
Pediatric Genetic Dystonias: Current Diagnostic Approaches and Treatment Options

 ,
, 

Abstract
1. Introduction
2. Methods
3. Isolated Dystonias
4. Combined Dystonias
4.1. Combined Dystonias with Parkinsonism
4.2. Combined Dystonias with Myoclonus
4.3. Combined Dystonias with Chorea
5. Complex Dystonias
5.1. Complex Dystonias in Combination with Spasticity
5.2. Complex Dystonias in Combination with Ataxia
5.3. Complex Dystonias in Combination with Parkinsonism
5.4. Complex Dystonias in Combination with Chorea
5.5. Complex Dystonias in Combination with Epilepsy
5.6. Complex Dystonias in Combination with Hearing Impairment
5.7. Complex Dystonias in Combination with Ocular Impairment
6. Paroxysmal Dyskinesia
7. Status Dystonicus
8. Treatment
9. Limitations
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Albanese, A.; Bhatia, K.; Bressman, S.B.; Delong, M.R.; Fahn, S.; Fung, V.S.; Hallett, M.; Jankovic, J.; Jinnah, H.A.; Klein, C.; et al. Phenomenology and classification of dystonia: A consensus update. Mov. Disord. Off. J. Mov. Disord. Soc. 2013, 28, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Marsden, C.D. Dystonia: The spectrum of the disease. Res. Publ. Assoc. Res. Nerv. Ment. Dis. 1976, 55, 351–367. [Google Scholar]
- Fahn, S.; Eldridge, R. Definition of dystonia and classification of the dystonic states. Adv. Neurol. 1976, 14, 1–5. [Google Scholar] [PubMed]
- Albanese, A.; Di Giovanni, M.; Lalli, S. Dystonia: Diagnosis and management. Eur. J. Neurol. 2019, 26, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Stephen, C.D. The Dystonias. Continuum 2022, 28, 1435–1475. [Google Scholar] [CrossRef]
- Herzog, R.; Weissbach, A.; Bäumer, T.; Münchau, A. Complex dystonias: An update on diagnosis and care. J. Neural Transm. 2021, 128, 431–445. [Google Scholar] [CrossRef]
- Di Fonzo, A.; Albanese, A.; Jinnah, H.A. The apparent paradox of phenotypic diversity and shared mechanisms across dystonia syndromes. Curr. Opin. Neurol. 2022, 35, 502–509. [Google Scholar] [CrossRef]
- Schirinzi, T.; Sciamanna, G.; Mercuri, N.B.; Pisani, A. Dystonia as a network disorder: A concept in evolution. Curr. Opin. Neurol. 2018, 31, 498–503. [Google Scholar] [CrossRef]
- Jinnah, H.A.; Sun, Y.V. Dystonia genes and their biological pathways. Neurobiol. Dis. 2019, 129, 159–168. [Google Scholar] [CrossRef]
- Jinnah, H.A.; Neychev, V.; Hess, E.J. The Anatomical Basis for Dystonia: The Motor Network Model. Tremor Other Hyperkinetic Mov. 2017, 7, 506. [Google Scholar] [CrossRef]
- Balint, B.; Mencacci, N.E.; Valente, E.M.; Pisani, A.; Rothwell, J.; Jankovic, J.; Vidailhet, M.; Bhatia, K.P. Dystonia. Nat. Rev. Dis. Primers 2018, 4, 25. [Google Scholar] [CrossRef] [PubMed]
- Niethammer, M.; Carbon, M.; Argyelan, M.; Eidelberg, D. Hereditary dystonia as a neurodevelopmental circuit disorder: Evidence from neuroimaging. Neurobiol. Dis. 2011, 42, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Weisheit, C.E.; Pappas, S.S.; Dauer, W.T. Inherited dystonias: Clinical features and molecular pathways. Handb. Clin. Neurol. 2018, 147, 241–254. [Google Scholar] [CrossRef]
- Kim, M.J.; Yum, M.S.; Seo, G.H.; Ko, T.S.; Lee, B.H. Phenotypic and Genetic Complexity in Pediatric Movement Disorders. Front. Genet. 2022, 13, 829558. [Google Scholar] [CrossRef]
- Dicanio, D.; Spoto, G.; Alibrandi, A.; Minutoli, R.; Nicotera, A.G.; Di Rosa, G. Long-term predictivity of early neurological assessment and developmental trajectories in low-risk preterm infants. Front. Neurol. 2022, 13, 958682. [Google Scholar] [CrossRef] [PubMed]
- Pearson, T.S.; Pons, R. Movement Disorders in Children. Continuum 2019, 25, 1099–1120. [Google Scholar] [CrossRef]
- Lalli, S.; Canavese, C.; Zorzi, G.; Nardocci, N.; Albanese, A. Diagnostic issues in childhood and adult dystonia. Expert Opin. Med. Diagn. 2011, 5, 483–500. [Google Scholar] [CrossRef]
- Meijer, I.A.; Pearson, T.S. The Twists of Pediatric Dystonia: Phenomenology, Classification, and Genetics. Semin. Pediatr. Neurol. 2018, 25, 65–74. [Google Scholar] [CrossRef]
- Steeves, T.D.; Day, L.; Dykeman, J.; Jette, N.; Pringsheim, T. The prevalence of primary dystonia: A systematic review and meta-analysis. Mov. Disord. Off. J. Mov. Disord. Soc. 2012, 27, 1789–1796. [Google Scholar] [CrossRef]
- Marras, C.; Lang, A.; van de Warrenburg, B.P.; Sue, C.M.; Tabrizi, S.J.; Bertram, L.; Mercimek-Mahmutoglu, S.; Ebrahimi-Fakhari, D.; Warner, T.T.; Durr, A.; et al. Nomenclature of genetic movement disorders: Recommendations of the International Parkinson and Movement Disorder Society task force. Mov. Disord. Off. J. Mov. Disord. Soc. 2017, 32, 724–725. [Google Scholar] [CrossRef]
- Zech, M.; Jech, R.; Boesch, S.; Škorvánek, M.; Weber, S.; Wagner, M.; Zhao, C.; Jochim, A.; Necpál, J.; Dincer, Y.; et al. Monogenic variants in dystonia: An exome-wide sequencing study. Lancet Neurol. 2020, 19, 908–918. [Google Scholar] [CrossRef] [PubMed]
- Stephen, C.D.; Dy-Hollins, M.; Gusmao, C.M.; Qahtani, X.A.; Sharma, N. Dystonias: Clinical Recognition and the Role of Additional Diagnostic Testing. Semin. Neurol. 2023, 43, 17–34. [Google Scholar] [CrossRef]
- Domingo, A.; Yadav, R.; Ozelius, L.J. Isolated dystonia: Clinical and genetic updates. J. Neural Transm. 2021, 128, 405–416. [Google Scholar] [CrossRef]
- Fan, Y.; Si, Z.; Wang, L.; Zhang, L. DYT-TOR1A dystonia: An update on pathogenesis and treatment. Front. Neurosci. 2023, 17, 1216929. [Google Scholar] [CrossRef]
- Zorzi, G.; Carecchio, M.; Zibordi, F.; Garavaglia, B.; Nardocci, N. Diagnosis and treatment of pediatric onset isolated dystonia. Eur. J. Paediatr. Neurol. 2018, 22, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Ozelius, L.J.; Hewett, J.W.; Page, C.E.; Bressman, S.B.; Kramer, P.L.; Shalish, C.; de Leon, D.; Brin, M.F.; Raymond, D.; Corey, D.P.; et al. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat. Genet. 1997, 17, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Lange, L.M.; Junker, J.; Loens, S.; Baumann, H.; Olschewski, L.; Schaake, S.; Madoev, H.; Petkovic, S.; Kuhnke, N.; Kasten, M.; et al. Genotype-Phenotype Relations for Isolated Dystonia Genes: MDSGene Systematic Review. Mov. Disord. Off. J. Mov. Disord. Soc. 2021, 36, 1086–1103. [Google Scholar] [CrossRef]
- Almasy, L.; Bressman, S.B.; Raymond, D.; Kramer, P.L.; Greene, P.E.; Heiman, G.A.; Ford, B.; Yount, J.; de Leon, D.; Chouinard, S.; et al. Idiopathic torsion dystonia linked to chromosome 8 in two Mennonite families. Ann. Neurol. 1997, 42, 670–673. [Google Scholar] [CrossRef]
- Djarmati, A.; Schneider, S.A.; Lohmann, K.; Winkler, S.; Pawlack, H.; Hagenah, J.; Brüggemann, N.; Zittel, S.; Fuchs, T.; Raković, A.; et al. Mutations in THAP1 (DYT6) and generalised dystonia with prominent spasmodic dysphonia: A genetic screening study. Lancet Neurol. 2009, 8, 447–452. [Google Scholar] [CrossRef]
- Gavarini, S.; Cayrol, C.; Fuchs, T.; Lyons, N.; Ehrlich, M.E.; Girard, J.P.; Ozelius, L.J. Direct interaction between causative genes of DYT1 and DYT6 primary dystonia. Ann. Neurol. 2010, 68, 549–553. [Google Scholar] [CrossRef]
- Kuipers, D.J.S.; Mandemakers, W.; Lu, C.S.; Olgiati, S.; Breedveld, G.J.; Fevga, C.; Tadic, V.; Carecchio, M.; Osterman, B.; Sagi-Dain, L.; et al. EIF2AK2 Missense Variants Associated with Early Onset Generalized Dystonia. Ann. Neurol. 2021, 89, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, M.; Lange, L.M.; Zech, M.; Lohmann, K. Genetics and Pathogenesis of Dystonia. Annu. Rev. Pathol. Mech. Dis. 2024, 19, 99–131. [Google Scholar] [CrossRef]
- Carecchio, M.; Invernizzi, F.; Gonzàlez-Latapi, P.; Panteghini, C.; Zorzi, G.; Romito, L.; Leuzzi, V.; Galosi, S.; Reale, C.; Zibordi, F.; et al. Frequency and phenotypic spectrum of KMT2B dystonia in childhood: A single-center cohort study. Mov. Disord. Off. J. Mov. Disord. Soc. 2019, 34, 1516–1527. [Google Scholar] [CrossRef]
- Ciolfi, A.; Foroutan, A.; Capuano, A.; Pedace, L.; Travaglini, L.; Pizzi, S.; Andreani, M.; Miele, E.; Invernizzi, F.; Reale, C.; et al. Childhood-onset dystonia-causing KMT2B vari-ants result in a distinctive genomic hypermethylation profile. Clin. Epigenet. 2021, 13, 157. [Google Scholar] [CrossRef]
- Zech, M.; Lam, D.D.; Winkelmann, J. Update on KMT2B-Related Dystonia. Curr. Neurol. Neurosci. Rep. 2019, 19, 92. [Google Scholar] [CrossRef] [PubMed]
- Keller Sarmiento, I.J.; Mencacci, N.E. Genetic Dystonias: Update on Classification and New Genetic Discoveries. Curr. Neurol. Neurosci. Rep. 2021, 21, 8. [Google Scholar] [CrossRef]
- Charlesworth, G.; Angelova, P.R.; Bartolomé-Robledo, F.; Ryten, M.; Trabzuni, D.; Stamelou, M.; Abramov, A.Y.; Bhatia, K.P.; Wood, N.W. Mutations in HPCA cause autosomal-recessive primary isolated dystonia. Am. J. Hum. Genet. 2015, 96, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Atasu, B.; Hanagasi, H.; Bilgic, B.; Pak, M.; Erginel-Unaltuna, N.; Hauser, A.K.; Guven, G.; Simón-Sánchez, J.; Heutink, P.; Gasser, T.; et al. HPCA confirmed as a genetic cause of DYT2-like dystonia phenotype. Mov. Disord. Off. J. Mov. Disord. Soc. 2018, 33, 1354–1358. [Google Scholar] [CrossRef]
- Magrinelli, F.; Bhatia, K.P.; Beiraghi Toosi, M.; Arab, F.; Karimiani, E.G.; Sedighzadeh, S.; Ansari, B.; Neshatdoust, M.; Rocca, C.; Houlden, H.; et al. Childhood-Onset Choreo-Dystonia Due to a Recurrent Novel Homozygous Nonsense HPCA Variant: Case Series and Literature Review. Mov. Disord. Clin. Pract. 2022, 10, 101–108. [Google Scholar] [CrossRef]
- Charlesworth, G.; Plagnol, V.; Holmström, K.M.; Bras, J.; Sheerin, U.M.; Preza, E.; Rubio-Agusti, I.; Ryten, M.; Schneider, S.A.; Stamelou, M.; et al. Mutations in ANO3 cause dominant craniocervical dystonia: Ion channel implicated in pathogenesis. Am. J. Hum. Genet. 2012, 91, 1041–1050. [Google Scholar] [CrossRef]
- MDSGENE. Available online: https://www.mdsgene.org/ (accessed on 17 February 2025).
- Jiang, L.T.; Li, L.X.; Liu, Y.; Zhang, X.L.; Pan, Y.G.; Wang, L.; Wan, X.H.; Jin, L.J. The expanding clinical and genetic spectrum of ANO3 dystonia. Neurosci. Lett. 2021, 746, 135590. [Google Scholar] [CrossRef] [PubMed]
- Stamelou, M.; Charlesworth, G.; Cordivari, C.; Schneider, S.A.; Kägi, G.; Sheerin, U.M.; Rubio-Agusti, I.; Batla, A.; Houlden, H.; Wood, N.W.; et al. The phenotypic spectrum of DYT24 due to ANO3 mutations. Mov. Disord. Off. J. Mov. Disord. Soc. 2014, 29, 928–934. [Google Scholar] [CrossRef]
- Wadon, M.E.; Fenner, E.; Kendall, K.M.; Bailey, G.A.; Sandor, C.; Rees, E.; Peall, K.J. Clinical and genotypic analysis in determining dystonia non-motor phenotypic heterogeneity: A UK Biobank study. J. Neurol. 2022, 269, 6436–6451. [Google Scholar] [CrossRef] [PubMed]
- Bally, J.F.; Camargos, S.; Oliveira Dos Santos, C.; Kern, D.S.; Lee, T.; Pereira da Silva-Junior, F.; Puga, R.D.; Cardoso, F.; Barbosa, E.R.; Yadav, R.; et al. DYT-TUBB4A (DYT4 Dystonia): New Clinical and Genetic Observations. Neurology 2021, 96, e1887–e1897. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, K.; Wilcox, R.A.; Winkler, S.; Ramirez, A.; Rakovic, A.; Park, J.S.; Arns, B.; Lohnau, T.; Groen, J.; Kasten, M.; et al. Whispering dysphonia (DYT4 dystonia) is caused by a mutation in the TUBB4 gene. Ann. Neurol. 2013, 73, 537–545. [Google Scholar] [CrossRef]
- Geoghegan, A.R.; Al Hussona, M.; Beauchamp, N.J.; Hutchinson, M.; Sean O’Riordan, M.B.; Lynch, T.; Webb, D. A novel GNAL mutation in familial dystonia presenting with childhood tremor and myoclonus. Mov. Disord. Off. J. Mov. Disord. Soc. 2019, 34, 923–924. [Google Scholar] [CrossRef]
- Magistrelli, L.; Contaldi, E.; Piola, B.; Caushi, F.; Carecchio, M.; D’Alfonso, S.; Corrado, L. Pediatric Onset of Generalized Dystonia, Cognitive Impairment, and Dys-morphic Features in a Patient Carrying Compound Heterozygous GNAL Mutations. Mov. Disord. Clin. Pract. 2024, 11, 1047–1048. [Google Scholar] [CrossRef]
- Masuho, I.; Fang, M.; Geng, C.; Zhang, J.; Jiang, H.; Özgul, R.K.; Yılmaz, D.Y.; Yal-nızoğlu, D.; Yüksel, D.; Yarrow, A.; et al. Homozygous GNAL mutation associated with familial childhood-onset generalized dystonia. Neurol. Genet. 2016, 2, e78. [Google Scholar] [CrossRef]
- Gorodetsky, C.; Fasano, A. VPS16 and VPS41: The List of Genes Causing Early-Onset Dystonia Keeps Expanding. Mov. Disord. Off. J. Mov. Disord. Soc. 2021, 36, 609. [Google Scholar] [CrossRef]
- Steel, D.; Zech, M.; Zhao, C.; Barwick, K.E.S.; Burke, D.; Demailly, D.; Kumar, K.R.; Zorzi, G.; Nardocci, N.; Kaiyrzhanov, R.; et al. Loss-of-Function Variants in HOPS Complex Genes VPS16 and VPS41 Cause Early Onset Dystonia Associated with Lysosomal Abnormalities. Ann. Neurol. 2020, 88, 867–877. [Google Scholar] [CrossRef]
- Monfrini, E.; Avanzino, L.; Palermo, G.; Bonato, G.; Brescia, G.; Ceravolo, R.; Cantarella, G.; Mandich, P.; Prokisch, H.; Storm Van’s Gravesande, K.; et al. Dominant VPS16 Pathogenic Variants: Not Only Isolated Dystonia. Mov. Disord. Clin. Pract. 2024, 11, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Ostrozovicova, M.; Jech, R.; Steel, D.; Pavelekova, P.; Han, V.; Gdovinova, Z.; Lichtner, P.; Kurian, M.A.; Wiethoff, S.; Houlden, H.; et al. A Recurrent VPS16 p.Arg187* Nonsense Variant in Early-Onset Generalized Dystonia. Mov. Disord. Off. J. Mov. Disord. Soc. 2021, 36, 1984–1985. [Google Scholar] [CrossRef] [PubMed]
- Waller, S.E.; Morales-Briceño, H.; Williams, L.; Mohammad, S.S.; Fellner, A.; Kumar, K.R.; Tchan, M.; Fung, V.S.C. Possible EIF2AK2-Associated Stress-Related Neu-rological Decompensation with Combined Dystonia and Striatal Lesions. Mov. Disord. Clin. Pract. 2021, 9, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Magrinelli, F.; Moualek, D.; Tazir, M.; Pacha, L.A.; Verghese, A.; Bhatia, K.P.; Maroofian, R.; Houlden, H. Heterozygous EIF2AK2 Variant Causes Adolescence-Onset Generalized Dystonia Partially Responsive to DBS. Mov. Disord. Clin. Pract. 2021, 9, 268–271. [Google Scholar] [CrossRef]
- Thomsen, M.; Lange, L.M.; Klein, C.; Lohmann, K. MDSGene: Extending the List of Isolated Dystonia Genes by VPS16, EIF2AK2, and AOPEP. Mov. Disord. Off. J. Mov. Disord. Soc. 2023, 38, 507–508. [Google Scholar] [CrossRef]
- Steinberger, D.; Korinthenberg, R.; Topka, H.; Berghäuser, M.; Wedde, R.; Müller, U.; German Dystonia Study Group. Dopa-responsive dystonia: Mutation analysis of GCH1 and analysis of therapeutic doses of L-dopa. Neurology 2000, 55, 1735–1737. [Google Scholar] [CrossRef]
- Furukawa, Y.; Lang, A.E.; Trugman, J.M.; Bird, T.D.; Hunter, A.; Sadeh, M.; Tagawa, T.; St George-Hyslop, P.H.; Guttman, M.; Morris, L.W.; et al. Gender-related penetrance and de novo GTP-cyclohydrolase I gene mutations in dopa-responsive dystonia. Neurology 1998, 50, 1015–1020. [Google Scholar] [CrossRef]
- Wijemanne, S.; Jankovic, J. Dopa-responsive dystonia—Clinical and genetic heterogeneity. Nat. Rev. Neurol. 2015, 11, 414–424. [Google Scholar] [CrossRef]
- Novelli, M.; Tolve, M.; Quiroz, V.; Carducci, C.; Bove, R.; Ricciardi, G.; Yang, K.; Manti, F.; Pisani, F.; Ebrahimi-Fakhari, D.; et al. Autosomal Recessive Guanosine Triphosphate Cyclohydrolase I Deficiency: Redefining the Phenotypic Spec-trum and Outcomes. Mov. Disord. Clin. Pract. 2024, 11, 1072–1084. [Google Scholar] [CrossRef]
- Yoshino, H.; Nishioka, K.; Li, Y.; Oji, Y.; Oyama, G.; Hatano, T.; Machida, Y.; Shimo, Y.; Hayashida, A.; Ikeda, A.; et al. GCH1 mutations in dopa-responsive dystonia and Parkinson’s disease. J. Neurol. 2018, 265, 1860–1870. [Google Scholar] [CrossRef]
- Erro, R.; Magrinelli, F.; Bhatia, K.P. Paroxysmal movement disorders: Parox-ysmal dyskinesia and episodic ataxia. Handb. Clin. Neurol. 2023, 196, 347–365. [Google Scholar] [CrossRef] [PubMed]
- Tolleson, C.; Claassen, D. The function of tyrosine hydroxylase in the normal and Parkinsonian brain. CNS Neurol. Disord. Drug Targets 2012, 11, 381–386. [Google Scholar] [CrossRef]
- Willemsen, M.A.; Verbeek, M.M.; Kamsteeg, E.J.; de Rijk-van Andel, J.F.; Aeby, A.; Blau, N.; Burlina, A.; Donati, M.A.; Geurtz, B.; Grattan-Smith, P.J.; et al. Tyrosine hydroxylase deficiency: A treatable disorder of brain catecholamine biosynthesis. Brain J. Neurol. 2010, 133 Pt 6, 1810–1822. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, G.F.; Assmann, B.; Bräutigam, C.; Dionisi-Vici, C.; Häussler, M.; de Klerk, J.B.; Naumann, M.; Steenbergen-Spanjers, G.C.; Strassburg, H.M.; Wevers, R.A. Tyrosine hydroxylase deficiency causes progressive encephalopathy and dopa-nonresponsive dystonia. Ann. Neurol. 2003, 54 (Suppl. S6), S56–S65. [Google Scholar] [CrossRef]
- Echenne, B.; Roubertie, A.; Assmann, B.; Lutz, T.; Penzien, J.M.; Thöny, B.; Blau, N.; Hoffmann, G.F. Sepiapterin reductase deficiency: Clinical presentation and evaluation of long-term therapy. Pediatr. Neurol. 2006, 35, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Camargos, S.; Scholz, S.; Simón-Sánchez, J.; Paisán-Ruiz, C.; Lewis, P.; Hernandez, D.; Ding, J.; Gibbs, J.R.; Cookson, M.R.; Bras, J.; et al. DYT16, a novel young-onset dystonia-parkinsonism disorder: Identification of a segregating mutation in the stress-response protein PRKRA. Lancet Neurol. 2008, 7, 207–215. [Google Scholar] [CrossRef]
- Zech, M.; Castrop, F.; Schormair, B.; Jochim, A.; Wieland, T.; Gross, N.; Lichtner, P.; Peters, A.; Gieger, C.; Meitinger, T.; et al. DYT16 revisited: Exome sequencing identifies PRKRA mutations in a European dystonia family. Mov. Disord. Off. J. Mov. Disord. Soc. 2014, 29, 1504–1510. [Google Scholar] [CrossRef]
- Seibler, P.; Djarmati, A.; Langpap, B.; Hagenah, J.; Schmidt, A.; Brüggemann, N.; Siebner, H.; Jabusch, H.C.; Altenmüller, E.; Münchau, A.; et al. A heterozygous frameshift mutation in PRKRA (DYT16) associated with generalised dystonia in a German patient. Lancet Neurol. 2008, 7, 380–381. [Google Scholar] [CrossRef]
- Himmelreich, N.; Montioli, R.; Garbade, S.F.; Kopesky, J.; Elsea, S.H.; Carducci, C.; Voltattorni, C.B.; Blau, N. Spectrum of DDC variants causing aromatic l-amino acid decarboxylase (AADC) deficiency and pathogenicity interpretation using ACMG-AMP/ACGS recommendations. Mol. Genet. Metab. 2022, 137, 359–381. [Google Scholar] [CrossRef]
- Mastrangelo, M.; Tolve, M.; Artiola, C.; Bove, R.; Carducci, C.; Carducci, C.; Angeloni, A.; Pisani, F.; Leuzzi, V. Phenotypes and Genotypes of Inherited Disorders of Biogenic Amine Neurotransmitter Metabolism. Genes 2023, 14, 263. [Google Scholar] [CrossRef]
- Manti, F.; Mastrangelo, M.; Battini, R.; Carducci, C.; Spagnoli, C.; Fusco, C.; Tolve, M.; Carducci, C.; Leuzzi, V. Long-term neurological and psychiatric outcomes in patients with aromatic l-amino acid decarboxylase deficiency. Park. Relat. Disord. 2022, 103, 105–111. [Google Scholar] [CrossRef]
- Rosewich, H.; Thiele, H.; Ohlenbusch, A.; Maschke, U.; Altmüller, J.; Frommolt, P.; Zirn, B.; Ebinger, F.; Siemes, H.; Nürnberg, P.; et al. Heterozygous de-novo mutations in ATP1A3 in patients with alternating hemiplegia of childhood: A whole-exome sequencing gene-identification study. Lancet Neurol. 2012, 11, 764–773. [Google Scholar] [CrossRef]
- Carecchio, M.; Zorzi, G.; Ragona, F.; Zibordi, F.; Nardocci, N. ATP1A3-related disorders: An update. Eur. J. Paediatr. Neurol. 2018, 22, 257–263. [Google Scholar] [CrossRef]
- Brashear, A.; Dobyns, W.B.; de Carvalho Aguiar, P.; Borg, M.; Frijns, C.J.; Gollamudi, S.; Green, A.; Guimaraes, J.; Haake, B.C.; Klein, C.; et al. The phenotypic spectrum of rapid-onset dystonia-parkinsonism (RDP) and mutations in the ATP1A3 gene. Brain J. Neurol. 2007, 130 Pt 3, 828–835. [Google Scholar] [CrossRef] [PubMed]
- Vandenbergh, D.J.; Persico, A.M.; Uhl, G.R. A human dopamine transporter cDNA predicts reduced glycosylation, displays a novel repetitive element and provides racially-dimorphic TaqI RFLPs. Brain research. Mol. Brain Res. 1992, 15, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Spaull, R.V.V.; Kurian, M.A. SLC6A3-Related Dopamine Transporter Deficiency Syndrome; Adam, M.P., Feldman, J., Mirzaa, G.M., Eds.; GeneReviews®. University of Washington: Seattle, WA, USA, 2017. [Google Scholar]
- Ng, J.; Barral, S.; Waddington, S.N.; Kurian, M.A. Dopamine Transporter Deficiency Syndrome (DTDS): Expanding the Clinical Phenotype and Precision Medicine Approaches. Cells 2023, 12, 1737. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, S.S.; Mahajan, S.K.; Shah, V.; Kashyap, K. Unmistakable Truncal Dystonia Mistaken as Psychogenic: A Case Report of VAC14-Related Neurodegeneration. Mov. Disord. Clin. Pract. 2023, 10 (Suppl. S3), S15–S20. [Google Scholar] [CrossRef]
- Lenk, G.M.; Szymanska, K.; Debska-Vielhaber, G.; Rydzanicz, M.; Walczak, A.; Bekiesinska-Figatowska, M.; Vielhaber, S.; Hallmann, K.; Stawinski, P.; Buehring, S.; et al. Biallelic Mutations of VAC14 in Pediatric-Onset Neurological Disease. Am. J. Hum. Genet. 2016, 99, 188–194. [Google Scholar] [CrossRef]
- Baumann, H.; Tunc, S.; Günther, A.; Münchau, A.; Lohmann, K.; Brüggemann, N. Altered homodimer formation and increased iron accumulation in VAC14-related disease: Case report and review of the literature. Park. Relat. Disord. 2020, 80, 41–46. [Google Scholar] [CrossRef]
- de Gusmao, C.M.; Stone, S.; Waugh, J.L.; Yang, E.; Lenk, G.M.; Rodan, L.H. VAC14 Gene-Related Parkinsonism-Dystonia With Response to Deep Brain Stimulation. Mov. Disord. Clin. Pract. 2019, 6, 494–497. [Google Scholar] [CrossRef]
- Graziola, F.; Danti, F.R.; Segre, G.; Fregonese, M.; Izzo, R.; Lamentea, E.; Ghezzi, D.; Ardissone, A.; Zorzi, G. Intrafamilial Variability in WARS2-Related Disorder: A Family Case. Mov. Disord. Clin. Pract. 2025; advance online publication. [Google Scholar] [CrossRef]
- Skorvanek, M.; Rektorova, I.; Mandemakers, W.; Wagner, M.; Steinfeld, R.; Orec, L.; Han, V.; Pavelekova, P.; Lackova, A.; Kulcsarova, K.; et al. WARS2 mutations cause dopa-responsive early-onset parkinsonism and progressive myoclonus ataxia. Park. Relat. Disord. 2022, 94, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Németh, A.H. The genetics of primary dystonias and related disorders. Brain J. Neurol. 2002, 125 Pt 4, 695–721. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, M.; Zimprich, A.; Lorenz-Depiereux, B.; Kalscheuer, V.; Asmus, F.; Gasser, T.; Meitinger, T.; Strom, T.M. The epsilon-sarcoglycan gene (SGCE), mutated in myoclonus-dystonia syndrome, is maternally imprinted. Eur. J. Hum. Genet. EJHG 2003, 11, 138–144. [Google Scholar] [CrossRef]
- Ritz, K.; van Schaik, B.D.; Jakobs, M.E.; van Kampen, A.H.; Aronica, E.; Tijssen, M.A.; Baas, F. SGCE isoform characterization and expression in human brain: Implications for myoclonus-dystonia pathogenesis? Eur. J. Hum. Genet. EJHG 2011, 19, 438–444. [Google Scholar] [CrossRef] [PubMed]
- De Francesch, V.; Cazurro-Gutiérrez, A.; Timmers, E.R.; Español-Martín, G.; Ferrero-Turrión, J.; Gómez-Andrés, D.; Marcé-Grau, A.; Dougherty-de Miguel, L.; González, V.; Moreno-Galdó, A.; et al. Natural history of SGCE-associated myoclonus dystonia in children and adolescents. Dev. Med. Child Neurol. 2024, 67, 740–749. [Google Scholar] [CrossRef]
- Asmus, F.; Zimprich, A.; Tezenas Du Montcel, S.; Kabus, C.; Deuschl, G.; Kupsch, A.; Ziemann, U.; Castro, M.; Kühn, A.A.; Strom, T.M.; et al. Myoclonus-dystonia syndrome: Epsilon-sarcoglycan mutations and phenotype. Ann. Neurol. 2002, 52, 489–492. [Google Scholar] [CrossRef]
- Valente, E.M.; Edwards, M.J.; Mir, P.; DiGiorgio, A.; Salvi, S.; Davis, M.; Russo, N.; Bozi, M.; Kim, H.T.; Pennisi, G.; et al. The epsilon-sarcoglycan gene in myoclonic syndromes. Neurology 2005, 64, 737–739. [Google Scholar] [CrossRef]
- Mencacci, N.E.; Rubio-Agusti, I.; Zdebik, A.; Asmus, F.; Ludtmann, M.H.; Ryten, M.; Plagnol, V.; Hauser, A.K.; Bandres-Ciga, S.; Bettencourt, C.; et al. A missense mutation in KCTD17 causes autosomal dominant myoclonus-dystonia. Am. J. Hum. Genet. 2015, 96, 938–947. [Google Scholar] [CrossRef]
- Marcé-Grau, A.; Correa, M.; Vanegas, M.I.; Muñoz-Ruiz, T.; Ferrer-Aparicio, S.; Baide, H.; Macaya, A.; Pérez-Dueñas, B. Childhood onset progressive myoclonic dystonia due to a de novo KCTD17 splicing mutation. Park. Relat. Disord. 2019, 61, 7–9. [Google Scholar] [CrossRef]
- Mencacci, N.E.; Brüggemann, N. KCTD17 is a confirmed new gene for dystonia, but is it responsible for SGCE-negative myoclonus-dystonia? Park. Relat. Disord. 2019, 61, 1–3. [Google Scholar] [CrossRef]
- Balint, B.; Guerreiro, R.; Carmona, S.; Dehghani, N.; Latorre, A.; Cordivari, C.; Bhatia, K.P.; Bras, J. KCNN2 mutation in autosomal-dominant tremulous myoclonus-dystonia. Eur. J. Neurol. 2020, 27, 1471–1477. [Google Scholar] [CrossRef] [PubMed]
- Mochel, F.; Rastetter, A.; Ceulemans, B.; Platzer, K.; Yang, S.; Shinde, D.N.; Helbig, K.L.; Lopergolo, D.; Mari, F.; Renieri, A.; et al. Variants in the SK2 channel gene (KCNN2) lead to dominant neu-rodevelopmental movement disorders. Brain J. Neurol. 2020, 143, 3564–3573. [Google Scholar] [CrossRef]
- Lavenstein, B.; McGurrin, P.; Attaripour, S.; Vial, F.; Hallett, M. KCNN2 Mutation in Pediatric Tremor Myoclonus Dystonia Syndrome with Electrophysiological Evaluation. Tremor Other Hyperkinetic Mov. 2022, 12, 2. [Google Scholar] [CrossRef] [PubMed]
- Fearon, C.; Grippe, T.C.; Chen, R.; Lang, A.E. Early-Onset Neurodevelop-mental Movement Disorder Secondary to Novel Mutation in KCNN2. Mov. Disord. Clin. Pract. 2022, 9 (Suppl. S2), S9–S12. [Google Scholar] [CrossRef]
- Pauly, M.G.; Thomsen, M.; Tadic, V.; Busch, H.; Depienne, C.; Lohmann, K.; Klein, C.; Brüggemann, N. Insufficient effect of deep brain stimulation in a patient with KCNN2-associated myoclonus-dystonia. Park. Relat. Disord. 2025, 131, 107260. [Google Scholar] [CrossRef]
- Matta, J.A.; Ashby, M.C.; Sanz-Clemente, A.; Roche, K.W.; Isaac, J.T. mGluR5 and NMDA receptors drive the experience- and activity-dependent NMDA receptor NR2B to NR2A subunit switch. Neuron 2011, 70, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Marmiesse, A.; Kusumoto, H.; Rekarte, S.; Roca, I.; Zhang, J.; Myers, S.J.; Traynelis, S.F.; Couce, M.L.; Gutierrez-Solana, L.; Yuan, H. A novel missense mutation in GRIN2A causes a nonepileptic neurodevelopmental disorder. Mov. Disord. Off. J. Mov. Disord. Soc. 2018, 33, 992–999. [Google Scholar] [CrossRef]
- Nicotera, A.G.; Calì, F.; Vinci, M.; Musumeci, S.A. GRIN2A: Involvement in movement disorders and intellectual disability without seizures. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2019, 40, 2405–2406. [Google Scholar] [CrossRef]
- Al Qahtani, X.; Multhaupt-Buell, T.; Sharma, N.; Dy-Hollins, M.E. Myoclonus-Dystonia in an Individual with a Mutation in the GRIN2A Gene. J. Pediatr. Neurol. 2023, 21, 437–439. [Google Scholar] [CrossRef]
- Strehlow, V.; Heyne, H.O.; Vlaskamp, D.R.M.; Marwick, K.F.M.; Rudolf, G.; de Bellescize, J.; Biskup, S.; Brilstra, E.H.; Brouwer, O.F.; Callenbach, P.M.C.; et al. GRIN2A study group GRIN2A-related disorders: Genotype and functional consequence predict phenotype. Brain J. Neurol. 2019, 142, 80–92. [Google Scholar] [CrossRef]
- Chawla, T.; Kumar, N.K.; Goyal, V. Heterozygous YY1 mutation—A mimicker of SGCE-myoclonus-dystonia. Park. Relat. Disord. 2023, 117, 105846. [Google Scholar] [CrossRef]
- Groen, J.L.; Andrade, A.; Ritz, K.; Jalalzadeh, H.; Haagmans, M.; Bradley, T.E.; Jongejan, A.; Verbeek, D.S.; Nürnberg, P.; Denome, S.; et al. CACNA1B mutation is linked to unique myoclonus-dystonia syndrome. Hum. Mol. Genet. 2015, 24, 987–993. [Google Scholar] [CrossRef] [PubMed]
- Mencacci, N.E.; R’bibo, L.; Bandres-Ciga, S.; Carecchio, M.; Zorzi, G.; Nardocci, N.; Garavaglia, B.; Batla, A.; Bhatia, K.P.; Pittman, A.M.; et al. The CACNA1B R1389H variant is not associated with myoclonus-dystonia in a large European multicentric cohort. Hum. Mol. Genet. 2015, 24, 5326–5329. [Google Scholar] [CrossRef]
- Wu, M.C.; Chang, Y.Y.; Lan, M.Y.; Chen, Y.F.; Tai, C.H.; Lin, Y.F.; Tsai, S.F.; Chen, P.L.; Lin, C.H. A Clinical and Integrated Genetic Study of Isolated and Combined Dystonia in Taiwan. J. Mol. Diagn. JMD 2022, 24, 262–273. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Harvey, S.; King, M.D.; Gorman, K.M. Paroxysmal Movement Disorders. Front. Neurol. 2021, 12, 659064. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Z.; Matsushita, M.M.; Robertson, P.; Rieder, M.; Girirajan, S.; Antonacci, F.; Lipe, H.; Eichler, E.E.; Nickerson, D.A.; Bird, T.D.; et al. Autosomal dominant familial dyskinesia and facial myokymia: Single exome sequencing identifies a mutation in adenylyl cyclase 5. Arch. Neurol. 2012, 69, 630–635. [Google Scholar] [CrossRef]
- Carecchio, M.; Mencacci, N.E.; Iodice, A.; Pons, R.; Panteghini, C.; Zorzi, G.; Zibordi, F.; Bonakis, A.; Dinopoulos, A.; Jankovic, J.; et al. ADCY5-related movement disorders: Frequency, disease course and phenotypic variability in a cohort of paediatric patients. Park. Relat. Disord. 2017, 41, 37–43. [Google Scholar] [CrossRef]
- Spoto, G.; Ceraolo, G.; Butera, A.; Di Rosa, G.; Nicotera, A.G. Exploring the Genetic Landscape of Chorea in Infancy and Early Childhood: Implications for Diagnosis and Treatment. Curr. Issues Mol. Biol. 2024, 46, 5632–5654. [Google Scholar] [CrossRef]
- Méneret, A.; Mohammad, S.S.; Cif, L.; Doummar, D.; DeGusmao, C.; Anheim, M.; Barth, M.; Damier, P.; Demonceau, N.; Friedman, J.; et al. Efficacy of Caffeine in ADCY5-Related Dyskinesia: A Retrospective Study. Mov. Disord. Off. J. Mov. Disord. Soc. 2022, 37, 1294–1298. [Google Scholar] [CrossRef]
- Domínguez Carral, J.; Reinhard, C.; Ebrahimi-Fakhari, D.; Dorison, N.; Galosi, S.; Garone, G.; Malenica, M.; Ravelli, C.; Serdaroglu, E.; van de Pol, L.A.; et al. Dyskinetic crisis in GNAO1-related disorders: Clinical perspectives and management strategies. Front. Neurol. 2024, 15, 1403815. [Google Scholar] [CrossRef]
- Feng, H.; Sjögren, B.; Karaj, B.; Shaw, V.; Gezer, A.; Neubig, R.R. Movement disorder in GNAO1 encephalopathy associated with gain-of-function mutations. Neurology 2017, 89, 762–770. [Google Scholar] [CrossRef]
- Muntean, B.S.; Masuho, I.; Dao, M.; Sutton, L.P.; Zucca, S.; Iwamoto, H.; Patil, D.N.; Wang, D.; Birnbaumer, L.; Blakely, R.D.; et al. Gαo is a major determinant of cAMP signaling in the pathophysiology of movement disorders. Cell Rep. 2021, 34, 108718. [Google Scholar] [CrossRef]
- Akamine, S.; Okuzono, S.; Yamamoto, H.; Setoyama, D.; Sagata, N.; Ohgidani, M.; Kato, T.A.; Ishitani, T.; Kato, H.; Masuda, K.; et al. GNAO1 organizes the cytoskeletal remodeling and firing of developing neurons. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 16601–16621. [Google Scholar] [CrossRef]
- Honey, C.M.; Malhotra, A.K.; Tarailo-Graovac, M.; van Karnebeek, C.D.M.; Horvath, G.; Sulistyanto, A. GNAO1 Mutation-Induced Pediatric Dystonic Storm Rescue With Pallidal Deep Brain Stimulation. J. Child Neurol. 2018, 33, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Schirinzi, T.; Garone, G.; Travaglini, L.; Vasco, G.; Galosi, S.; Rios, L.; Castiglioni, C.; Barassi, C.; Battaglia, D.; Gambardella, M.L.; et al. Phenomenology and clinical course of movement disorder in GNAO1 variants: Results from an analytical review. Park. Relat. Disord. 2019, 61, 19–25. [Google Scholar] [CrossRef] [PubMed]
- de Gusmao, C.M.; Kok, F.; Casella, E.B.; Waugh, J.L. Benign hereditary chorea related to NKX2-1 with ataxia and dystonia. Neurol. Genet. 2015, 2, e40. [Google Scholar] [CrossRef]
- Gras, D.; Jonard, L.; Roze, E.; Chantot-Bastaraud, S.; Koht, J.; Motte, J.; Rodriguez, D.; Louha, M.; Caubel, I.; Kemlin, I.; et al. Benign hereditary chorea: Phenotype, prognosis, therapeutic outcome and long term follow-up in a large series with new mutations in the TITF1/NKX2-1 gene. J. Neurol. Neurosurg. Psychiatry 2012, 83, 956–962. [Google Scholar] [CrossRef]
- di Biase, L.; Di Santo, A.; Caminiti, M.L.; Pecoraro, P.M.; Di Lazzaro, V. Classification of Dystonia. Life 2022, 12, 206. [Google Scholar] [CrossRef]
- Klein, C.; Münchau, A. Progressive dystonia. Handb. Clin. Neurol. 2013, 113, 1889–1897. [Google Scholar] [CrossRef]
- Crow, Y.J.; Chase, D.S.; Lowenstein Schmidt, J.; Szynkiewicz, M.; Forte, G.M.; Gornall, H.L.; Oojageer, A.; Anderson, B.; Pizzino, A.; Helman, G.; et al. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am. J. Med. Genet. Part A 2015, 167A, 296–312. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, A.; Calì, F.; Scuderi, C.; Vinci, M.; Vitello, G.A.; Musumeci, S.A.; Chiavetta, V.; Federico, C.; Amore, G.; Saccone, S.; et al. Identification of a Novel Missense Mutation of POLR3A Gene in a Cohort of Sicilian Patients with Leukodystrophy. Biomedicines 2022, 10, 2276. [Google Scholar] [CrossRef]
- Saraf, U.; Chandarana, M.; Puthenveedu, D.K.; Kesavapisharady, K.; Krishnan, S.; Kishore, A. Childhood-Onset Dystonia Attributed to Aicardi-Goutières Syndrome and Responsive to Deep Brain Stimulation. Mov. Disord. Clin. Pract. 2021, 8, 613–615. [Google Scholar] [CrossRef]
- Gregory, A.; Hayflick, S. Neurodegeneration with Brain Iron Accumulation Disorders Overview; Adam, M.P., Feldman, J., Mirzaa, G.M., Eds.; GeneReviews®; University of Washington: Seattle, WA, USA, 2013. [Google Scholar]
- Baumeister, F.A.; Auer, D.P.; Hörtnagel, K.; Freisinger, P.; Meitinger, T. The eye-of-the-tiger sign is not a reliable disease marker for Hallervorden-Spatz syndrome. Neuropediatrics 2005, 36, 221–222. [Google Scholar] [CrossRef]
- Kurian, M.A.; Hayflick, S.J. Pantothenate kinase-associated neurodegeneration (PKAN) and PLA2G6-associated neurodegeneration (PLAN): Review of two major neurodegeneration with brain iron accumulation (NBIA) phenotypes. Int. Rev. Neurobiol. 2013, 110, 49–71. [Google Scholar] [CrossRef] [PubMed]
- Akçakaya, N.H.; Haryanyan, G.; Mercan, S.; Sozer, N.; Ali, A.; Tombul, T.; Ozbek, U.; Uğur İşeri, S.A.; Yapıcı, Z. Clinical and genetic spectrum of an orphan disease MPAN: A series with new variants and a novel phenotype. Neurol. Neurochir. Pol. 2019, 53, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Hartig, M.B.; Iuso, A.; Haack, T.; Kmiec, T.; Jurkiewicz, E.; Heim, K.; Roeber, S.; Tarabin, V.; Dusi, S.; Krajewska-Walasek, M.; et al. Absence of an orphan mitochondrial protein, c19orf12, causes a distinct clinical subtype of neurodegeneration with brain iron accumulation. Am. J. Hum. Genet. 2011, 89, 543–550. [Google Scholar] [CrossRef]
- Hogarth, P.; Gregory, A.; Kruer, M.C.; Sanford, L.; Wagoner, W.; Natowicz, M.R.; Egel, R.T.; Subramony, S.H.; Goldman, J.G.; Berry-Kravis, E.; et al. New NBIA subtype: Genetic, clinical, pathologic, and radiographic features of MPAN. Neurology 2013, 80, 268–275. [Google Scholar] [CrossRef]
- Edvardson, S.; Hama, H.; Shaag, A.; Gomori, J.M.; Berger, I.; Soffer, D.; Korman, S.H.; Taustein, I.; Saada, A.; Elpeleg, O. Mutations in the fatty acid 2-hydroxylase gene are associated with leukodystrophy with spastic paraparesis and dystonia. Am. J. Hum. Genet. 2008, 83, 643–648. [Google Scholar] [CrossRef]
- Kruer, M.C.; Paisán-Ruiz, C.; Boddaert, N.; Yoon, M.Y.; Hama, H.; Gregory, A.; Malandrini, A.; Woltjer, R.L.; Munnich, A.; Gobin, S.; et al. Defective FA2H leads to a novel form of neurodegeneration with brain iron accumulation (NBIA). Ann. Neurol. 2010, 68, 611–618. [Google Scholar] [CrossRef]
- Kim, M.; Kim, A.R.; Park, J.; Kim, J.S.; Ahn, J.H.; Park, W.Y.; Kim, N.K.D.; Lee, C.; Kim, N.S.; Cho, J.W.; et al. Clinical characteristics of ataxia-telangiectasia presenting dystonia as a main manifestation. Clin. Neurol. Neurosurg. 2020, 199, 106267. [Google Scholar] [CrossRef] [PubMed]
- Zaki-Dizaji, M.; Tajdini, M.; Kiaee, F.; Shojaaldini, H.; Badv, R.S.; Abolhassani, H.; Aghamohammadi, A. Dystonia in Ataxia Telangiectasia: A Case Report with Novel Mutations. Oman Med. J. 2020, 35, e93. [Google Scholar] [CrossRef]
- Kuiper, A.; Eggink, H.; Tijssen, M.A.; de Koning, T.J. Neurometabolic disorders are treatable causes of dystonia. Rev. Neurol. 2016, 172, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, R.; Mahale, R.R.; Sindhu, D.M.; Stezin, A.; Kamble, N.; Holla, V.V.; Netravathi, M.; Yadav, R.; Pal, P.K. Spectrum of Movement Disorders in Niemann-Pick Disease Type C. Tremor Other Hyperkinetic Mov. 2022, 12, 28. [Google Scholar] [CrossRef] [PubMed]
- Vanier, M.T. Niemann-Pick disease type C. Orphanet J. Rare Dis. 2010, 5, 16. [Google Scholar] [CrossRef]
- Okada, S.; O’Brien, J.S. Generalized gangliosidosis: Beta-galactosidase deficiency. Science 1968, 160, 1002–1004. [Google Scholar] [CrossRef]
- Roze, E.; Paschke, E.; Lopez, N.; Eck, T.; Yoshida, K.; Maurel-Ollivier, A.; Doummar, D.; Caillaud, C.; Galanaud, D.; Billette de Villemeur, T.; et al. Dystonia and parkinsonism in GM1 type 3 gangliosidosis. Mov. Disord. Off. J. Mov. Disord. Soc. 2005, 20, 1366–1369. [Google Scholar] [CrossRef]
- Bidchol, A.M.; Dalal, A.; Trivedi, R.; Shukla, A.; Nampoothiri, S.; Sankar, V.H.; Danda, S.; Gupta, N.; Kabra, M.; Hebbar, S.A.; et al. Recurrent and novel GLB1 mutations in India. Gene 2015, 567, 173–181. [Google Scholar] [CrossRef]
- Stepien, K.M.; Ciara, E.; Jezela-Stanek, A. Fucosidosis-Clinical Manifestation, Long-Term Outcomes, and Genetic Profile—Review and Case Series. Genes 2020, 11, 1383. [Google Scholar] [CrossRef]
- Willems, P.J.; Gatti, R.; Darby, J.K.; Romeo, G.; Durand, P.; Dumon, J.E.; O’Brien, J.S. Fucosidosis revisited: A review of 77 patients. Am. J. Med. Genet. 1991, 38, 111–131. [Google Scholar] [CrossRef]
- Gautschi, M.; Merlini, L.; Calza, A.M.; Hayflick, S.; Nuoffer, J.M.; Fluss, J. Late diagnosis of fucosidosis in a child with progressive fixed dystonia, bilateral pallidal lesions and red spots on the skin. Eur. J. Paediatr. Neurol. 2014, 18, 516–519. [Google Scholar] [CrossRef]
- Zubarioglu, T.; Kiykim, E.; Zeybek, C.A.; Cansever, M.S.; Benbir, G.; Aydin, A.; Yalcinkaya, C. Clinical and neuroradiological approach to fucosidosis in a child with atypical presentation. Ann. Indian Acad. Neurol. 2015, 18, 471–474. [Google Scholar] [CrossRef] [PubMed]
- Wali, G.; Wali, G.M.; Sue, C.M.; Kumar, K.R. A Novel Homozygous Mutation in the FUCA1 Gene Highlighting Fucosidosis as a Cause of Dystonia: Case Report and Literature Review. Neuropediatrics 2019, 50, 248–252. [Google Scholar] [CrossRef] [PubMed]
- Karkheiran, S.; Shahidi, G.A.; Walker, R.H.; Paisán-Ruiz, C. PLA2G6-associated Dystonia-Parkinsonism: Case Report and Literature Review. Tremor Other Hyperkinetic Mov. 2015, 5, 317. [Google Scholar] [CrossRef]
- Haack, T.B.; Ignatius, E.; Calvo-Garrido, J.; Iuso, A.; Isohanni, P.; Maffezzini, C.; Lönnqvist, T.; Suomalainen, A.; Gorza, M.; Kremer, L.S.; et al. Absence of the Autophagy Adaptor SQSTM1/p62 Causes Childhood-Onset Neurodegeneration with Ataxia, Dystonia, and Gaze Palsy. Am. J. Hum. Genet. 2016, 99, 735–743. [Google Scholar] [CrossRef]
- Muto, V.; Flex, E.; Kupchinsky, Z.; Primiano, G.; Galehdari, H.; Dehghani, M.; Cecchetti, S.; Carpentieri, G.; Rizza, T.; Mazaheri, N.; et al. Biallelic SQSTM1 mutations in early-onset, variably progressive neurodegeneration. Neurology 2018, 91, e319–e330. [Google Scholar] [CrossRef] [PubMed]
- Garg, D.; Kapoor, H.; Ahmad, I.; Goel, D.; Zahra, S.; Sharma, P.; Srivastava, A.K.; Faruq, M. Cognitive Impairment, Ataxia, Dystonia, and Gaze Palsy Due to a Novel Variant in SQSTM1: New Lessons. Mov. Disord. Off. J. Mov. Disord. Soc. 2024, 39, 445–447. [Google Scholar] [CrossRef]
- Zúñiga-Ramírez, C.; de Oliveira, L.M.; Kramis-Hollands, M.; Algarni, M.; Soto-Escageda, A.; Sáenz-Farret, M.; González-Usigli, H.A.; Fasano, A. Beyond dystonia and ataxia: Expanding the phenotype of SQSTM1 mutations. Park. Relat. Disord. 2019, 62, 192–195. [Google Scholar] [CrossRef]
- Ortiz, J.F.; Morillo Cox, Á.; Tambo, W.; Eskander, N.; Wirth, M.; Valdez, M.; Niño, M. Neurological Manifestations of Wilson’s Disease: Pathophysiology and Localization of Each Component. Cureus 2020, 12, e11509. [Google Scholar] [CrossRef]
- Taly, A.B.; Meenakshi-Sundaram, S.; Sinha, S.; Swamy, H.S.; Arunodaya, G.R. Wilson disease: Description of 282 patients evaluated over 3 decades. Medicine 2007, 86, 112–121. [Google Scholar] [CrossRef]
- Machado, A.; Chien, H.F.; Deguti, M.M.; Cançado, E.; Azevedo, R.S.; Scaff, M.; Barbosa, E.R. Neurological manifestations in Wilson’s disease: Report of 119 cases. Mov. Disord. Off. J. Mov. Disord. Soc. 2006, 21, 2192–2196. [Google Scholar] [CrossRef] [PubMed]
- Lorincz, M.T. Neurologic Wilson’s disease. Ann. N. Y. Acad. Sci. 2010, 1184, 173–187. [Google Scholar] [CrossRef]
- Ebrahimi-Fakhari, D.; Van Karnebeek, C.; Münchau, A. Movement Disorders in Treatable Inborn Errors of Metabolism. Mov. Disord. Off. J. Mov. Disord. Soc. 2019, 34, 598–613. [Google Scholar] [CrossRef] [PubMed]
- Quadri, M.; Federico, A.; Zhao, T.; Breedveld, G.J.; Battisti, C.; Delnooz, C.; Severijnen, L.A.; Di Toro Mammarella, L.; Mignarri, A.; Monti, L.; et al. Mutations in SLC30A10 cause parkinsonism and dystonia with hypermanganesemia, polycythemia, and chronic liver disease. Am. J. Hum. Genet. 2012, 90, 467–477. [Google Scholar] [CrossRef]
- Tavasoli, A.; Arjmandi Rafsanjani, K.; Hemmati, S.; Mojbafan, M.; Zarei, E.; Hosseini, S. A case of dystonia with polycythemia and hypermanganesemia caused by SLC30A10 mutation: A treatable inborn error of manganese metabolism. BMC Pediatr. 2019, 19, 229. [Google Scholar] [CrossRef] [PubMed]
- Jagadish, S.; Howard, L.; Thati Ganganna, S. Atypical presentation of SLC30A10 gene mutation with hypermanganesemia, seizures and polycythemia. Epilepsy Behav. Rep. 2021, 16, 100505. [Google Scholar] [CrossRef]
- Tuschl, K.; Clayton, P.T.; Gospe, S.M.; Jr Gulab, S.; Ibrahim, S.; Singhi, P.; Aulakh, R.; Ribeiro, R.T.; Barsottini, O.G.; Zaki, M.S.; et al. Syndrome of hepatic cirrhosis, dystonia, polycythemia, and hypermanganesemia caused by mutations in SLC30A10, a manganese transporter in man. Am. J. Hum. Genet. 2012, 90, 457–466. [Google Scholar] [CrossRef]
- Marti-Sanchez, L.; Ortigoza-Escobar, J.D.; Darling, A.; Villaronga, M.; Baide, H.; Molero-Luis, M.; Batllori, M.; Vanegas, M.I.; Muchart, J.; Aquino, L.; et al. Hypermanganesemia due to mutations in SLC39A14: Further insights into Mn deposition in the central nervous system. Orphanet J. Rare Dis. 2018, 13, 28. [Google Scholar] [CrossRef]
- Rodichkin, A.N.; Guilarte, T.R. Hereditary Disorders of Manganese Metabolism: Pathophysiology of Childhood-Onset Dystonia-Parkinsonism in SLC39A14 Mutation Carriers and Genetic Animal Models. Int. J. Mol. Sci. 2022, 23, 12833. [Google Scholar] [CrossRef]
- Schneider, S.A.; Bird, T. Huntington’s Disease, Huntington’s Disease Look-Alikes, and Benign Hereditary Chorea: What’s New? Mov. Disord. Clin. Pract. 2016, 3, 342–354. [Google Scholar] [CrossRef]
- Yang, K.; Quiroz, V.; Tam, A.; Srouji, R.; Villanueva, X.; Amarales, C.; Ebrahi-mi-Fakhari, D. Juvenile-onset Huntington’s disease—Spectrum and evolution of presenting movement disorders. Ann. Clin. Transl. Neurol. 2024, 11, 2805–2810. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.A. Neurodegeneration with Brain Iron Accumulation. Curr. Neurol. Neurosci. Rep. 2016, 16, 9. [Google Scholar] [CrossRef]
- Hogarth, P. Neurodegeneration with brain iron accumulation: Diagnosis and management. J. Mov. Disord. 2015, 8, 1–13. [Google Scholar] [CrossRef]
- Gurram, S.; Holla, V.V.; Kumari, R.; Dhar, D.; Kamble, N.; Yadav, R.; Muthusamy, B.; Pal, P.K. Dystonic Opisthotonus in Kufor-Rakeb Syndrome: Expanding the Phenotypic and Genotypic Spectrum. J. Mov. Disord. 2023, 16, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Nanagiri, A.; Shabbir, N. Lesch-Nyhan Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Visser, J.E.; Bär, P.R.; Jinnah, H.A. Lesch-Nyhan disease and the basal ganglia. Brain Res. Brain Res. Rev. 2000, 32, 449–475. [Google Scholar] [CrossRef]
- Jinnah, H.A.; Visser, J.E.; Harris, J.C.; Verdu, A.; Larovere, L.; Ceballos-Picot, I.; Gonzalez-Alegre, P.; Neychev, V.; Torres, R.J.; Dulac, O.; et al. Lesch-Nyhan Disease International Study Group Delineation of the motor disorder of Lesch-Nyhan disease. Brain J. Neurol. 2006, 129 Pt 5, 1201–1217. [Google Scholar] [CrossRef] [PubMed]
- Wajner, M. Neurological manifestations of organic acidurias. Nat. Rev. Neurol. 2019, 15, 253–271. [Google Scholar] [CrossRef]
- Di Rosa, G.; Nicotera, A.G.; Lenzo, P.; Spanò, M.; Tortorella, G. Long-term neuropsychiatric follow-up in hyperprolinemia type I. Psychiatr. Genet. 2014, 24, 172–175. [Google Scholar] [CrossRef]
- Papandreou, A.; Danti, F.R.; Spaull, R.; Leuzzi, V.; Mctague, A.; Kurian, M.A. The expanding spectrum of movement disorders in genetic epilepsies. Dev. Med. Child Neurol. 2020, 62, 178–191. [Google Scholar] [CrossRef]
- Dzinovic, I.; Škorvánek, M.; Necpál, J.; Boesch, S.; Švantnerová, J.; Wagner, M.; Havránková, P.; Pavelekova, P.; Haň, V.; Janzarik, W.G.; et al. Dystonia as a prominent presenting feature in developmental and epileptic encephalopathies: A case series. Park. Relat. Disord. 2021, 90, 73–78. [Google Scholar] [CrossRef]
- Di Rosa, G.; Dicanio, D.; Nicotera, A.G.; Mondello, P.; Cannavò, L.; Gitto, E. Efficacy of Intravenous Hydrocortisone Treatment in Refractory Neonatal Seizures: A Report on Three Cases. Brain Sci. 2020, 10, 885. [Google Scholar] [CrossRef]
- Nicotera, A.G.; Dicanio, D.; Pironti, E.; Bonsignore, M.; Cafeo, A.; Efthymiou, S.; Mondello, P.; Salpietro, V.; Houlden, H.; Di Rosa, G. De novo mutation in SLC25A22 gene: Expansion of the clinical and electroencephalographic phenotype. J. Neurogenet. 2021, 35, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Dicanio, D.; Nicotera, A.G.; Cucinotta, F.; Di Rosa, G. Perampanel treatment in Early-onset Epileptic Encephalopathy with infantile movement disorders associated with a de novo GRIN1 gene mutation: A 3-year follow-up. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2021, 42, 1627–1629. [Google Scholar] [CrossRef]
- Oliver, K.L.; Trivisano, M.; Mandelstam, S.A.; De Dominicis, A.; Francis, D.I.; Green, T.E.; Muir, A.M.; Chowdhary, A.; Hertzberg, C.; Goldhahn, K.; et al. WWOX developmental and epileptic encephalopathy: Understanding the epileptology and the mortality risk. Epilepsia 2023, 64, 1351–1367. [Google Scholar] [CrossRef]
- Yuan, M.; Wang, X.; Yang, Z.; Luo, H.; Gan, J.; Luo, R. Advances in genetic developmental and epileptic encephalopathies with movement disorders. Acta Epileptol. 2025, 7, 9. [Google Scholar] [CrossRef]
- Kojovic, M.; Pareés, I.; Lampreia, T.; Pienczk-Reclawowicz, K.; Xiromerisiou, G.; Rubio-Agusti, I.; Kramberger, M.; Carecchio, M.; Alazami, A.M.; Brancati, F.; et al. The syndrome of deafness-dystonia: Clinical and genetic heterogeneity. Mov. Disord. Off. J. Mov. Disord. Soc. 2013, 28, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Ha, A.D.; Parratt, K.L.; Rendtorff, N.D.; Lodahl, M.; Ng, K.; Rowe, D.B.; Sue, C.M.; Hayes, M.W.; Tranebjaerg, L.; Fung, V.S. The phenotypic spectrum of dystonia in Mohr-Tranebjaerg syndrome. Mov. Disord. Off. J. Mov. Disord. Soc. 2012, 27, 1034–1040. [Google Scholar] [CrossRef] [PubMed]
- Alazami, A.M.; Schneider, S.A.; Bonneau, D.; Pasquier, L.; Carecchio, M.; Kojovic, M.; Steindl, K.; de Kerdanet, M.; Nezarati, M.M.; Bhatia, K.P.; et al. C2orf37 mutational spectrum in Woodhouse-Sakati syndrome patients. Clin. Genet. 2010, 78, 585–590. [Google Scholar] [CrossRef]
- Zavala, L.; Ziegler, G.; Morón, D.G.; Garretto, N. Dystonia-Deafness Syndrome: ACTB Pathogenic Variant in an Argentinean Family. Mov. Disord. Clin. Pract. 2021, 9, 122–124. [Google Scholar] [CrossRef]
- Vittal, P.; Hall, D.A.; Dames, S.; Mao, R.; Berry-Kravis, E. BCAP31 Mutation Causing a Syndrome of Congenital Dystonia, Facial Dysorphism and Central Hypomyelination Discovered Using Exome Sequencing. Mov. Disord. Clin. Pract. 2015, 3, 197–199. [Google Scholar] [CrossRef]
- Rinaldi, B.; Van Hoof, E.; Corveleyn, A.; Van Cauter, A.; de Ravel, T. BCAP31-related syndrome: The first de novo report. Eur. J. Med. Genet. 2020, 63, 103732. [Google Scholar] [CrossRef] [PubMed]
- Cacciagli, P.; Sutera-Sardo, J.; Borges-Correia, A.; Roux, J.C.; Dorboz, I.; Desvignes, J.P.; Badens, C.; Delepine, M.; Lathrop, M.; Cau, P.; et al. Mutations in BCAP31 cause a severe X-linked phenotype with deafness, dystonia, and central hypomyelination and disorganize the Golgi apparatus. Am. J. Hum. Genet. 2013, 93, 579–586. [Google Scholar] [CrossRef]
- Tranchant, C.; Anheim, M. Movement disorders in mitochondrial diseases. Rev. Neurol. 2016, 172, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.S.; Harvey, J.P.; Gilhooley, M.J.; Jurkute, N.; Yu-Wai-Man, P. Mitochondria and the eye-manifestations of mitochondrial diseases and their management. Eye 2023, 37, 2416–2425. [Google Scholar] [CrossRef] [PubMed]
- Martikainen, M.H.; Ng, Y.S.; Gorman, G.S.; Alston, C.L.; Blakely, E.L.; Schaefer, A.M.; Chinnery, P.F.; Burn, D.J.; Taylor, R.W.; McFarland, R.; et al. Clinical, Genetic, and Radiological Features of Extrapyramidal Movement Disorders in Mitochondrial Disease. JAMA Neurol. 2016, 73, 668–674. [Google Scholar] [CrossRef]
- Baertling, F.; Rodenburg, R.J.; Schaper, J.; Smeitink, J.A.; Koopman, W.J.; Mayatepek, E.; Morava, E.; Distelmaier, F. A guide to diagnosis and treatment of Leigh syndrome. J. Neurol. Neurosurg. Psychiatry 2014, 85, 257–265. [Google Scholar] [CrossRef]
- Nikoskelainen, E.K.; Marttila, R.J.; Huoponen, K.; Juvonen, V.; Lamminen, T.; Sonninen, P.; Savontaus, M.L. Leber’s “plus”: Neurological abnormalities in patients with Leber’s hereditary optic neuropathy. J. Neurol. Neurosurg. Psychiatry 1995, 59, 160–164. [Google Scholar] [CrossRef]
- Flønes, I.H.; Tzoulis, C. Movement disorders in mitochondrial disease: A clinicopathological correlation. Curr. Opin. Neurol. 2018, 31, 472–483. [Google Scholar] [CrossRef]
- Heimer, G.; Kerätär, J.M.; Riley, L.G.; Balasubramaniam, S.; Eyal, E.; Pietikäinen, L.P.; Hiltunen, J.K.; Marek-Yagel, D.; Hamada, J.; Gregory, A.; et al. MECR Mutations Cause Childhood-Onset Dystonia and Optic Atrophy, a Mitochondrial Fatty Acid Synthesis Disorder. Am. J. Hum. Genet. 2016, 99, 1229–1244. [Google Scholar] [CrossRef]
- Defazio, G.; Abbruzzese, G.; Livrea, P.; Berardelli, A. Epidemiology of primary dystonia. Lancet Neurol. 2004, 3, 673–678. [Google Scholar] [CrossRef]
- Garg, D.; Mohammad, S.; Shukla, A.; Sharma, S. Genetic Links to Episodic Movement Disorders: Current Insights. Appl. Clin. Genet. 2023, 16, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Balint, B.; Bhatia, K.P. Isolated and combined dystonia syndromes—An update on new genes and their phenotypes. Eur. J. Neurol. 2015, 22, 610–617. [Google Scholar] [CrossRef]
- Chen, W.J.; Lin, Y.; Xiong, Z.Q.; Wei, W.; Ni, W.; Tan, G.H.; Guo, S.L.; He, J.; Chen, Y.F.; Zhang, Q.J.; et al. Exome sequencing identifies truncating mutations in PRRT2 that cause paroxysmal kinesigenic dyskinesia. Nat. Genet. 2011, 43, 1252–1255. [Google Scholar] [CrossRef]
- You, D.D.; Huang, Y.M.; Wang, X.Y.; Li, W.; Li, F. Long-term low-dose lamotrigine for paroxysmal kinesigenic dyskinesia: A two-year investigation of cognitive function in children. Front. Psychiatry 2024, 15, 1368289. [Google Scholar] [CrossRef] [PubMed]
- Spoto, G.; Valentini, G.; Saia, M.C.; Butera, A.; Amore, G.; Salpietro, V.; Nicotera, A.G.; Di Rosa, G. Synaptopathies in Developmental and Epileptic Encephalopathies: A Focus on Pre-synaptic Dysfunction. Front. Neurol. 2022, 13, 826211. [Google Scholar] [CrossRef]
- Erro, R.; Sheerin, U.M.; Bhatia, K.P. Paroxysmal dyskinesias revisited: A review of 500 genetically proven cases and a new classification. Mov. Disord. Off. J. Mov. Disord. Soc. 2014, 29, 1108–1116. [Google Scholar] [CrossRef]
- Garone, G.; Capuano, A.; Travaglini, L.; Graziola, F.; Stregapede, F.; Zanni, G.; Vigevano, F.; Bertini, E.; Nicita, F. Clinical and Genetic Overview of Paroxysmal Movement Disorders and Episodic Ataxias. Int. J. Mol. Sci. 2020, 21, 3603. [Google Scholar] [CrossRef]
- Chen, Y.L.; Chen, D.F.; Li, H.F.; Wu, Z.Y. Features Differ Between Paroxysmal Kinesigenic Dyskinesia Patients with PRRT2 and TMEM151A Variants. Mov. Disord. Off. J. Mov. Disord. Soc. 2022, 37, 608–613. [Google Scholar] [CrossRef] [PubMed]
- Li, H.F.; Chen, Y.L.; Zhuang, L.; Chen, D.F.; Ke, H.Z.; Luo, W.J.; Liu, G.L.; Wu, S.N.; Zhou, W.H.; Xiong, Z.Q.; et al. TMEM151A variants cause paroxysmal kinesigenic dyskinesia. Cell Discov. 2021, 7, 83. [Google Scholar] [CrossRef]
- Necpál, J.; Zech, M.; Valachová, A.; Sedláček, Z.; Bendová, Š.; Hančárová, M.; Okáľová, K.; Winkelmann, J.; Jech, R. Severe paroxysmal dyskinesias without epilepsy in a RHOBTB2 mutation carrier. Park. Relat. Disord. 2020, 77, 87–88. [Google Scholar] [CrossRef]
- de Pedro Baena, S.; Sariego Jamardo, A.; Castro, P.; López González, F.J.; Sánchez Carpintero, R.; Cerisola, A.; Troncoso, M.; Witting, S.; Barrios, A.; Fons, C.; et al. Exploring the Spectrum of RHOBTB2 Variants Associated with Developmental Encephalopathy 64: A Case Series and Literature Review. Mov. Disord. Clin. Pract. 2023, 10, 1671–1679. [Google Scholar] [CrossRef] [PubMed]
- Gardella, E.; Møller, R.S. Phenotypic and genetic spectrum of SCN8A-related disorders, treatment options, and outcomes. Epilepsia 2019, 60 (Suppl. S3), S77–S85. [Google Scholar] [CrossRef] [PubMed]
- Gordon, N.; Newton, R.W. Glucose transporter type1 (GLUT-1) deficiency. Brain Dev. 2003, 25, 477–480. [Google Scholar] [CrossRef]
- Schneider, S.A.; Paisan-Ruiz, C.; Garcia-Gorostiaga, I.; Quinn, N.P.; Weber, Y.G.; Lerche, H.; Hardy, J.; Bhatia, K.P. GLUT1 gene mutations cause sporadic paroxysmal exercise-induced dyskinesias. Mov. Disord. Off. J. Mov. Disord. Soc. 2009, 24, 1684–1688. [Google Scholar] [CrossRef] [PubMed]
- Baroni, M.G.; Oelbaum, R.S.; Pozzilli, P.; Stocks, J.; Li, S.R.; Fiore, V.; Galton, D.J. Polymorphisms at the GLUT1 (HepG2) and GLUT4 (muscle/adipocyte) glucose transporter genes and non-insulin-dependent diabetes mellitus (NIDDM). Hum. Genet. 1992, 88, 557–561. [Google Scholar] [CrossRef]
- Pons, R.; Collins, A.; Rotstein, M.; Engelstad, K.; De Vivo, D.C. The spectrum of movement disorders in Glut-1 deficiency. Mov. Disord. Off. J. Mov. Disord. Soc. 2010, 25, 275–281. [Google Scholar] [CrossRef]
- Weber, Y.G.; Kamm, C.; Suls, A.; Kempfle, J.; Kotschet, K.; Schüle, R.; Wuttke, T.V.; Maljevic, S.; Liebrich, J.; Gasser, T.; et al. Paroxysmal choreoathetosis/spasticity (DYT9) is caused by a GLUT1 defect. Neurology 2011, 77, 959–964. [Google Scholar] [CrossRef]
- Suls, A.; Dedeken, P.; Goffin, K.; Van Esch, H.; Dupont, P.; Cassiman, D.; Kempfle, J.; Wuttke, T.V.; Weber, Y.; Lerche, H.; et al. Paroxysmal exercise-induced dyskinesia and epilepsy is due to mutations in SLC2A1, encoding the glucose transporter GLUT1. Brain J. Neurol. 2008, 131 Pt 7, 1831–1844. [Google Scholar] [CrossRef]
- Delorme, C.; Giron, C.; Bendetowicz, D.; Méneret, A.; Mariani, L.L.; Roze, E. Current challenges in the pathophysiology, diagnosis, and treatment of paroxysmal movement disorders. Expert Rev. Neurother. 2021, 21, 81–97. [Google Scholar] [CrossRef]
- Steel, D.; Heim, J.; Kruer, M.C.; Sanchis-Juan, A.; Raymond, L.F.; Eunson, P.; Kurian, M.A. Biallelic mutations of TBC1D24 in exercise-induced paroxysmal dystonia. Mov. Disord. Off. J. Mov. Disord. Soc. 2020, 35, 372–373. [Google Scholar] [CrossRef]
- Zimmern, V.; Riant, F.; Roze, E.; Ranza, E.; Lehmann-Horn, F.; de Bellescize, J.; Ville, D.; Lesca, G.; Korff, C.M. Infantile-Onset Paroxysmal Movement Disorder and Episodic Ataxia Associated with a TBC1D24 Mutation. Neuropediatrics 2019, 50, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Illsinger, S.; Korenke, G.C.; Boesch, S.; Nocker, M.; Karall, D.; Nuoffer, J.M.; Laugwitz, L.; Mayr, J.A.; Scholl-Bürgi, S.; Freisinger, P.; et al. Paroxysmal and non-paroxysmal dystonia in 3 patients with biallelic ECHS1 variants: Expanding the neurological spectrum and therapeutic approaches. Eur. J. Med. Genet. 2020, 63, 104046. [Google Scholar] [CrossRef] [PubMed]
- Olgiati, S.; Skorvanek, M.; Quadri, M.; Minneboo, M.; Graafland, J.; Breedveld, G.J.; Bonte, R.; Ozgur, Z.; van den Hout, M.C.; Schoonderwoerd, K.; et al. Paroxysmal exercise-induced dystonia within the phenotypic spectrum of ECHS1 deficiency. Mov. Disord. Off. J. Mov. Disord. Soc. 2016, 31, 1041–1048. [Google Scholar] [CrossRef]
- Shen, Y.; Lee, H.Y.; Rawson, J.; Ojha, S.; Babbitt, P.; Fu, Y.H.; Ptácek, L.J. Mutations in PNKD causing paroxysmal dyskinesia alters protein cleavage and stability. Hum. Mol. Genet. 2011, 20, 2322–2332. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.B.; Tian, M.Q.; Gao, K.; Jiang, Y.W.; Wu, Y. De novo KCNMA1 mutations in children with early-onset paroxysmal dyskinesia and developmental delay. Mov. Disord. Off. J. Mov. Disord. Soc. 2015, 30, 1290–1292. [Google Scholar] [CrossRef]
- Guerrini, R.; Sanchez-Carpintero, R.; Deonna, T.; Santucci, M.; Bhatia, K.P.; Moreno, T.; Parmeggiani, L.; Bernardina, B.D. Early-onset absence epilepsy and paroxysmal dyskinesia. Epilepsia 2002, 43, 1224–1229. [Google Scholar] [CrossRef]
- Alkubaisi, A.; Sandhu, M.K.; Polyhronopoulos, N.E.; Honey, C.R. Deep brain stimulation as a rescue for pediatric dystonic storm. Case reports and literature review. Interdiscip. Neurosurg. 2022, 30, 101654. [Google Scholar] [CrossRef]
- Vogt, L.M.; Yan, H.; Santyr, B.; Breitbart, S.; Anderson, M.; Germann, J.; Lizarraga, K.J.; Hewitt, A.L.; Fasano, A.; Ibrahim, G.M.; et al. Deep Brain Stimulation for Refractory Status Dystonicus in Children: Multicenter Case Series and Systematic Review. Ann. Neurol. 2023, 95, 156–173. [Google Scholar] [CrossRef]
- Vogt, L.M.; Yang, K.; Tse, G.; Quiroz, V.; Zaman, Z.; Wang, L.; Srouji, R.; Tam, A.; Estrella, E.; Manzi, S.; et al. Recommendations for the Management of Initial and Refractory Pediatric Status Dystonicus. Mov. Disord. 2024, 39, 1435–1445. [Google Scholar] [CrossRef]
- Lumsden, D.E.; Cif, L.; Capuano, A.; Allen, N.M. The changing face of reported status dystonicus—A systematic review. Park. Relat. Disord. 2023, 112, 105438. [Google Scholar] [CrossRef]
- Goswami, J.N.; Roy, S.; Patnaik, S.K. Pediatric Dystonic Storm: A Hospital-Based Study. Neurol. Clin. Pract. 2021, 11, e645–e653. [Google Scholar] [CrossRef]
- Kalenahalli, J.K.; Gopalkrishna, M.V.; Nandish, H.R.; Akshaya, A.S. Status dystonicus: A rare and underdiagnosed complication of dystonia. J. Pediatr. Res. 2024, 11, 254–258. [Google Scholar] [CrossRef]
- Novelli, M.; Galosi, S.; Zorzi, G.; Martinelli, S.; Capuano, A.; Nardecchia, F.; Granata, T.; Pollini, L.; Di Rocco, M.; Marras, C.E.; et al. GNAO1-related movement disorder: An update on phenomenology, clinical course, and response to treatments. Park. Relat. Disord. 2023, 111, 105405. [Google Scholar] [CrossRef] [PubMed]
- Waak, M.; Mohammad, S.S.; Coman, D.; Sinclair, K.; Copeland, L.; Silburn, P.; Coyne, T.; McGill, J.; O’Regan, M.; Selway, R.; et al. GNAO1-related movement disorder with life-threatening exacerbations: Movement phenomenology and response to DBS. J. Neurol. Neurosurg. Psychiatry 2018, 89, 221–222. [Google Scholar] [CrossRef]
- Lucena-Valera, A.; Ruz-Zafra, P.; Ampuero, J. Wilson’s disease: Overview. Enfermedad de Wilson. Med. Clin. 2023, 160, 261–267. [Google Scholar] [CrossRef]
- Ferreira, C.R.; Gahl, W.A. Disorders of metal metabolism. Transl. Sci. Rare Dis. 2017, 2, 101–139. [Google Scholar] [CrossRef] [PubMed]
- Iankova, V.; Karin, I.; Klopstock, T.; Schneider, S.A. Emerging Disease-Modifying Therapies in Neurodegeneration With Brain Iron Accumulation (NBIA) Disorders. Front. Neurol. 2021, 12, 629414. [Google Scholar] [CrossRef]
- Klopstock, T.; Tricta, F.; Neumayr, L.; Karin, I.; Zorzi, G.; Fradette, C.; Kmieć, T.; Büchner, B.; Steele, H.E.; Horvath, R.; et al. Safety and efficacy of deferiprone for pantothenate kinase-associated neurodegeneration: A randomised, double-blind, controlled trial and an open-label extension study. Lancet Neurol. 2019, 18, 631–642. [Google Scholar] [CrossRef]
- Pineda, M.; Walterfang, M.; Patterson, M.C. Miglustat in Niemann-Pick disease type C patients: A review. Orphanet J. Rare Dis. 2018, 13, 140. [Google Scholar] [CrossRef]
- European Medicine Agency. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/upstaza (accessed on 26 April 2025).
- FDA Approves First Gene Therapy for Treatment of Aromatic L-amino Acid Decarboxylase Deficiency. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-gene-therapy-treatment-aromatic-l-amino-acid-decarboxylase-deficiency (accessed on 26 April 2025).
- Tai, C.H.; Lee, N.C.; Chien, Y.H.; Byrne, B.J.; Muramatsu, S.I.; Tseng, S.H.; Hwu, W.L. Long-term efficacy and safety of eladocagene exuparvovec in patients with AADC deficiency. Mol. Ther. J. Am. Soc. Gene Ther. 2022, 30, 509–518. [Google Scholar] [CrossRef]
- Kojima, K.; Nakajima, T.; Taga, N.; Miyauchi, A.; Kato, M.; Matsumoto, A.; Ikeda, T.; Nakamura, K.; Kubota, T.; Mizukami, H.; et al. Gene therapy improves motor and mental function of aromatic l-amino acid decarboxylase deficiency. Brain J. Neurol. 2019, 142, 322–333. [Google Scholar] [CrossRef]
- Pearson, T.S.; Gupta, N.; San Sebastian, W.; Imamura-Ching, J.; Viehoever, A.; Grijal-vo-Perez, A.; Fay, A.J.; Seth, N.; Lundy, S.M.; Seo, Y.; et al. Gene therapy for aromatic L-amino acid decarboxylase deficiency by MR-guided direct delivery of AAV2-AADC to midbrain dopaminergic neurons. Nat. Commun. 2021, 12, 4251. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J. Medical treatment of dystonia. Mov. Disord. Off. J. Mov. Disord. Soc. 2013, 28, 1001–1012. [Google Scholar] [CrossRef] [PubMed]
- Rasera, A.; Squintani, G.M.; Cerruto, M.A. A Systematic Review of Botulinum Toxin Injection in Pediatric Dystonia. Toxins 2024, 16, 289. [Google Scholar] [CrossRef] [PubMed]
- Nicotera, A.G.; Di Rosa, G.; Turriziani, L.; Costanzo, M.C.; Stracuzzi, E.; Vitello, G.A.; Rando, R.G.; Musumeci, A.; Vinci, M.; Musumeci, S.A.; et al. Role of COMT V158M Polymorphism in the Development of Dystonia after Administration of Antipsychotic Drugs. Brain Sci. 2021, 11, 1293. [Google Scholar] [CrossRef]
- Koptielow, J.; Szyłak, E.; Szewczyk-Roszczenko, O.; Roszczenko, P.; Kochanowicz, J.; Kułakowska, A.; Chorąży, M. Genetic Update and Treatment for Dystonia. Int. J. Mol. Sci. 2024, 25, 3571. [Google Scholar] [CrossRef]
- DeArias, A.L.; Bamford, N.S. Levodopa for Dystonia in Children: A Case Series and Review of the Literature. Pediatr. Neurol. 2024, 152, 16–19. [Google Scholar] [CrossRef]
- Spoto, G.; Accetta, A.S.; Grella, M.; Di Modica, I.; Nicotera, A.G.; Di Rosa, G. Respiratory Comorbidities and Complications of Cerebral Palsy. Dev. Neurorehabilit. 2024, 27, 194–203. [Google Scholar] [CrossRef]
- Elkaim, L.M.; Alotaibi, N.M.; Sigal, A.; Alotaibi, H.M.; Lipsman, N.; Kalia, S.K.; Fehlings, D.L.; Lozano, A.M.; Ibrahim, G.M.; North American Pediatric DBS Collaboration. Deep brain stimulation for pediatric dystonia: A meta-analysis with individual participant data. Dev. Med. Child Neurol. 2019, 61, 49–56. [Google Scholar] [CrossRef]
- Chudy, D.; Raguž, M.; Vuletić, V.; Rački, V.; Papić, E.; Nenadić Baranašić, N.; Barišić, N. GPi DBS treatment outcome in children with monogenic dystonia: A case series and review of the literature. Front. Neurol. 2023, 14, 1151900. [Google Scholar] [CrossRef]
- Latorre, A.; Bhatia, K.P. Treatment of Paroxysmal Dyskinesia. Neurol. Clin. 2020, 38, 433–447. [Google Scholar] [CrossRef] [PubMed]
- Di Rosa, G.; Lenzo, P.; Parisi, E.; Neri, M.; Guerrera, S.; Nicotera, A.; Alibrandi, A.; Germanò, E.; Caccamo, D.; Spanò, M.; et al. Role of plasma homocysteine levels and MTHFR polymorphisms on IQ scores in children and young adults with epilepsy treated with antiepileptic drugs. Epilepsy Behav. EB 2013, 29, 548–551. [Google Scholar] [CrossRef] [PubMed]
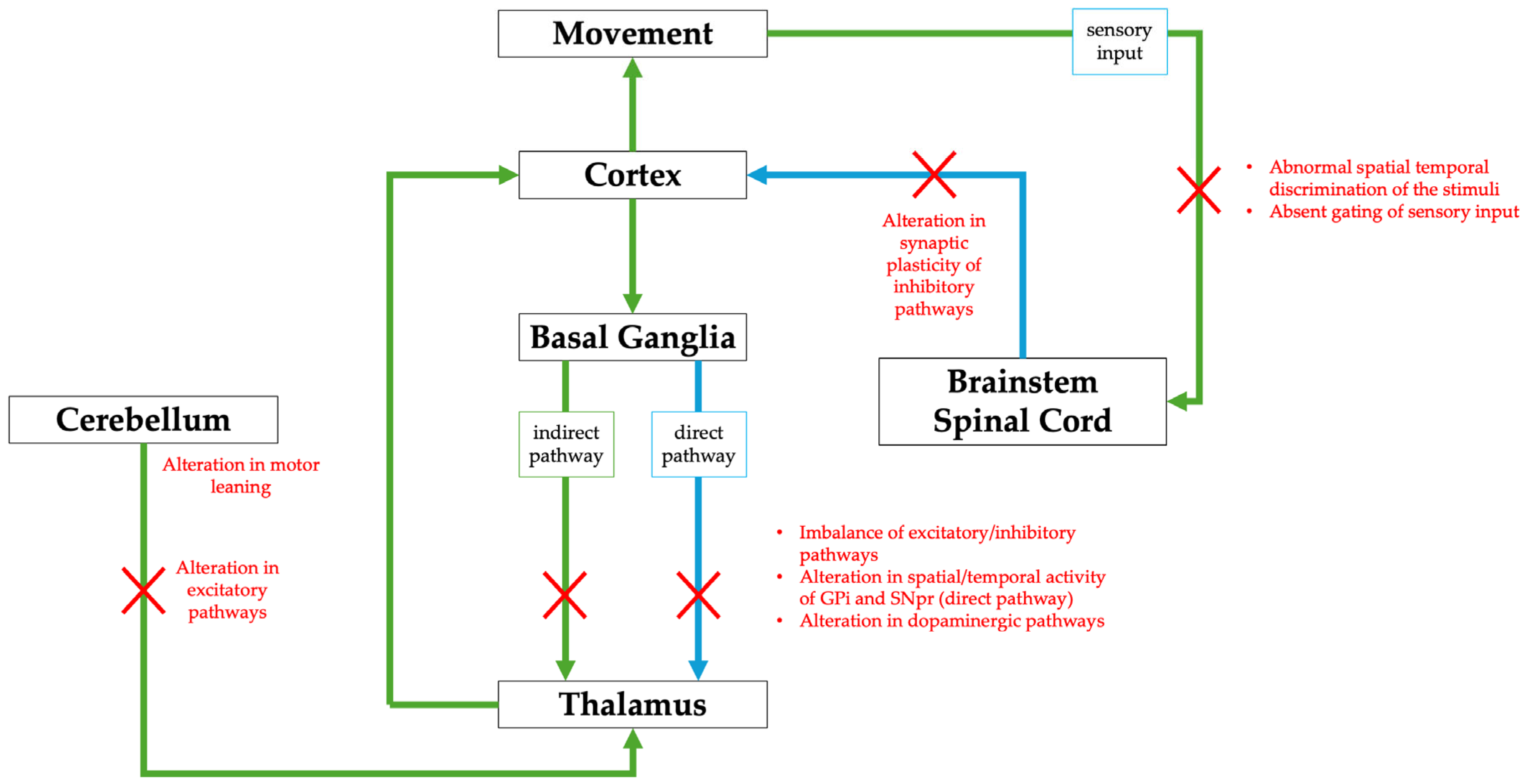
- Steel, D.; Kurian, M.A. Recent genetic advances in early-onset dystonia. Curr. Opin. Neurol. 2020, 33, 500–507. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Gene | Inheritance | Age at Onset | Dystonia Onset | Course of Dystonia | Dystonia Progression | Cognitive Development | Neuropsychiatric Features | Brain MRI | Treatment Options |
|---|---|---|---|---|---|---|---|---|---|
| TOR1A | AD | Childhood | Focal | Progressive | Onset in lower limbs and generalizing with sparing of larynx and neck | Normal | Gait difficulties | Nonspecific findings | Symptomatic GPi DBS |
| THAP1 | AD | Childhood to adulthood | Segmental | Progressive | Onset in cranio-cervical region/upper limbs and generalizing with larynx involvement | NR | Speech impairment, dysarthria, dysphonia | GP hypointensities | Symptomatic GPi DBS |
| KMT2B | AD | Childhood | Focal | Progressive | Onset in lower limbs with caudocranial generalization | DD/IDD | Microcephaly, seizures, and mixed MD with choreoathetosis or myoclonus | GP hypointensities | Symptomatic GPi DBS |
| HPCA | AR | Childhood | Focal | Slowly progressive | Onset distally and generalizing with prominent cranio-cervical involvement | Normal or DD/IDD | Mood disorder, seizures, mixed MD with choreoathetosis | Normal | Symptomatic |
| ANO3 | AD | Childhood to adulthood | Multifocal/ segmental | Progressive | Cranio-cervical onset with generalizing to upper limbs | Normal, occasionally ID | Anxiety, depression, sleep disturbance, seizures, mixed MD with chorea, myoclonus | Normal | Symptomatic GPi-DBS |
| TUBB4A | AD | Early childhood to adulthood | Focal | Progressive | Laryngeal onset with generalizing to neck, face, and limbs | DD | Dysarthria, whispered dysphonia, mixed MD with choreoathetosis, ataxia, spasticity | H-ABC | Symptomatic |
| GNAL | AD/AR | Childhood to adulthood | Focal/ Segmental; rarely generalized | Progressive | Onset in laryngeal and cranio-cervical regions, occasionally generalizing | Normal, occasionally IDD | Frequent mixed MD with tremor or parkinsonism; in biallelic variants, dysmorphic features, sensorineural hearing | Normal | Symptomatic GPi-DBS |
| VPS16 | AD/AR | Childhood | Generalized | Slowly progressive | Onset in oromandibular, bulbar, cervical, upper limb regions, and generalization | Normal or DD/IDD | Anxiety, depression, and emotional lability, neurodevelopmental disorders | Normal or BG hypointensities (iron deposition) | Symptomatic GPi-DBS |
| VPS41 | AR | Infancy | Generalized | Progressive | NR | DD/IDD | Motor dysfunction with ataxia, nystagmus, speech delay, optic atrophy, and axonal neuropathy | Normal or cerebellar vermis atrophy | NR |
| EIF2AK2 | AD/AR | Childhood | Generalized | Progressive | Onset either in the upper or lower limbs or trunk, and generalizing | Normal or DD/IDD | MD, spasticity, seizures, speech impairment, and neurological regression in the context of febrile illness | Normal or hypomyelination/delayed myelination, thin CC, lower medullary lesions | Symptomatic |
| Gene | Inheritance | Age at Onset | Dystonia Onset | Course of Dystonia | Dystonia Progression | Other MDs | Cognitive Development | Neuropsychiatric Features | Brain MRI | Treatment Option |
|---|---|---|---|---|---|---|---|---|---|---|
| GCH1 | AD | Infancy (atypical form) to childhood | Focal limb dystonia | Progressive | Rostrally spreading until generalized | Parkinsonism | Normal | Anxiety, depression | Normal | Levodopa |
| AR | Neonatal to early childhood | Dystonic dysarthria, focal limb or cervical dystonia | Progressive | Neurological deterioration and generalized dystonia | Parkinsonism | Normal to severe IDD | NR | Normal | Levodopa, Phe-restricted diet | |
| TH | AR | Neonatal period/infancy to childhood | Focal lower limb dystonia | Progressive | From one leg to the other leg, arms, trunk, face, and oropharyngeal musculature | Parkinsonism, tremors, myoclonus, OGC | DD/IDD | Behavioral disturbances, encephalopathy | Normal | Levodopa |
| SPR | AR | Infancy | Focal and generalized | Progressive | NR | Parkinsonism, tremors, dysarthria, distal hypertonia, OGC | DD/IDD | Psychiatric disorders, sleep disturbances | Normal or nonspecific findings | Levodopa |
| PRKRA | AR | Childhood to adolescence | Focal limb or cervical dystonia | Progressive | Focal limb or cervical dystonia spreading until generalized | Parkinsonism, tremors | Normal, DD/IDD | Language delay, behavioral disturbances | Normal | Mild to non-response to levodopa |
| DDC | AR | Infancy | Focal limb or cervical dystonia, OGC | Progressive | Generalized | Parkinsonism, OGC, dysarthria, ptosis, choreoathetosis, myoclonic startles | DD/IDD | Anxiety, depression, OCD, ODD, ADHD | Normal | Intraputaminal infusions of eladocagene exuparvovec |
| ATP1A3 | AD | Adolescence to early adulthood | Asymmetrical focal dystonia | Slowly progressive with abrupt exacerbations | Rostro-caudal spreading | Parkinsonism, dysarthria, dysphagia, ocular apraxia | IDD | Mood disorders, psychosis, schizoid personality disorder | Normal | Symptomatic |
| SLC6A3 | AR | Typical: infancy (6 months) Atypical: childhood to adulthood | Generalized | Progressive | Hyperkinetic disorder that progresses to dystonia–parkinsonism | Parkinsonism, chorea, ballism, orolingual dyskinesia, tremors | Normal | ASD, ADHD, bipolar disorder | Normal | Symptomatic |
| VAC14 | AR | Infancy to childhood | Generalized | Progressive | Progression in severity | Parkinsonism | DD/IDD | NR | Normal or T2 hyperintensities of the putamen and caudate nucleus and T2 hypointensity of the pallidum and substantia nigra | Symptomatic or GPi-DBS |
| WARS2 | AR | Childhood | Focal limb dystonia | Progressive | Generalized | Parkinsonism, tremors, ballism, myoclonus | Normal to IDD | Aggressive behavior, anxiety, depression, psychosis, epilepsy | Normal | Levodopa |
| SGCE | AD | Childhood | Focal limb or cervical dystonia | Progressive | Increased action dystonia (writer’s cramp) | Myoclonus | Normal to mild IDD | Panic disorder, depression, anxiety disorder, OCD, ADHD | Normal | Symptomatic |
| KCTD17 | AD | Childhood to adolescence | Focal upper limb dystonia | Progressive | Spreading to cranio-cervical regions, limbs, trunk, and oromandibular and laryngeal muscles | Myoclonus, distal choreic movements | Normal to mild DD/IDD | Anxiety and social phobia, obsessive traits, depression | Normal | GPi-DBS |
| KCNN2 | AD | Childhood | Focal upper limb dystonia (writer’s cramp) | Static | NR | Myoclonus, tremor, ataxia, chorea, tics, nystagmus | Normal to mild IDD | ASD, anxiety, epilepsy, depression, ADHD | Normal to periventricular hyperintensities | Symptomatic |
| GRIN2A | AD | Infancy to childhood | Generalized | Progressive | Progression in severity | Myoclonus, chorea, tremor, ataxia | DD/IDD | Speech and language disorder, epilepsy, ADHD, ASD | Normal or unspecific findings | Symptomatic |
| YY1 | AD | Childhood | Focal upper limb dystonia, laterocollis | Static | NR | Myoclonus | Normal | ADHD | Normal | Symptomatic |
| ADCY5 | AD/AR | Infancy to childhood | Generalized | Static to mild improvement | Progression in severity | Myoclonus, chorea | Normal to mild IDD | OCD, depression, anxiety, and phobias | Normal or nonspecific findings | Caffeine, acetazolamide, clonazepam, methylphenidate, and DBS |
| GNAO1 | AD | Infancy to early childhood | Generalized | Progressive | Progression in severity, frequent status dystonicus | Chorea, ballism, myoclonus | DD/IDD | Epilepsy | Diffuse cortical atrophy, cerebellar atrophy, thinning of CC, focal abnormalities of the BG | Symptomatic, DBS |
| NKX2-1 | AD | Infancy | Focal limb dystonia | Static | Improvement during adolescence | Chorea, ataxia | Normal to IDD | Hypotonia, ASD, ADHD, anxiety | Normal | Symptomatic |
| Gene | Inheritance | Age at Onset | Dystonia Onset and/or Distribution | Other MD | Course of Dystonia | Other Systemic Features/Associated Diseases | Cognitive Development | Neuropsychiatric Features | Brain MRI | Treatment Option |
|---|---|---|---|---|---|---|---|---|---|---|
| ADAR, TREX1 | AR/AD | Infancy | Generalized | Spasticity, ataxia, spastic paraparesis | Slowly progressive | AGS, chilblain, sterile pyrexias, hepatosplenomegaly, cortical blindness | IDD | Irritability, microcephaly, seizures, encephalopathy | Calcification of the putamen, GP, and thalamus | Symptomatic |
| RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, IFIH1 | AR | |||||||||
| PANK2 | AR | Classic PKAN: infancy | Focal progressing to generalized | Parkinsonism, spasticity, choreoathetosis | Progressive | PKAN, pigmentary retinopathy, acanthocytosis | DD/IDD | Dyspraxia, dysarthria, speech disorder | “Eye of the tiger” sign | Symptomatic, DBS, iron chelation |
| Atypical PKAN: childhood | Slowly progressive | OCD, dysarthria, Tourette disorder | ||||||||
| C19orf12 | AR/AD | Childhood to early adulthood | Focal/Multifocal progressing generalized | Spasticity, parkinsonism | Progressive | MPAN, optic atrophy, peripheral neuropathy, bladder and/or bowel incontinence | IDD | Depression, anxiety, OCD, hallucinations, ADHD, dysarthria, dysphagia | Iron deposition in BG and SN | Symptomatic |
| FA2H | AR | Early childhood | Focal progressing to generalized | Ataxia, spasticity | Progressive | FAHN, SPG35, exotropia, optic atrophy | IDD | Seizures, leukodystrophy, mood disorder | Iron deposition in GP pontocerebellar atrophy, thinning of CC | Symptomatic and ablative pallidotomy or thalamotomy |
| Gene | Inheritance | Age at Onset | Dystonia Onset and/or Distribution | Other MD | Course of Dystonia | Other Systemic Features/Associated Diseases | Cognitive Development | Neuropsychiatric Features | Brain MRI | Treatment Option |
|---|---|---|---|---|---|---|---|---|---|---|
| ATM | AR | Early childhood to adulthood | Focal/segmental progressing to generalized | Ataxia, myoclonus, chorea, parkinsonism, postural and rest kinetic tremor | Progressive | Ataxia–telangiectasia, immunodeficiency, predisposition to malignancy | Cognitive deterioration | Peripheral neuropathy, oculomotor apraxia, dysarthria | Cerebellar atrophy | Symptomatic |
| NPC1, NPC2 | AR | Infancy to adulthood | Generalized | Ataxia | Progressive | Niemann–Pick disease, hepatosplenomegaly | DD/IDD | Dysarthria, dysphagia, seizures, gelastic cataplexy, vertical supranuclear gaze palsy | Atrophy of the cerebellar vermis, thinning of the CC, mild cerebral atrophy | Miglustat, symptomatic |
| GLB1 | AR | Late childhood to adulthood | Multifocal progressing to generalized | Ataxia, akinetic– rigid parkinsonism, prominent orofacial dystonia | Progressive | GM1 gangliosidosis, skeletal abnormalities, short stature, corneal clouding, facial dysmorphism | IDD | Behavioral/psychiatric disorders | General cerebral atrophy, ventriculomegaly, and/or a “wish bone” pattern of iron accumulation | Symptomatic |
| FUCA1 | AR | Childhood | Focal progressing to generalized | Dystonic posturing, spasticity | Progressive | Coarse facial features, delayed growth, sinopulmonary infections, visceromegaly, angiokeratoma, dysostosis | Cognitive deterioration | Seizures | Hyperintensity in the BG | Symptomatic |
| PLA2G6 | AR | Infancy to early adulthood | Focal progressing to generalized | Parkinsonism, spasticity, nystagmus | Variable | INAD, ANAD, strabismus, optic atrophy | IDD | Speech delay, ASD, ADHD, emotional lability, seizures | Cerebellar atrophy, iron deposition in GP | Symptomatic |
| SQSTM1 | AR | Childhood | Facial and lower limb dystonia progressing to generalized | Ataxia, gaze palsy, myoclonus | Progressive | Dysautonomia, delayed growth | Cognitive deterioration | Cerebellar and pyramidal signs, dysarthria, oculomotor involvement | Normal or iron accumulation in the BG | Symptomatic |
| DNM1L | AR/AD | Childhood | Paroxysmal, generalized, or action dystonia | Ataxia, spasticity, nystagmus | Progressive | Hyperlactacidemia optic atrophy | IDD | Epilepsy, dysarthria, microcephaly, sensory and motor axonal neuropathy, neurodegenerative disorder | Nonspecific findings | Symptomatic |
| Gene | Inheritance | Age at Onset | Dystonia Onset and/or Distribution | Other MD | Course of Dystonia | Other systemic Features/Associated Diseases | Cognitive Development | Neuropsychiatric Features | Brain MRI | Treatment Option |
|---|---|---|---|---|---|---|---|---|---|---|
| ATP7B | AR | Childhood to adulthood | Focal/generalized | Face involvement with risus sardonicus “flapping” tremor, parkinsonism, choreoathetosis | Progressive | WD, Liver disease, Kayser-Fleischer corneal rings, low serum copper, and ceruloplasmin | IDD | Depression, bipolar spectrum disorder, personality changes, psychosis | “Face of the giant panda” sign | Chelation therapy |
