
The Hidden Price of Plenty: Oxidative Stress and Calorie-Induced Cardiometabolic Dysfunction
 ,
,  , , , ,
, , , ,  , and
, and 
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Oxygen—A Medal with Two Sides
3. Nutritional Excess and Oxidative Stress—A Pathophysiological Perspective
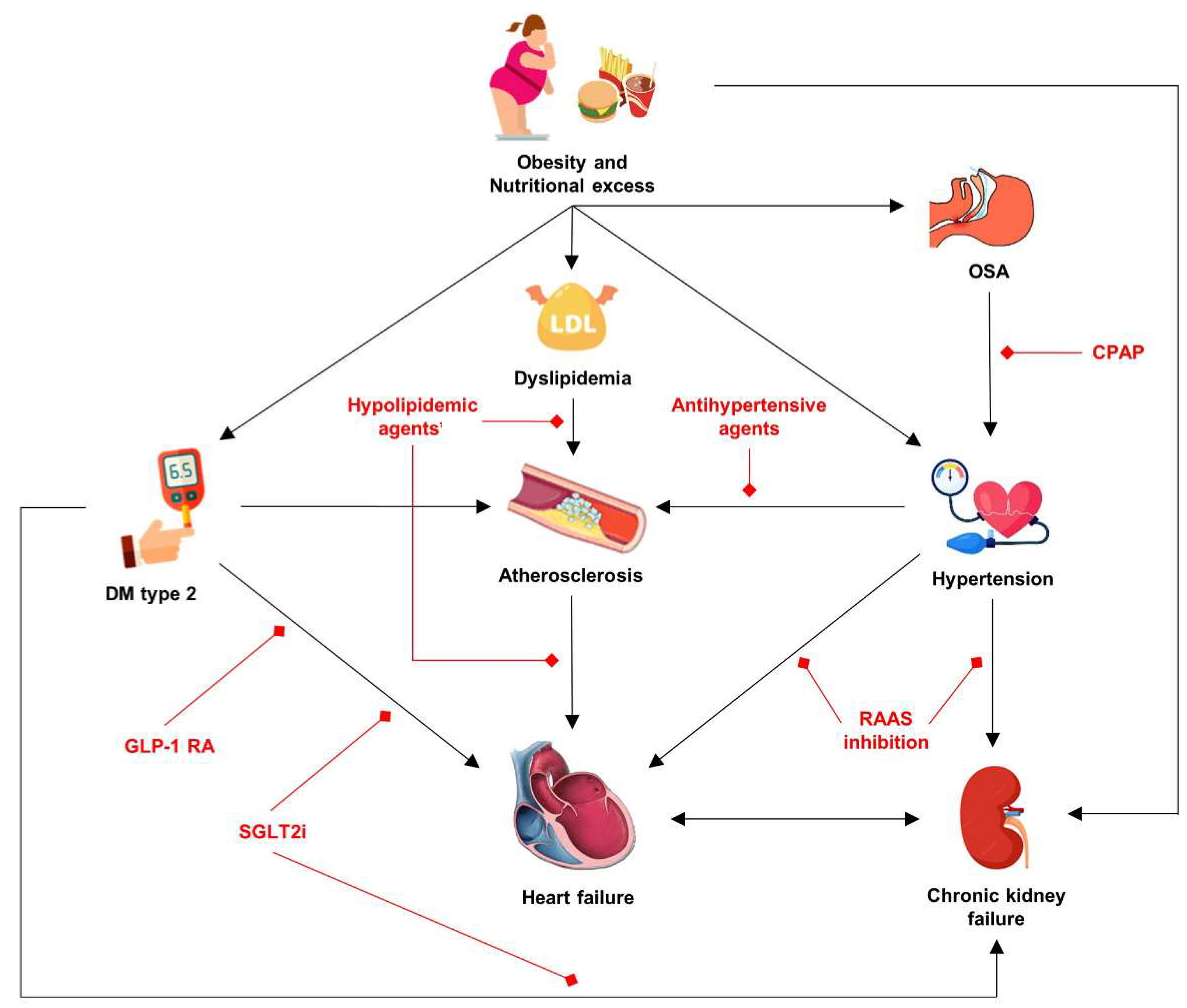
4. Overfeeding, Oxidative Stress, and Cardiometabolic Diseases—Connecting the Dots
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Who.int. Obesity and Overweight. Available online: http://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 25 April 2025).
- Jiang, S.; Liu, H.; Li, C. Dietary regulation of oxidative stress in chronic metabolic diseases. Foods 2021, 10, 1854. [Google Scholar] [CrossRef]
- Halliwell, B.; Cross, C.E. Oxygen-derived species: Their relation to human disease and environmental stress. Environ. Health Perspect. 1994, 102 (Suppl. S10), 5–12. [Google Scholar]
- Martin, W.F.; Sousa, F.L. Early microbial evolution: The age of anaerobes. Cold Spring Harb. Perspect. Biol. 2015, 8, a018127. [Google Scholar] [CrossRef] [PubMed]
- Decker, K.; Jungermann, K.; Thauer, R.K. Energy production in anaerobic organisms. Angew. Chem. Int. Ed. Engl. 1970, 9, 138–158. [Google Scholar] [CrossRef] [PubMed]
- Kennelly, P.J. The Biochemistry of Aging. In Harper’s Illustrated Biochemistry, 31st ed.; Rodwell, V.W., Bender, D.A., Botham, K.M., Kennelly, P.J., Weil, P.A., Eds.; McGraw-Hill Education: New York, NY, USA, 2018. [Google Scholar]
- Gerschman, R.; Gilbert, D.L.; Nye, S.W.; Dwyer, P.; Fenn, W.O. Oxygen poisoning and x-irradiation: A mechanism in common. Science 1954, 119, 623–626. [Google Scholar] [CrossRef]
- Ávila-Escalante, M.L.; Coop-Gamas, F.; Cervantes-Rodríguez, M.; Méndez-Iturbide, D.; Aranda-González, I.I. The effect of diet on oxidative stress and metabolic diseases-Clinically controlled trials. J. Food Biochem. 2020, 44, e13191. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Lobo, V.; Patil, A.; Phatak, A.; Chandra, N. Free radicals, antioxidants and functional foods: Impact on human health. Pharmacogn. Rev. 2010, 4, 118–126. [Google Scholar] [CrossRef]
- Diamanti-Kandarakis, E.; Papalou, O.; Kandaraki, E.A.; Kassi, G. Mechanisms in Endocrinology: Nutrition as a mediator of oxidative stress in metabolic and reproductive disorders in women. Eur. J. Endocrinol. 2017, 176, R79–R99. [Google Scholar] [CrossRef]
- Lushchak, V.I. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem. Biol. Interact. 2014, 224, 164–175. [Google Scholar] [CrossRef]
- Srivastava, K.K.; Kumar, R. Stress, oxidative injury and disease. Indian J. Clin. Biochem. 2015, 30, 3–10. [Google Scholar] [CrossRef] [PubMed]
- McMullan, A.; Zwierzynski, J.B.; Jain, N.; Haneline, L.S.; Shou, W.; Kua, K.L.; Hota, S.K.; Durbin, M.D. Role of maternal obesity in offspring cardiovascular development and congenital heart defects. J. Am. Heart Assoc. 2025, 14, e039684. [Google Scholar] [CrossRef]
- Kowalczyk, P.; Sulejczak, D.; Kleczkowska, P.; