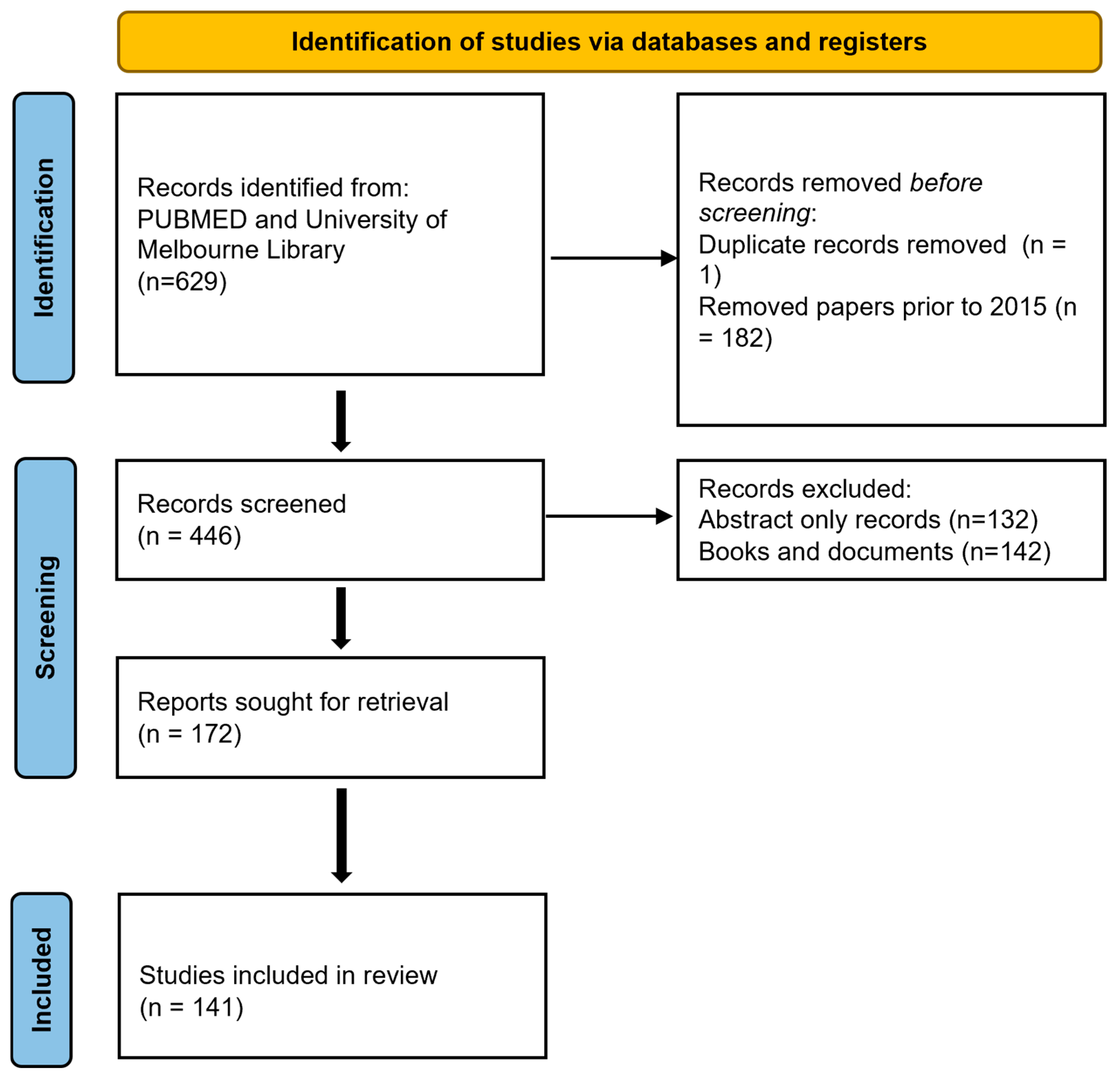
2. Methodology
3. Neuropathophysiology of Parkinson’s Disease—What We Know
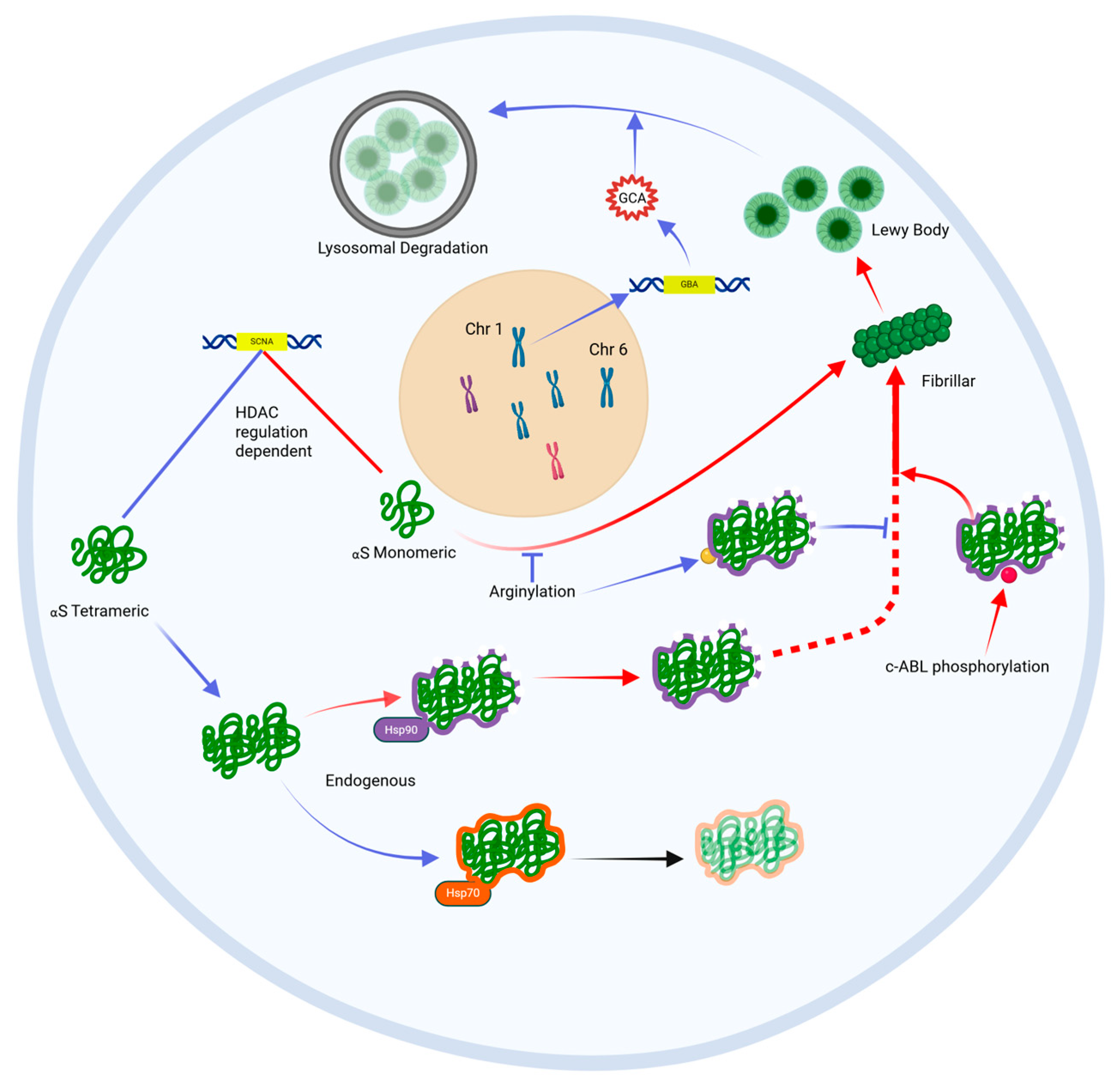
3.1. Alpha-Synuclein Aggregates, Known as Lewy Bodies, Are the Histopathological Hallmark of PD
3.2. Oxidative Stress and Mitochondrial Dysfunction Induce Innate Immune Processes That Drive Cell Death in PD
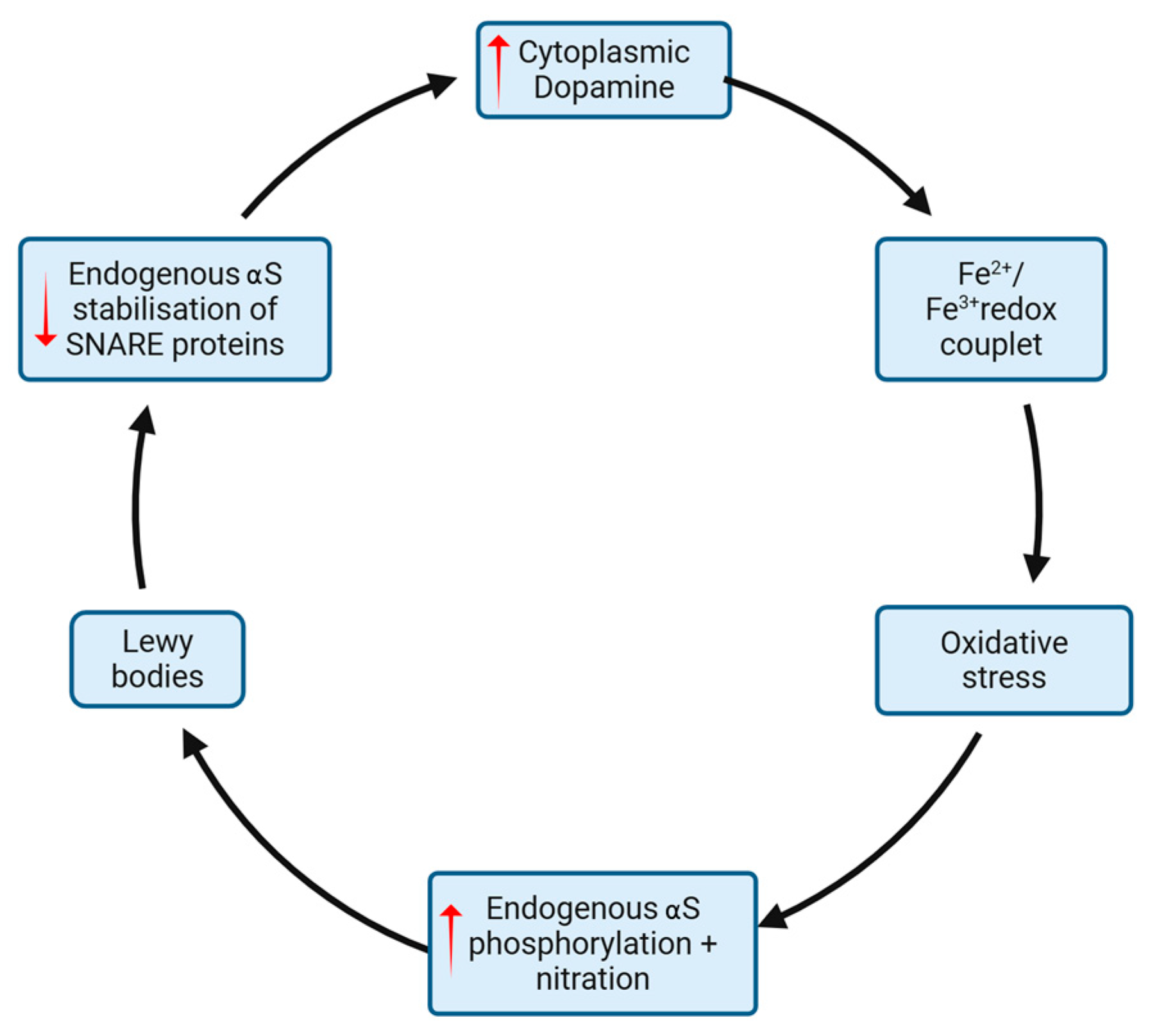
3.3. Reciprocal Reinforcement of Oxidative Stress and Alpha-Synuclein Aggregation
4. Targets for Disease Modification (See Table 1)
4.1. Prevention and Attenuation of Alpha-Synuclein Aggregation
4.2. Modification of Oxidative Stress, Mitochondrial Dysfunction, and Neuroinflammation
4.3. Regeneration of Lost Neurons
4.4. Repurposing Old Drugs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Intervention/Agent | Mechanism of Action | Model of Disease | Outcome/Status | Reference |
|---|---|---|---|---|
| Alpha-SynucleinTargeted | ||||
| ABT-888 | PARP-1 inhibitor | dSTR aS injected C57BL/6j mice | Suppression of aS fibrillar formation | [40] |
| Salbutamol | Beta-2 receptor mediated SNCA gene suppression | Norwegian population association study (4 million people) | PD incidence in regular salbutamol users vs. non-users, OR = 0.66 (CI 0.58 to 0.76) | [55] |
| SIFTO | m6A-dependent regulation of ATM mRNA | MPTP mouse model | Suppression of aS upregulation and reduced dopaminergic neuron death | [56] |
| GELDENAMYCIN | HSP70 induction | MPTP mouse model | Reduced dopaminergic neuron loss | [62] |
| Nanoca | Upregulation of tfEB mediated aS clearance | MPP+ mouse model | Reduced dopaminergic neuron loss | [64] |
| GYY4137 | Reduced oxidative stress and reduced aS nitration | MPTP mouse model | Reduced dopaminergic neuron loss | [67] |
| NILOTINIB | C-abl inhibitor | MPTP mouse model | Reduced dopaminergic neuron loss | [68] |
| Oxidative Stress/Mitochondria | ||||
| NAC (ORAL + INTRAVENOUS) | Anti-oxidant | 6-OHDA mouse model | Enhanced dopaminergic neuron viability | [82] |
| CELASTROL | Anti-oxidant, NrF2 pathway activation | MPTP mouse model | Enhanced dopaminergic neuron viability | [87] |
| AEOL11207 + AEOL11114 (METALLOPORPHYRINS) | Anti-oxidant | 6-OHDA mouse model | Reduced cytokine production, reduced microglial activity and enhanceddopaminergic neuron viability | [88] |
| RAPAMYCIN + COENZYME Q10 | Anti-oxidant | Ex vivo stem cells with LRRK2 or PINK-1 mutation | Mitochondrial protection | [89] |
| UFP-512 | Delta-opioid receptor agonist | Ex vivo PC12 cells under oxidative stress | Upregulated PARKN mediated mitophagy | [90] |
| HYDROCORTISONE | PARKN upregulation | 6OHDA mouse model | Enhanced dopaminergic neuron viability | [91] |
| Immune Modulation | ||||
| C-DIM12 | Microglial modification | MPTP mouse model | Enhanced dopaminergic neuron viability | [94] |
| LYPOSOME VECTOR DELIVERY OF HYDROCORTISONE | Macrophage modificatoin | 6-OHDA mouse model | Enhanced dopaminergic neuron viability | [95] |
| MDG548 | PPAR-gamma mediated immunomodulation | MPTP mouse model | Enhanced dopaminergic neuron viability | [96] |
| PHLOROGLUCINOL DERIVATIVES | Src/phosphatase mediated immunomodulation | MPTP mouse model | Reduced neuroinflammation | [97] |
| FINGOLIMOD | S1p modulation | 6-OHDA | Enhanced dopaminergic neuron viability | [119] |
| Intervention/Agent | Mechanism of Action | Trial Phase | Outcome | Reference |
|---|---|---|---|---|
| Alpha-Synuclein | ||||
| UCB0599 | Inhibition of aS misfolding and aggregation | Phase 1/1b | Good tolerability amongst 94 volunteers | [60] |
| NILOTINIB | c-abl tyrosine kinase inhibitor | Phase 1 | 75 PD patients—well tolerated, agent detectible in CSF | [70] |
| VODOBATINIB | c-abl tyrosine kinase inhibitor | Phase 1 | Good tolerability, superior CSF penetrance to nilotinib | [72] |
| PDO1A | Active immunistation against aS | Phase 1 | 32 participants with mild PD showed reduced CSF aS concentrations | [73] |
| PRASINEZUMAB | Passive immunotherapy against aS | Phases 1 and 2 | 316 mild PD participants—good tolerability, failed to reach primary endpoint | [75,76] |
| CINPANEMAB | Passive immunotherapy against aS | Phase 2 | 357 mild PD participants—failed to reach primary endpoint | [77] |
| Oxidative Stress/Mitochondria | ||||
| VITAMIN E | Termination of oxidative chain reactions | Cohort meta-analysis Phase 2 (DATATOP) | Reduced IPD incidence in vitamin E users 800 early PD participants randomised— Modest reduction in UPDRS score at 12 weeks compared with placebo | [78,80] |
| IV + ORAL NAC | Anti-oxidant | Phase 1 | 42 participants with early PD showed improved DAT binding in caudate and putamen | [83] |
| INTRANASAL GLUTATHIONE | Anti-oxidant | Phase 2 | 45 participants HY stage 1–3 PD randomised —no improvement in UPDRS from placebo over three month treatment | [84] |
| INOSINE | Induction of urate-mediated anti-oxidant properties | Phase 1 Phase 2 (SURE-PD3) | Increased CSF urate, well tolerated 298 participants with PD randomised—failed to reach primary endpoint | [85,86] |
| INTRAPUTAMENAL INJECTION OF CDNF | Neurotrophic stimulation of putamenal stem cells | Phase 1 | Goold tolerability | [106] |
| ISRADIPINE | ?blockade of Ca2+ channel induced oxidative stress | Phase 3 | 336 participants with early PD randomised—failed to reach primary endpoitn | [112] |
| NLY01 (EXENATIDE) | GLP-1 agonist -Modification of microglia | Phase 2 | 255 participants with early PD randomised—failed to reach primary endpoint | [117] |
5. COVID-19: A Unique Window into the Pathogenesis of IPD?
6. Conclusions and ‘Where to from Here’
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACE2 | Angiotensin converting enzyme 2 | MPTP | Methyl-phenyl-tetrahydro-pyridine |
| AD | Alzheimer’s disease | mRNA | messenger ribose nucleic acid |
| ADP | Adenosine diphosphate | MSA | Multisystem atrophy |
| AIF1 | Apoptosis-inducing factor | mtPTP | Mitochondrial permeability transition pore |
| aS | alpha-synuclein | NAC | N-acetyl cysteine |
| BAX | BCL2-associated X protein | NLR | Neutrophil-to-lymphocyte ratio |
| BDNF | Brain derived neurotrophic factor | Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| Ca2+ | Calcium cations | PARP | Poly ADP-ribose polymerase |
| c-abl | cellular Abelson murine leukaemia | PC12 | Phaechromocytoma cell line |
| viral oncogene homolog 1 | PD | Parkinson’s Disease | |
| CBD | Cannabidiol | PDD | Parkinson’s Disease Dementia |
| CDNF | Cerebral dopamine neurotrophic factor | PEP | Post-encephalitic parkinsonism |
| Chr | Chromosome | PET | Positron emission tomography |
| CNS | Central nervous system | PPR | Peroxisome proliferator-activated receptor |
| COVID | Coronavirus disease | REM | Rapid eye movement |
| CSF | cerebrospinal fluid | ROS | reactive oxygen species |
| Cyt c | Cytochrome C | RT-QuiC | Real-time quaking-induced conversion |
| DAT | Dopamine transporter | SNCA/SCNA | synuclein alpha |
| DLB | Dementia with lewy bodies | SNpc | Substantia nigra pars compacta |
| Fe2+/3+ | Iron cations | SPECT | Single photon emission computed tomography |
| GBA/GCA | Glucocerebrosidase | SWEDD | Scans without evidence of dopamine deficiency |
| GLB | Glucagon-like peptide | S1P | Sphingosine-1-phosphate |
| HAT | Histone acetyltransferase | S129 | Serine 129 |
| HDAC | Histone deacetylatse | THC | etrahydrocannabinoid |
| HSF-1 | Heat shock factor 1 | TLR | Toll like receptor |
| Hsp | Heat shock protein | TNF | Tumour necrosis factor |
| H2S | Hydrogen sulfide | VEGF | Vascular endothelial growth factor |
| IFN | interferon | VMAT | Vesicular monoamine transporter |
| IL | Interleukin | VPS35 | Vacuolar Protein Sorting 35 |
| LRRK2 | Leucine-rich repeat kinase 2 | 6-OHDA | 6-hydroxydopamine |
| MAO | Monoamine oxidase | ||
| MAPT | Microtubule-associated protein tau | ||
| MDS-UPDRS | Movement disorders society-unified Parkinson’s disease rating scale | ||
| MPP | methyl-phenyl-pyridinium |
References
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- Hung, A.Y.; Schwarzschild, M.A. Approaches to Disease Modification for Parkinson’s Disease: Clinical Trials and Lessons Learned. Neurotherapeutics 2020, 17, 1393–1405. [Google Scholar] [CrossRef]
- Grenn, F.P.; Kim, J.J.; Makarious, M.B.; Iwaki, H.; Illarionova, A.; Brolin, K.; Kluss, J.H.; Schumacher-Schuh, A.F.; Leonard, H.; Faghri, F.; et al. The Parkinson’s Disease Genome-Wide Association Study Locus Browser. Mov. Disord. 2020, 35, 2056–2067. [Google Scholar] [CrossRef]
- Kim, C.Y.; Alcalay, R.N. Genetic Forms of Parkinson’s Disease. Semin. Neurol. 2017, 37, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Fearnley, J.M.; Lees, A.J. Ageing and Parkinson’s disease: Substantia nigra regional selectivity. Brain 1991, 114 Pt 5, 2283–2301. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.; Seppi, K.; Marini, K.; Mahlknecht, P. New hopes for disease modification in Parkinson’s Disease. Neuropharmacology 2020, 171, 108085. [Google Scholar] [CrossRef] [PubMed]
- Jackson, H.; Anzures-Cabrera, J.; Simuni, T.; Postuma, R.B.; Marek, K.; Pagano, G. Identifying prodromal symptoms at high specificity for Parkinson’s disease. Front. Aging Neurosci. 2023, 15, 1232387. [Google Scholar] [CrossRef]
- Fengler, S.; Liepelt-Scarfone, I.; Brockmann, K.; Schaffer, E.; Berg, D.; Kalbe, E. Cognitive changes in prodromal Parkinson’s disease: A review. Mov. Disord. 2017, 32, 1655–1666. [Google Scholar] [CrossRef]
- Fang, C.; Lv, L.; Mao, S.; Dong, H.; Liu, B. Cognition Deficits in Parkinson’s Disease: Mechanisms and Treatment. Park. Dis. 2020, 2020, 2076942. [Google Scholar] [CrossRef]
- Poewe, W.; Antonini, A.; Zijlmans, J.C.; Burkhard, P.R.; Vingerhoets, F. Levodopa in the treatment of Parkinson’s disease: An old drug still going strong. Clin. Interv. Aging 2010, 5, 229–238. [Google Scholar] [CrossRef]
- Barbagallo, G.; Quattrone, A. Does levodopa have a disease-modifying effect in Parkinson’s disease? Evidence from a delayed-start trial. Mov. Disord. 2019, 34, 820. [Google Scholar] [CrossRef]
- Fahn, S.; Oakes, D.; Shoulson, I.; Kieburtz, K.; Rudolph, A.; Lang, A.; Olanow, C.W.; Tanner, C.; Marek, K.; Parkinson Study Group. Levodopa and the Progression of Parkinson’s Disease. N. Engl. J. Med. 2004, 351, 2498–2508. [Google Scholar]
- Marsden, C.D. Problems with long-term levodopa therapy for Parkinson’s disease. Clin. Neuropharmacol. 1994, 17 (Suppl. S2), S32–S44. [Google Scholar] [CrossRef]
- Kordower, J.H.; Burke, R.E. Disease Modification for Parkinson’s Disease: Axonal Regeneration and Trophic Factors. Mov. Disord. 2018, 33, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Kalia, S.K.; Lang, A.E. Disease-modifying strategies for Parkinson’s disease. Mov. Disord. 2015, 30, 1442–1450. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, E.; Gomes, M.D.M. Lewy and his inclusion bodies: Discovery and rejection. Dement. Neuropsychol. 2017, 11, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K.; Rub, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Hilker, R.; Schweitzer, K.; Coburger, S.; Ghaemi, M.; Weisenbach, S.; Jacobs, A.H.; Rudolf, J.; Herholz, K.; Heiss, W.-D. Nonlinear progression of Parkinson disease as determined by serial positron emission tomographic imaging of striatal fluorodopa F 18 activity. Arch. Neurol. 2005, 62, 378–382. [Google Scholar] [CrossRef]
- Athauda, D.; Foltynie, T. Challenges in detecting disease modification in Parkinson’s disease clinical trials. Park. Relat. Disord. 2016, 32, 1–11. [Google Scholar] [CrossRef]
- Burre, J.; Sharma, M.; Sudhof, T.C. Cell Biology and Pathophysiology of alpha-Synuclein. Cold Spring Harb. Perspect. Med. 2018, 8, a024091. [Google Scholar] [CrossRef]
- Melki, R. Alpha-synuclein and the prion hypothesis in Parkinson’s disease. Rev. Neurol. 2018, 174, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Mazzocchi, M.; Collins, L.M.; Sullivan, A.M.; O’Keeffe, G.W. The class II histone deacetylases as therapeutic targets for Parkinson’s disease. Neuronal Signal. 2020, 4, Ns20200001. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.A.; D’Mello, S.R. Complex neuroprotective and neurotoxic effects of histone deacetylases. J. Neurochem. 2018, 145, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Park, G.; Tan, J.; Garcia, G.; Kang, Y.; Salvesen, G.; Zhang, Z. Regulation of Histone Acetylation by Autophagy in Parkinson Disease. J. Biol. Chem. 2016, 291, 3531–3540. [Google Scholar] [CrossRef]
- Friesen, E.L.; De Snoo, M.L.; Rajendran, L.; Kalia, L.V.; Kalia, S.K. Chaperone-Based Therapies for Disease Modification in Parkinson’s Disease. Park. Dis. 2017, 2017, 5015307. [Google Scholar] [CrossRef]
- Tao, J.; Berthet, A.; Citron, Y.R.; Tsiolaki, P.L.; Stanley, R.; Gestwicki, J.E.; Agard, D.A.; McConlogue, L. Hsp70 chaperone blocks alpha-synuclein oligomer formation via a novel engagement mechanism. J. Biol. Chem. 2021, 296, 100613. [Google Scholar] [CrossRef]
- Riboldi, G.M.; Di Fonzo, A.B. GBA, Gaucher Disease, and Parkinson’s Disease: From Genetic to Clinic to New Therapeutic Approaches. Cells 2019, 8, 364. [Google Scholar] [CrossRef]
- Gan-Or, Z.; Bar-Shira, A.; Gurevich, T.; Giladi, N.; Orr-Urtreger, A. Homozygosity for the MTX1 c.184T>A (p.S63T) alteration modifies the age of onset in GBA-associated Parkinson’s disease. Neurogenetics 2011, 12, 325–332. [Google Scholar] [CrossRef]
- Glajch, K.E.; Moors, T.E.; Chen, Y.; Bechade, P.A.; Nam, A.Y.; Rajsombath, M.M.; McCaffery, T.D.; Dettmer, U.; Weihofen, A.; Hirst, W.D.; et al. Wild-type GBA1 increases the alpha-synuclein tetramer-monomer ratio, reduces lipid-rich aggregates, and attenuates motor and cognitive deficits in mice. Proc. Natl. Acad. Sci. USA 2021, 118, e2103425118. [Google Scholar] [CrossRef]
- Pan, B.; Kamo, N.; Shimogawa, M.; Huang, Y.; Kashina, A.; Rhoades, E.; Petersson, E.J. Effects of Glutamate Arginylation on alpha-Synuclein: Studying an Unusual Post-Translational Modification through Semisynthesis. J. Am. Chem. Soc. 2020, 142, 21786–21798. [Google Scholar] [CrossRef]
- Mahul-Mellier, A.L.; Fauvet, B.; Gysbers, A.; Dikiy, I.; Oueslati, A.; Georgeon, S.; Lamontanara, A.J.; Bisquertt, A.; Eliezer, D.; Masliah, E.; et al. c-Abl phosphorylates alpha-synuclein and regulates its degradation: Implication for alpha-synuclein clearance and contribution to the pathogenesis of Parkinson’s disease. Hum. Mol. Genet. 2014, 23, 2858–2879. [Google Scholar] [CrossRef]
- Anderson, J.P.; Walker, D.E.; Goldstein, J.M.; de Laat, R.; Banducci, K.; Caccavello, R.J.; Barbour, R.; Huang, J.; Kling, K.; Lee, M.; et al. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 2006, 281, 29739–29752. [Google Scholar] [CrossRef] [PubMed]
- Arawaka, S.; Sato, H.; Sasaki, A.; Koyama, S.; Kato, T. Mechanisms underlying extensive Ser129-phosphorylation in alpha-synuclein aggregates. Acta Neuropathol. Commun. 2017, 5, 48. [Google Scholar] [CrossRef] [PubMed]
- Ghanem, S.S.; Majbour, N.K.; Vaikath, N.N.; Ardah, M.T.; Erskine, D.; Jensen, N.M.; Fayyad, M.; Sudhakaran, I.P.; Vasili, E.; Melachroinou, K.; et al. α-Synuclein phosphorylation at serine 129 occurs after initial protein deposition and inhibits seeded fibril formation and toxicity. Proc. Natl. Acad. Sci. USA 2022, 119, e2109617119. [Google Scholar] [CrossRef] [PubMed]
- Irwin, D.J.; Lee, V.M.; Trojanowski, J.Q. Parkinson’s disease dementia: Convergence of alpha-synuclein, tau and amyloid-beta pathologies. Nat. Rev. Neurosci. 2013, 14, 626–636. [Google Scholar] [CrossRef]
- van der Gaag, B.L.; Deshayes, N.A.C.; Breve, J.J.P.; Bol, J.; Jonker, A.J.; Hoozemans, J.J.M.; Courade, J.-P.; van de Berg, W.D.J. Distinct tau and alpha-synuclein molecular signatures in Alzheimer’s disease with and without Lewy bodies and Parkinson’s disease with dementia. Acta Neuropathol. 2024, 147, 14. [Google Scholar] [CrossRef]
- Skaper, S.D.; Facci, L.; Zusso, M.; Giusti, P. An Inflammation-Centric View of Neurological Disease: Beyond the Neuron. Front. Cell. Neurosci. 2018, 12, 72. [Google Scholar] [CrossRef]
- Bjorklund, G.; Dadar, M.; Anderson, G.; Chirumbolo, S.; Maes, M. Preventive treatments to slow substantia nigra damage and Parkinson’s disease progression: A critical perspective review. Pharmacol. Res. 2020, 161, 105065. [Google Scholar] [CrossRef]
- Park, H.A.; Ellis, A.C. Dietary Antioxidants and Parkinson’s Disease. Antioxidants 2020, 9, 570. [Google Scholar] [CrossRef]
- Hastings, L.; Sokratian, A.; Apicco, D.J.; Stanhope, C.M.; Smith, L.; Hirst, W.D.; West, A.B.; Kelly, K. Evaluation of ABT-888 in the amelioration of alpha-synuclein fibril-induced neurodegeneration. Brain Commun. 2022, 4, fcac042. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef]
- Wallings, R.; Manzoni, C.; Bandopadhyay, R. Cellular processes associated with LRRK2 function and dysfunction. FEBS J. 2015, 282, 2806–2826. [Google Scholar] [CrossRef]
- Le, W.; Wu, J.; Tang, Y. Protective Microglia and Their Regulation in Parkinson’s Disease. Front. Mol. Neurosci. 2016, 9, 89. [Google Scholar] [CrossRef]
- Reynolds, A.; Laurie, C.; Mosley, R.L.; Gendelman, H.E. Oxidative stress and the pathogenesis of neurodegenerative disorders. Int. Rev. Neurobiol. 2007, 82, 297–325. [Google Scholar]
- Schwab, A.D.; Thurston, M.J.; Machhi, J.; Olson, K.E.; Namminga, K.L.; Gendelman, H.E.; Mosley, R.L. Immunotherapy for Parkinson’s disease. Neurobiol. Dis. 2020, 137, 104760. [Google Scholar] [CrossRef] [PubMed]
- Grillo, P.; Sancesario, G.M.; Bovenzi, R.; Zenuni, H.; Bissacco, J.; Mascioli, D.; Simonetta, C.; Forti, P.; Degoli, G.R.; Pieri, M.; et al. Neutrophil-to-lymphocyte ratio and lymphocyte count reflect alterations in central neurodegeneration-associated proteins and clinical severity in Parkinson Disease patients. Park. Relat. Disord. 2023, 112, 105480. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhao, Y.; Zhu, G. The role of sphingosine-1-phosphate in the development and progression of Parkinson’s disease. Front. Cell. Neurosci. 2023, 17, 1288437. [Google Scholar] [CrossRef] [PubMed]
- Schwedhelm, E.; Englisch, C.; Niemann, L.; Lezius, S.; von Lucadou, M.; Marmann, K.; Böger, R.; Peine, S.; Daum, G.; Gerloff, C.; et al. Sphingosine-1-Phosphate, Motor Severity, and Progression in Parkinson’s Disease (MARK-PD). Mov. Disord. 2021, 36, 2178–2182. [Google Scholar] [CrossRef]
- Barrett, P.J.; Timothy Greenamyre, J. Post-translational modification of alpha-synuclein in Parkinson’s disease. Brain Res. 2015, 1628 Pt B, 247–253. [Google Scholar] [CrossRef]
- Pezzella, A.; d’Ischia, M.; Napolitano, A.; Misuraca, G.; Prota, G. Iron-mediated generation of the neurotoxin 6-hydroxydopamine quinone by reaction of fatty acid hydroperoxides with dopamine: A possible contributory mechanism for neuronal degeneration in Parkinson’s disease. J. Med. Chem. 1997, 40, 2211–2216. [Google Scholar] [CrossRef]
- Barbosa, J.H.; Santos, A.C.; Tumas, V.; Liu, M.; Zheng, W.; Haacke, E.M.; Salmon, C.E.G. Quantifying brain iron deposition in patients with Parkinson’s disease using quantitative susceptibility mapping, R2 and R2. Magn. Reason. Imaging 2015, 33, 559–565. [Google Scholar] [CrossRef]
- De Franceschi, G.; Fecchio, C.; Sharon, R.; Schapira, A.H.V.; Proukakis, C.; Bellotti, V.; de Laureto, P.P. alpha-Synuclein structural features inhibit harmful polyunsaturated fatty acid oxidation, suggesting roles in neuroprotection. J. Biol. Chem. 2017, 292, 6927–6937. [Google Scholar] [CrossRef]
- Abdelmotilib, H.; West, A.B. Breathing new life into an old target: Pulmonary disease drugs for Parkinson’s disease therapy. Genome Med. 2017, 9, 88. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; Bjørnevik, K.; Im, D.S.; Flierl, A.; Dong, X.; Locascio, J.J.; Abo, K.M.; Long, E.; Jin, M.; Xu, B.; et al. β2-Adrenoreceptor is a regulator of the α-synuclein gene driving risk of Parkinson’s disease. Science 2017, 357, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Hopfner, F.; Höglinger, G.U.; Kuhlenbäumer, G.; Pottegård, A.; Wod, M.; Christensen, K.; Tanner, C.M.; Deuschl, G. β-adrenoreceptors and the risk of Parkinson’s disease. Lancet Neurol. 2020, 19, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Long, X.; Zhang, Y.; Wang, Y.; You, G.; Guo, W.; Zhuang, G.; Zhang, Y.; Cheng, X.; Yuan, Z.; et al. FTO-targeted siRNA delivery by MSC-derived exosomes synergistically alleviates dopaminergic neuronal death in Parkinson’s disease via m6A-dependent regulation of ATM mRNA. J. Transl. Med. 2023, 21, 652. [Google Scholar] [CrossRef]
- den Heijer, J.M.; Kruithof, A.C.; Moerland, M.; Walker, M.; Dudgeon, L.; Justman, C.; Solomini, I.; Splitalny, L.; Leymarie, N.; Khatri, K.; et al. A Phase 1B Trial in GBA1-Associated Parkinson’s Disease of BIA-28-6156, a Glucocerebrosidase Activator. Mov. Disord. 2023, 38, 1197–1208. [Google Scholar] [CrossRef]
- Wang, Q.; Luo, Y.; Ray Chaudhuri, K.; Reynolds, R.; Tan, E.K.; Pettersson, S. The role of gut dysbiosis in Parkinson’s disease: Mechanistic insights and therapeutic options. Brain 2021, 144, 2571–2593. [Google Scholar] [CrossRef]
- Sun, M.F.; Zhu, Y.L.; Zhou, Z.L.; Jia, X.B.; Xu, Y.D.; Yang, Q.; Cui, C.; Shen, Y.Q. Neuroprotective effects of fecal microbiota transplantation on MPTP-induced Parkinson’s disease mice: Gut microbiota, glial reaction and TLR4/TNF-α signaling pathway. Brain Behav. Immun. 2018, 70, 48–60. [Google Scholar] [CrossRef]
- Smit, J.W.; Basile, P.; Prato, M.K.; Detalle, L.; Mathy, F.X.; Schmidt, A.; Lalla, M.; Germani, M.; Domange, C.; Biere, A.L.; et al. Phase 1/1b Studies of UCB0599, an Oral Inhibitor of α-Synuclein Misfolding, Including a Randomized Study in Parkinson’s Disease. Mov. Disord. 2022, 37, 2045–2056. [Google Scholar] [CrossRef]
- Török, N.; Majláth, Z.; Szalárdy, L.; Vécsei, L. Investigational α-synuclein aggregation inhibitors: Hope for Parkinson’s disease. Expert Opin. Investig. Drugs 2016, 25, 1281–1294. [Google Scholar] [CrossRef]
- Shen, H.Y.; He, J.C.; Wang, Y.; Huang, Q.-Y.; Chen, J.-F. Geldanamycin induces heat shock protein 70 and protects against MPTP-induced dopaminergic neurotoxicity in mice. J. Biol. Chem. 2005, 280, 39962–39969. [Google Scholar] [CrossRef]
- Sardoiwala, M.N.; Karmakar, S.; Choudhury, S.R. Chitosan nanocarrier for FTY720 enhanced delivery retards Parkinson’s disease via PP2A-EzH2 signaling in vitro and ex vivo. Carbohydr. Polym. 2021, 254, 117435. [Google Scholar] [CrossRef]
- Liu, J.; Liu, C.; Zhang, J.; Zhang, Y.; Liu, K.; Song, J.X.; Sreenivasmurthy, S.G.; Wang, Z.; Shi, Y.; Chu, C. A Self-Assembled alpha-Synuclein Nanoscavenger for Parkinson’s Disease. ACS Nano 2020, 14, 1533–1549. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, D.I.; Billings, J.L.; Adlard, P.A.; Ayton, S.; Sedjahtera, A.; Masters, C.L.; Wilkins, S.; Shackleford, D.M.; Charman, S.A.; Bal, W.; et al. The novel compound PBT434 prevents iron mediated neurodegeneration and alpha-synuclein toxicity in multiple models of Parkinson’s disease. Acta Neuropathol. Commun. 2017, 5, 53. [Google Scholar] [CrossRef] [PubMed]
- Devos, D.; Labreuche, J.; Rascol, O.; Corvol, J.-C.; Duhamel, A.; Delannoy, P.G.; Poewe, W.; Compta, Y.; Pavese, N.; Růžička, E.; et al. Trial of Deferiprone in Parkinson’s Disease. N. Engl. J. Med. 2022, 387, 2045–2055. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Yuan, Y.; Sheng, Y.; Yuan, B.; Wang, Y.; Zheng, J.; Liu, C.F.; Zhang, X.; Hu, L.F. GYY4137, an H(2)S Slow-Releasing Donor, Prevents Nitrative Stress and alpha-Synuclein Nitration in an MPTP Mouse Model of Parkinson’s Disease. Front. Pharmacol. 2017, 8, 741. [Google Scholar] [CrossRef]
- Karuppagounder, S.S.; Brahmachari, S.; Lee, Y.; Dawson, V.L.; Dawson, T.M.; Ko, H.S. The c-Abl inhibitor, Nilotinib, protects dopaminergic neurons in a preclinical animal model of Parkinson’s disease. Sci. Rep. 2014, 4, 4874. [Google Scholar] [CrossRef]
- Pagan, F.; Hebron, M.; Valadez, E.H.; Torres-Yaghi, Y.; Huang, X.; Mills, R.R.; Wilmarth, B.M.; Howard, H.; Dunn, C.; Carlson, A.; et al. Nilotinib Effects in Parkinson’s disease and Dementia with Lewy bodies. J. Park. Dis. 2016, 6, 503–517. [Google Scholar] [CrossRef]
- Pagan, F.L.; Hebron, M.L.; Wilmarth, B.; Torres-Yaghi, Y.; Lawler, A.; Mundel, E.E.; Yusuf, N.; Starr, N.J.; Anjum, M.; Arellano, J.; et al. Nilotinib Effects on Safety, Tolerability, and Potential Biomarkers in Parkinson Disease: A Phase 2 Randomized Clinical Trial. JAMA Neurol. 2020, 77, 309–317. [Google Scholar] [CrossRef]
- Werner, M.H.; Olanow, C.W. Parkinson’s Disease Modification Through Abl Kinase Inhibition: An Opportunity. Mov. Disord. 2022, 37, 6–15. [Google Scholar] [CrossRef]
- Walsh, R.R.; Damle, N.K.; Mandhane, S.; Piccoli, S.P.; Talluri, R.S.; Love, D.; Yao, S.L.; Ramanathan, V.; Hurko, O. Plasma and cerebrospinal fluid pharmacokinetics of vodobatinib, a neuroprotective c-Abl tyrosine kinase inhibitor for the treatment of Parkinson’s disease. Park. Relat. Disord. 2023, 108, 105281. [Google Scholar] [CrossRef] [PubMed]
- Volc, D.; Poewe, W.; Kutzelnigg, A.; Lührs, P.; Thun-Hohenstein, C.; Schneeberger, A.; Galabova, G.; Majbour, N.; Vaikath, N.; El-Agnaf, O.; et al. Safety and immunogenicity of the alpha-synuclein active immunotherapeutic PD01A in patients with Parkinson’s disease: A randomised, single-blinded, phase 1 trial. Lancet Neurol. 2020, 19, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J.; Goodman, I.; Safirstein, B.; Marmon, T.K.; Schenk, D.B.; Koller, M.; Zago, W.; Ness, D.K.; Griffith, S.G.; Grundman, M.; et al. Safety and Tolerability of Multiple Ascending Doses of PRX002/RG7935, an Anti-alpha-Synuclein Monoclonal Antibody, in Patients with Parkinson Disease: A Randomized Clinical Trial. JAMA Neurol. 2018, 75, 1206–1214. [Google Scholar] [CrossRef]
- Pagano, G.; Boess, F.G.; Taylor, K.I.; Ricci, B.; Mollenhauer, B.; Poewe, W.; Boulay, A.; Anzures-Cabrera, J.; Vogt, A.; Marchesi, M.; et al. A Phase II Study to Evaluate the Safety and Efficacy of Prasinezumab in Early Parkinson’s Disease (PASADENA): Rationale, Design, and Baseline Data. Front. Neurol. 2021, 12, 705407. [Google Scholar] [CrossRef] [PubMed]
- Pagano, G.; Taylor, K.I.; Anzures-Cabrera, J.; Marchesi, M.; Simuni, T.; Marek, K.; Postuma, R.B.; Pavese, N.; Stocchi, F.; Azulay, J.-P.; et al. Trial of Prasinezumab in Early-Stage Parkinson’s Disease. N. Engl. J. Med. 2022, 387, 421–432. [Google Scholar] [CrossRef]
- Lang, A.E.; Siderowf, A.D.; Macklin, E.A.; Poewe, W.; Brooks, D.J.; Fernandez, H.H.; Rascol, O.; Giladi, N.; Stocchi, F.; Tanner, C.M.; et al. Trial of Cinpanemab in Early Parkinson’s Disease. N. Engl. J. Med. 2022, 387, 408–420. [Google Scholar] [CrossRef]
- Etminan, M.; Gill, S.S.; Samii, A. Intake of vitamin E, vitamin C, and carotenoids and the risk of Parkinson’s disease: A meta-analysis. Lancet Neurol. 2005, 4, 362–365. [Google Scholar] [CrossRef]
- Parkinson Study, G. Effects of tocopherol and deprenyl on the progression of disability in early Parkinson’s disease. N. Engl. J. Med. 1993, 328, 176–183. [Google Scholar]
- Taghizadeh, M.; Tamtaji, O.R.; Dadgostar, E.; Kakhaki, R.D.; Bahmani, F.; Abolhassani, J.; Aarabi, M.H.; Kouchaki, E.; Memarzadeh, M.R.; Asemi, Z. The effects of omega-3 fatty acids and vitamin E co-supplementation on clinical and metabolic status in patients with Parkinson’s disease: A randomized, double-blind, placebo-controlled trial. Neurochem. Int. 2017, 108, 183–189. [Google Scholar] [CrossRef]
- Tardiolo, G.; Bramanti, P.; Mazzon, E. Overview on the Effects of N-Acetylcysteine in Neurodegenerative Diseases. Molecules 2018, 23, 3305. [Google Scholar] [CrossRef]
- Virel, A.; Johansson, J.; Axelsson, J.; Ericsson, M.; Laterveer, R.; Ögren, M.; Orädd, G.; Mo, S.J.; Bjerkén, S.A. N-acetylcysteine decreases dopamine transporter availability in the non-lesioned striatum of the 6-OHDA hemiparkinsonian rat. Neurosci. Lett. 2022, 770, 136420. [Google Scholar] [CrossRef]
- Monti, D.A.; Zabrecky, G.; Kremens, D.; Liang, T.-W.; Wintering, N.A.; Bazzan, A.J.; Zhong, L.; Bowens, B.K.; Chervoneva, I.; Intenzo, C.; et al. N-Acetyl Cysteine Is Associated with Dopaminergic Improvement in Parkinson’s Disease. Clin. Pharmacol. Ther. 2019, 106, 884–890. [Google Scholar] [CrossRef]
- Mischley, L.K.; Lau, R.C.; Shankland, E.G.; Wilbur, T.K.; Padowski, J.M. Phase IIb Study of Intranasal Glutathione in Parkinson’s Disease. J. Park. Dis. 2017, 7, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Crotty, G.F.; Schwarzschild, M.A. Chasing Protection in Parkinson’s Disease: Does Exercise Reduce Risk and Progression? Front. Aging Neurosci. 2020, 12, 186. [Google Scholar] [CrossRef] [PubMed]
- Investigators, T.P.S.G.S.-P. Effect of Urate-Elevating Inosine on Early Parkinson Disease Progression: The SURE-PD3 Randomized Clinical Trial. JAMA 2021, 326, 926–939. [Google Scholar]
- Zhang, C.; Zhao, M.; Wang, B.; Su, Z.; Guo, B.; Qin, L.; Zhang, W.; Zheng, R. The Nrf2-NLRP3-caspase-1 axis mediates the neuroprotective effects of Celastrol in Parkinson’s disease. Redox Biol. 2021, 47, 102134. [Google Scholar] [CrossRef]
- Liang, L.P.; Fulton, R.; Bradshaw-Pierce, E.L.; Pearson-Smith, J.; Day, B.J.; Patel, M. Optimization of Lipophilic Metalloporphyrins Modifies Disease Outcomes in a Rat Model of Parkinsonism. J. Pharmacol. Exp. Ther. 2021, 377, 1–10. [Google Scholar] [CrossRef]
- Cooper, O.; Seo, H.; Andrabi, S.; Guardia-Laguarta, C.; Graziotto, J.; Sundberg, M.; McLean, J.R.; Carrillo-Reid, L.; Xie, Z.; Osborn, T.; et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci. Transl. Med. 2012, 4, 141ra90. [Google Scholar] [CrossRef]
- Xu, Y.; Zhi, F.; Mao, J.; Peng, Y.; Shao, N.; Balboni, G.; Yang, Y.; Xia, Y. delta-opioid receptor activation protects against Parkinson’s disease-related mitochondrial dysfunction by enhancing PINK1/Parkin-dependent mitophagy. Aging 2020, 12, 25035–25059. [Google Scholar] [CrossRef]
- Ham, S.; Lee, Y.I.; Jo, M.; Kim, H.; Kang, H.; Jo, A.; Lee, G.H.; Mo, Y.J.; Park, S.C.; Lee, Y.S.; et al. Hydrocortisone-induced parkin prevents dopaminergic cell death via CREB pathway in Parkinson’s disease model. Sci. Rep. 2017, 7, 525. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Shen, J.; Ke, K.; Gu, X. Clinical potential and current progress of mesenchymal stem cells for Parkinson’s disease: A systematic review. Neurol. Sci. 2020, 41, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Cooray, R.; Gupta, V.; Suphioglu, C. Current Aspects of the Endocannabinoid System and Targeted THC and CBD Phytocannabinoids as Potential Therapeutics for Parkinson’s and Alzheimer’s Diseases: A Review. Mol. Neurobiol. 2020, 57, 4878–4890. [Google Scholar] [CrossRef] [PubMed]
- Hammond, S.L.; Popichak, K.A.; Li, X.; Hunt, L.G.; Richman, E.H.; Damale, P.U.; Chong, E.K.P.; Backos, D.S.; Safe, S.; Tjalkens, R.B. The Nurr1 Ligand,1,1-bis(3′-Indolyl)-1-(p-Chlorophenyl)Methane, Modulates Glial Reactivity and Is Neuroprotective in MPTP-Induced Parkinsonism. J. Pharmacol. Exp. Ther. 2018, 365, 636–651. [Google Scholar] [CrossRef]
- Tentillier, N.; Etzerodt, A.; Olesen, M.N.; Rizalar, F.S.; Jacobsen, J.; Bender, D.; Moestrup, S.K.; Romero-Ramos, M. Anti-Inflammatory Modulation of Microglia via CD163-Targeted Glucocorticoids Protects Dopaminergic Neurons in the 6-OHDA Parkinson’s Disease Model. J. Neurosci. 2016, 36, 9375–9390. [Google Scholar] [CrossRef]
- Lecca, D.; Nevin, D.K.; Mulas, G.; Casu, M.; Diana, A.; Rossi, D.; Sacchetti, G.; Carta, A. Neuroprotective and anti-inflammatory properties of a novel non-thiazolidinedione PPARγ agonist in vitro and in MPTP-treated mice. Neuroscience 2015, 302, 23–35. [Google Scholar] [CrossRef]
- Wang, Y.D.; Bao, X.Q.; Xu, S.; Yu, W.-W.; Cao, S.-N.; Hu, J.-P.; Li, Y.; Wang, X.-L.; Zhang, D.; Yu, S.-S. A Novel Parkinson’s Disease Drug Candidate with Potent Anti-neuroinflammatory Effects through the Src Signaling Pathway. J. Med. Chem. 2016, 59, 9062–9079. [Google Scholar] [CrossRef]
- Ross, G.W.; Petrovitch, H.; Abbott, R.D.; Nelson, J.; Markesbery, W.; Davis, D.; Hardman, J.; Launer, L.; Masaki, K.; Tanner, C.M.; et al. Parkinsonian signs and substantia nigra neuron density in decendents elders without PD. Ann. Neurol. 2004, 56, 532–539. [Google Scholar] [CrossRef]
- Kirkeby, A.; Parmar, M.; Barker, R.A. Strategies for bringing stem cell-derived dopamine neurons to the clinic: A European approach (STEM-PD). Prog. Brain Res. 2017, 230, 165–190. [Google Scholar]
- Unnisa, A.; Dua, K.; Kamal, M.A. Mechanism of Mesenchymal Stem Cells as a Multitarget Disease- Modifying Therapy for Parkinson’s Disease. Curr. Neuropharmacol. 2023, 21, 988–1000. [Google Scholar] [CrossRef]
- Song, J.J.; Oh, S.M.; Kwon, O.C.; Wulansari, N.; Lee, H.-S.; Chang, M.-Y.; Lee, E.; Sun, W.; Chang, S.; An, H.; et al. Cografting astrocytes improves cell therapeutic outcomes in a Parkinson’s disease model. J. Clin. Investig. 2018, 128, 463–482. [Google Scholar] [CrossRef]
- Marsili, L.; Sharma, J.; Outeiro, T.F.; Colosimo, C. Stem Cell Therapies in Movement Disorders: Lessons from Clinical Trials. Biomedicines 2023, 11, 505. [Google Scholar] [CrossRef] [PubMed]
- Stoddard-Bennett, T.; Pera, R.R. Stem cell therapy for Parkinson’s disease: Safety and modeling. Neural. Regen. Res. 2020, 15, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Virachit, S.; Mathews, K.J.; Cottam, V.; Werry, E.; Galli, E.; Rappou, E.; Lindholm, P.; Saarma, M.; Halliday, G.M. Levels of glial cell line-derived neurotrophic factor are decreased, but fibroblast growth factor 2 and cerebral dopamine neurotrophic factor are increased in the hippocampus in Parkinson’s disease. Brain Pathol. 2019, 29, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Kirik, D.; Cederfjäll, E.; Halliday, G.; Petersén, Å. Gene therapy for Parkinson’s disease: Disease modification by GDNF family of ligands. Neurobiol. Dis. 2017, 97 Pt B, 179–188. [Google Scholar] [CrossRef]
- Huttunen, H.J.; Booms, S.; Sjogren, M.; Kerstens, V.; Johansson, J.; Holmnäs, R.; Koskinen, J.; Kulesskaya, N.; Fazio, P.; Woolley, M.; et al. Intraputamenal Cerebral Dopamine Neurotrophic Factor in Parkinson’s Disease: A Randomized, Double-Blind, Multicenter Phase 1 Trial. Mov. Disord. 2023, 38, 1209–1222. [Google Scholar] [CrossRef]
- Francardo, V.; Geva, M.; Bez, F.; Denis, Q.; Steiner, L.; Hayden, M.R.; Cenci, M.A. Pridopidine Induces Functional Neurorestoration Via the Sigma-1 Receptor in a Mouse Model of Parkinson’s Disease. Neurotherapeutics 2019, 16, 465–479. [Google Scholar] [CrossRef]
- Fischer, D.L.; Sortwell, C.E. BDNF provides many routes toward STN DBS-mediated disease modification. Mov. Disord. 2019, 34, 22–34. [Google Scholar] [CrossRef]
- Hauser, R.A.; Li, R.; Pérez, A.; Ren, X.; Weintraub, D.; Elm, J.; Goudreau, J.L.; Morgan, J.C.; Fang, J.Y.; Aminoff, M.J.; et al. Longer Duration of MAO-B Inhibitor Exposure is Associated with Less Clinical Decline in Parkinson’s Disease: An Analysis of NET-PD LS1. J. Park. Dis. 2017, 7, 117–127. [Google Scholar] [CrossRef]
- Guo, B.; Hu, S.; Zheng, C.; Wang, H.; Luo, F.; Li, H.; Cui, W.; Yang, X.; Cui, G.; Mak, S.; et al. Substantial protection against MPTP-associated Parkinson’s neurotoxicity in vitro and in vivo by anti-cancer agent SU4312 via activation of MEF2D and inhibition of MAO-B. Neuropharmacology 2017, 126, 12–24. [Google Scholar] [CrossRef]
- Mullapudi, A.; Gudala, K.; Boya, C.S.; Bansal, D. Risk of Parkinson’s Disease in the Users of Antihypertensive Agents: An Evidence from the Meta-Analysis of Observational Studies. J. Neurodegener. Dis. 2016, 2016, 5780809. [Google Scholar] [CrossRef]
- Parkinson Study Group STEADY-PD III Investigators. Isradipine Versus Placebo in Early Parkinson Disease: A Randomized Trial. Ann. Intern. Med. 2020, 172, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Dietiker, C.; Kim, S.; Zhang, Y.; Christine, C.W. Characterization of Vitamin B12 Supplementation and Correlation with Clinical Outcomes in a Large Longitudinal Study of Early Parkinson’s Disease. J. Mov. Disord. 2019, 12, 91–96. [Google Scholar] [CrossRef]
- Athauda, D.; Foltynie, T. Insulin resistance and Parkinson’s disease: A new target for disease modification? Prog. Neurobiol. 2016, 145–146, 98–120. [Google Scholar] [CrossRef] [PubMed]
- Athauda, D.; Wyse, R.; Brundin, P.; Foltynie, T. Is Exenatide a Treatment for Parkinson’s Disease? J. Park. Dis. 2017, 7, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Bu, L.L.; Liu, Y.Q.; Shen, Y.; Fan, Y.; Yu, W.-B.; Jiang, D.-L.; Tang, Y.-L.; Yang, Y.-J.; Wu, P.; Zuo, C.-T.; et al. Neuroprotection of Exendin-4 by Enhanced Autophagy in a Parkinsonian Rat Model of α-Synucleinopathy. Neurotherapeutics 2021, 18, 962–978. [Google Scholar] [CrossRef]
- McGarry, A.; Rosanbalm, S.; Leinonen, M.; Olanow, C.W.; To, D.; Bell, A.; Lee, D.; Chang, J.; Dubow, J.; Dhall, R.; et al. Safety, tolerability, and efficacy of NLY01 in early untreated Parkinson’s disease: A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2024, 23, 37–45. [Google Scholar] [CrossRef]
- Schmitt, M.; Dehay, B.; Bezard, E.; Garcia-Ladona, F.J. Harnessing the trophic and modulatory potential of statins in a dopaminergic cell line. Synapse 2016, 70, 71–86. [Google Scholar] [CrossRef]
- Zhao, P.; Yang, X.; Yang, L.; Li, M.; Wood, K.; Liu, Q.; Zhu, X. Neuroprotective effects of fingolimod in mouse models of Parkinson’s disease. FASEB J. 2017, 31, 172–179. [Google Scholar] [CrossRef]
- Roy, D.; Ghosh, R.; Dubey, S.; Dubey, M.J.; Benito-León, J.; Ray, B.K. Neurological and Neuropsychiatric Impacts of COVID-19 Pandemic. Can. J. Neurol. Sci. 2021, 48, 9–24. [Google Scholar] [CrossRef]
- Sulzer, D.; Antonini, A.; Leta, V.; Nordvig, A.; Smeyne, R.J.; Goldman, J.E.; Al-Dalahmah, O.; Zecca, L.; Sette, A.; Bubacco, L.; et al. COVID-19 and possible links with Parkinson’s disease and parkinsonism: From bench to bedside. NPJ Park. Dis. 2020, 6, 18. [Google Scholar] [CrossRef] [PubMed]
- Brundin, P.; Nath, A.; Beckham, J.D. Is COVID-19 a Perfect Storm for Parkinson’s Disease? Trends Neurosci. 2020, 43, 931–933. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Boltz, D.; Sturm-Ramirez, K.; Shepherd, K.R.; Jiao, Y.; Webster, R.; Smeyne, R.J. Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration. Proc. Natl. Acad. Sci. USA 2009, 106, 14063–14068. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, L.A.; Vilensky, J.A. Encephalitis lethargica: 100 years after the epidemic. Brain 2017, 140, 2246–2251. [Google Scholar] [CrossRef]
- Victorino, D.B.; Guimarães-Marques, M.; Nejm, M.; Scorza, F.A.; Scorza, C.A. COVID-19 and Parkinson’s Disease: Are We Dealing with Short-term Impacts or Something Worse? J. Park. Dis. 2020, 10, 899–902. [Google Scholar] [CrossRef]
- Sadasivan, S.; Zanin, M.; O’Brien, K.; Schultz-Cherry, S.; Smeyne, R.J. Induction of microglia activation after infection with the non-neurotropic A/CA/04/2009 H1N1 influenza virus. PLoS ONE 2015, 10, e0124047. [Google Scholar] [CrossRef]
- Jellinger, K.A. Absence of alpha-synuclein pathology in postencephalitic parkinsonism. Acta Neuropathol. 2009, 118, 371–379. [Google Scholar] [CrossRef]
- Pavel, A.; Murray, D.K.; Stoessl, A.J. COVID-19 and selective vulnerability to Parkinson’s disease. Lancet Neurol. 2020, 19, 719. [Google Scholar] [CrossRef]
- Chaná-Cuevas, P.; Salles-Gándara, P.; Rojas-Fernandez, A.; Salinas-Rebolledo, C.; Milán-Solé, A. The Potential Role of SARS-COV-2 in the Pathogenesis of Parkinson’s Disease. Front. Neurol. 2020, 11, 1044. [Google Scholar] [CrossRef]
- Desforges, M.; Le Coupanec, A.; Dubeau, P.; Bourgouin, A.; Lajoie, L.; Dubé, M.; Talbot, P.J. Human Coronaviruses and Other Respiratory Viruses: Underestimated Opportunistic Pathogens of the Central Nervous System? Viruses 2019, 12, 14. [Google Scholar] [CrossRef]
- Chaudhry, Z.L.; Klenja, D.; Janjua, N.; Cami-Kobeci, G.; Ahmed, B.Y. COVID-19 and Parkinson’s Disease: Shared Inflammatory Pathways Under Oxidative Stress. Brain Sci. 2020, 10, 807. [Google Scholar] [CrossRef]
- Högl, B.; Stefani, A.; Videnovic, A. Idiopathic REM sleep behaviour disorder and neurodegeneration—An update. Nat. Rev. Neurol. 2018, 14, 40–55. [Google Scholar] [CrossRef] [PubMed]
- Fairfoul, G.; McGuire, L.I.; Pal, S.; Ironside, J.W.; Neumann, J.; Christie, S.; Joachim, C.; Esiri, M.; Evetts, S.G.; Rolinski, M.; et al. Alpha-synuclein RT-QuIC in the CSF of patients with alpha-synucleinopathies. Ann. Clin. Transl. Neurol. 2016, 3, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Okuzumi, A.; Hatano, T.; Matsumoto, G.; Nojiri, S.; Ueno, S.-I.; Imamichi-Tatano, Y.; Kimura, H.; Kakuta, S.; Kondo, A.; Fukuhara, T.; et al. Propagative α-synuclein seeds as serum biomarkers for synucleinopathies. Nat. Med. 2023, 29, 1448–1455. [Google Scholar] [CrossRef] [PubMed]
- Iranzo, A.; Mammana, A.; Munoz-Lopetegi, A.; Dellavalle, S.; Mayà, G.; Rossi, M.; Serradell, M.; Baiardi, S.; Arqueros, A.; Quadalti, C.; et al. Misfolded alpha-Synuclein Assessment in the Skin and CSF by RT-QuIC in Isolated REM Sleep Behavior Disorder. Neurology 2023, 100, e1944–e1954. [Google Scholar] [CrossRef]
- Vijiaratnam, N.; Simuni, T.; Bandmann, O.; Morris, H.R.; Foltynie, T. Progress towards therapies for disease modification in Parkinson’s disease. Lancet Neurol. 2021, 20, 559–572. [Google Scholar] [CrossRef]
- Gonzalez-Robles, C.; Bartlett, M.; Burnell, M.; Clarke, C.S.; Haar, S.; Hu, M.T.; Huxford, B.; Jha, A.; Lawton, M.; Noyce, A.; et al. Embedding Patient Input in Outcome Measures for Long-Term Disease-Modifying Parkinson Disease Trials. Mov. Disord. 2024, 39, 433–438. [Google Scholar] [CrossRef]
- Lenka, A.; Jankovic, J. How should future clinical trials be designed in the search for disease-modifying therapies for Parkinson’s disease? Expert Rev. Neurother. 2023, 23, 107–122. [Google Scholar] [CrossRef]
- Ko, W.K.D.; Bezard, E. Experimental animal models of Parkinson’s disease: A transition from assessing symptomatology to alpha-synuclein targeted disease modification. Exp. Neurol. 2017, 298 Pt B, 172–179. [Google Scholar] [CrossRef]
- Gao, L.; Tang, H.; Nie, K.; Wang, L.; Zhao, J.; Gan, R.; Huang, J.; Zhu, R.; Feng, S.; Duan, Z.; et al. Cerebrospinal fluid alpha-synuclein as a biomarker for Parkinson’s disease diagnosis: A systematic review and meta-analysis. Int. J. Neurosci. 2015, 125, 645–654. [Google Scholar] [CrossRef]
- McGhee, D.J.; Ritchie, C.W.; Zajicek, J.P.; Counsell, C.E. A review of clinical trial designs used to detect a disease-modifying effect of drug therapy in Alzheimer’s disease and Parkinson’s disease. BMC Neurol. 2016, 16, 92. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barber, D.; Wijeratne, T.; Singh, L.; Barnham, K.; Masters, C.L. The Search for Disease Modification in Parkinson’s Disease—A Review of the Literature. Life 2025, 15, 1169. https://doi.org/10.3390/life15081169
Barber D, Wijeratne T, Singh L, Barnham K, Masters CL. The Search for Disease Modification in Parkinson’s Disease—A Review of the Literature. Life. 2025; 15(8):1169. https://doi.org/10.3390/life15081169
Chicago/Turabian StyleBarber, Daniel, Tissa Wijeratne, Lakshman Singh, Kevin Barnham, and Colin L. Masters. 2025. "The Search for Disease Modification in Parkinson’s Disease—A Review of the Literature" Life 15, no. 8: 1169. https://doi.org/10.3390/life15081169
APA StyleBarber, D., Wijeratne, T., Singh, L., Barnham, K., & Masters, C. L. (2025). The Search for Disease Modification in Parkinson’s Disease—A Review of the Literature. Life, 15(8), 1169. https://doi.org/10.3390/life15081169