| SLC30A10 | AR | Childhood to adolescence | Focal progressing to generalized | “Cock-walk gait”, fine tremor, bradykinesia, dysdiadochokinesis | Progressive | Hypermanganesemia, polycythemia, liver disease, darker skin tone | Normal | PICA, dysarthria | Hyperintensity in the BG and cerebellum | Chelation therapy |
| SLC39A14 | AR | Infancy to early childhood | Focal progressing to generalized | Spasticity, bulbar dysfunction, parkinsonism | Progressive | Hypermanganesemia, low serum iron, iron deficiency anemia | IDD | NR | Hyperintensity of the BG and cerebellum | Chelation therapy |
| HTT | AD (CAG repeat expansion) | Childhood to adulthood | Focal | Cervical dystonia, parkinsonism, myoclonus, rigidity, bradykinesia, and chorea | NR | Huntington’s disease | DD/IDD | Learning disabilities, epilepsy, and behavioral and psychiatric manifestations | Nonspecific findings | Symptomatic |
| COASY | AR | Infancy | Focal | Oromandibular dystonia, parkinsonism, spasticity, dysarthria | Progressive | CoPAN, peripheral neuropathy, optic atrophy/pigmentary retinopathy | IDD | Gait impairment, learning difficulties, epilepsy | Iron deposition in GP, Pontocerebellar hypoplasia | NR |
| ATP13A2 | AR | Late childhood to adulthood | Focal progressing to generalized | Oromandibular dystonia, parkinsonism spasticity, ataxia, OGC, myoclonus | NR | Kufor Rakeb disease, visual loss | Cognitive deterioration | Epilepsy, ASD, psychosis | Nonspecific findings | Symptomatic |
| Gene | Inheritance | Age at Onset | Dystonia Onset and/or Distribution | Other MD | Course of Dystonia | Other Systemic Features/Associated Diseases | Cognitive Development | Neuropsychiatric Features | Brain MRI | Treatment Option |
|---|---|---|---|---|---|---|---|---|---|---|
| HPRT1 | XL | Infancy | Generalized, action dystonia | Opisthotonos, choreoathetosis, occasionally ballism, cerebral palsy | Progressive | LND, delayed growth and puberty | IDD | Self-injurious behavior, oppositional defiance, seizures, gait impairment | Nonspecific findings | Symptomatic |
| MMUT, MMAA, MMAB, MMADHC, MCEE | AR | Infancy, childhood, or adolescence | Generalized dystonia | Chorea, spasticity, ataxia | Progressive | Methylmalonic acidemia, acute metabolic crises, hematology alteration, | DD/IDD | Seizures, lethargy, hypotonia | BG injury (GP) | Symptomatic, dietary restriction, L-carnitine |
| PCCA, PCCB | AR | Infancy | Focal upper limb dystonia or generalized dystonia after crisis | Choreoathetosis, spasticity | Progressive | Propionic acidemia, acute metabolic crises, ketoacidosis, hyperammonemia, cardiomyopathy, hematology alteration, short stature, gastrointestinal disturbation | DD | Seizure, lethargy, hypotonia, | BG injury (caudate and putamen) | Symptomatic, dietary restriction, L-carnitine |
| GCDH | AR | Infancy | Generalized dystonia | Choreoathetosis, parkinsonism, spasticity, orofacial dyskinesia | Progressive | Glutaric acidemia type 1, acute metabolic crises, macrocephaly, hepatomegaly | IDD | Hypotonia, seizures | BG degeneration, striatal necrosis, frontotemporal atrophy, widening of cortical sulci, symmetrical progressive demyelination | Symptomatic, dietary restriction, L-carnitine |
| ETHE1 | AR | Infancy | Generalized dystonia | Chorea, ataxia | Progressive | Ethylmalonic encephalopathy, chronic diarrhea, petechiae, orthostatic acrocyanosis | DD/IDD | Hypotonia, seizures | Hyperintense lesions in the BG | Symptomatic |
| Gene | Inheritance | Age at Onset | Dystonia Onset and/or Distribution | Other MD | Course of Dystonia | Other Systemic Features/Associated Diseases | Cognitive Development | Neuropsychiatric Features | Brain MRI | Treatment Option |
|---|---|---|---|---|---|---|---|---|---|---|
| SCN1A | AD | Infancy | NR | Chorea, ballism, myoclonus, hand stereotypies | Progressive | Dravet Syndrome, dysmorphic features | DD/IDD | Epileptic encephalopathy, inability to speak, and walk | Progressive cortical and WM atrophy, thinning of the CC, impaired myelination | Symptomatic |
| UBA5 | AR | Infancy | Generalized and status dystonicus | Spasticity, dystonic, or athetoid movements | Progressive | Delayed growth | DD/IDD | Microcephaly, DEE, axial hypotonia with peripheral hypertonia, inability to speak | Nonspecific findings | Symptomatic |
| WWOX | AR | Early childhood | NR | Spasticity, dyskinetic component, bradykinesia | Progressive | Visual impairment, short stature | DD/IDD | DEE, microcephaly, inability to speak, and walk | Nonspecific findings | Symptomatic |
| FOXG1 | AD | Early childhood | Generalized, focal, paroxysmal | Craniofacial involvement, choreoathetosis, stereotypies, akinetic-rigid parkinsonism | Progressive | Rett Syndrome | DD/IDD | Microcephaly, epilepsy, ASD | Nonspecific findings | Symptomatic |
| GABRA1 | AD | Infancy | Generalized | Choreoathetosis, spasticity, cerebral palsy | NR | NR | DD/IDD | DEE, seizures, speech impairment, strabismus | Nonspecific findings | Symptomatic |
| GRIN1 | AD/AR | Infancy | NR | Mixed MD chorea and dystonia, OGC | NR | Dysmorphic features | DD/IDD | Microcephaly, prominent hypotonia, epilepsy, complex stereotypies, sleep disturbances | Nonspecific findings | Symptomatic |
| SPTAN1 | AD | Infancy to early childhood | NR | Ataxia, myoclonus, abnormal ocular movements | NR | NR | DD/IDD | DEE, HSP, ASD | Cerebellar atrophy, delayed myelination, thin CC | Symptomatic |
| DNM1 | AD/AR | Childhood | Focal or segmental | Choreoathetosis, spasticity | NR | Visual impairment | Mild to profound DD/IDD | Epilepsy, encephalopathy, ASD, ADHD, aggressive behaviour | Nonspecific findings | Symptomatic |
| Gene | Inheritance | Age at Onset | Dystonia Onset and/or Distribution | Other MD | Course of Dystonia | Other Systemic Features/Associated Diseases | Cognitive Development | Neuropsychiatric Features | Brain MRI | Treatment Option |
|---|---|---|---|---|---|---|---|---|---|---|
| TIMM8A | XL | Late childhood to adulthood | Focal progressing to generalized | Spasticity, tremor, and postural instability | Progressive | Mohr–Tranebjaerg syndrome, deafness, and visual impairment | Cognitive deterioration | Psychiatric disturbance, dysarthria, and dysphagia | Brain and caudate nuclei atrophy, iron deposition in GP and SN | Symptomatic, DBS |
| DCAF17 | AR | Adolescence | Focal progressing to generalized | Dystonic spasms with dystonic posturing, chorea | Progressive | NBIA, Woodhouse–Sakati syndrome, ↓ IGF-1 | Mild IDD | Dysarthria and dysphagia | Iron deposition in the GP, SN, and red nucleus | Symptomatic or DBS |
| GAMT, SERAC1, SUCLA2 | AR | Infancy to childhood | Focal/generalized | Ataxia, chorea, athetosis | Progressive | SNHL, deafness, failure to thrive, and early death | IDD | Hyperactivity, ASD, self-injurious behavior, seizures, speech disorder | Pathologic intensities in the BG, hypomyelination of WM | Symptomatic |
| SLC6A8, BCAP31 | XL | |||||||||
| ACTB | AD | Adolescence | Focal progressing to generalized | Bulbar dysfunction | Progressive | Congenital deafness, skeletal abnormalities | DD/IDD | Dysarthria and dysphagia | Nonspecific findings | Symptomatic, DBS |
| Gene | Inheritance | Age at Onset | Dystonia Onset and/or Distribution | Other MD | Course of Dystonia | Other Systemic Features/Associated Diseases | Cognitive Development | Neuropsychiatric Features | Brain MRI | Treatment Option |
|---|---|---|---|---|---|---|---|---|---|---|
| POLG | AR/AD | Adolescence to adulthood | Focal/generalized | Ataxia, myoclonus, choreoathetosis, parkinsonism | Progressive | External ophthalmoplegia, liver, gastrointestinal, and renal diseases, hearing loss | DD | Seizures, dysarthria, mood disorder, sleep disorders | Nonspecific findings | Symptomatic |
| SLC19A3 | AR | Childhood | Generalized | Ataxia, rigidity | Progressive | External ophthalmoplegia, quadriparesis, coma, and death | DD/IDD | Acute encephalopathy, dysarthria, dysphagia, loss of developmental milestones, and seizures | Symmetric and bilateral necrosis in the BG with severe edema | Thiamine and/or biotin therapy |
| MECR | AR | Infancy to childhood | Generalized | NR | Progressive | Optic atrophy | Normal | Epilepsy and dysarthria | Symmetric and bilateral BG abnormalities | Symptomatic, DBS |
| Gene | Inheritance | Age at Onset | Dystonia/Dyskinesia Features | Type of PxMD | Cognitive Development | Neuropsychiatric Features | Brain MRI | Treatment Option |
|---|---|---|---|---|---|---|---|---|
| PRRT2 | AD | Childhood, adolescence | Dystonic postures, chorea, or athetosis | PKD | Normal, mild DD/IDD | Epilepsy, anxiety, depression, sleep disorders, ADHD | Normal | SCB |
| TMEM151A | AD | Childhood, adolescence | Dystonia with facial involvement | PKD | NR | Occasional migraine or tremor, epilepsy | Normal | SCB |
| RHOBTB2 | AD | Infancy, childhood | Choreodystonia, ataxia, and/or stereotypies | PKD | Severe DD/IDD | ASD | Normal or unspecific findings | SCB |
| SCN8A | AD | Childhood | Orobuccolingual dyskinesia, choreiform movements, tremor | PKD | DD/IDD | Epilepsy, ASD, ADHD | Unspecific findings | SCB, KD |
| SLC2A1 | AD | Childhood | Foot dystonia, paroxysmal choreoathetosis, progressive spastic paraplegia | PED | DD/IDD | Epilepsy | Normal | KD, triheptanoin |
| TBC1D24 | AR | Childhood | Dystonic movements, myoclonus, ataxia, dysmetria, dysarthria | PED | Normal, DD/IDD | Epilepsy | Non-progressive pontocerebellar hypoplasia | SCB, Acetazolamide |
| ECHS1 | AR | Childhood | Generalized dystonia, hemydystonia, focal limb dystonia, torticollis | PED | IDD | Leigh-like syndrome | Bilateral hyperintensity of the GP | Dietary protein restriction and symptomatic supplementation |
| PNKD | AD | Childhood, early adolescence | Dystonia, chorea, athetosis | PNKD | Normal | Tourette syndrome, Tic disorder, anxiety, depression | Normal | BZD |
| KCNMA1 | AD | Infancy, early childhood | Dystonic movements and ocular signs | PNKD | DD/IDD | Epilepsy | Normal | Symptomatic |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ceraolo, G.; Spoto, G.; Consoli, C.; Modafferi, E.; Di Rosa, G.; Nicotera, A.G. Pediatric Genetic Dystonias: Current Diagnostic Approaches and Treatment Options. Life 2025, 15, 992. https://doi.org/10.3390/life15070992
Ceraolo G, Spoto G, Consoli C, Modafferi E, Di Rosa G, Nicotera AG. Pediatric Genetic Dystonias: Current Diagnostic Approaches and Treatment Options. Life. 2025; 15(7):992. https://doi.org/10.3390/life15070992
Chicago/Turabian StyleCeraolo, Graziana, Giulia Spoto, Carla Consoli, Elena Modafferi, Gabriella Di Rosa, and Antonio Gennaro Nicotera. 2025. "Pediatric Genetic Dystonias: Current Diagnostic Approaches and Treatment Options" Life 15, no. 7: 992. https://doi.org/10.3390/life15070992
APA StyleCeraolo, G., Spoto, G., Consoli, C., Modafferi, E., Di Rosa, G., & Nicotera, A. G. (2025). Pediatric Genetic Dystonias: Current Diagnostic Approaches and Treatment Options. Life, 15(7), 992. https://doi.org/10.3390/life15070992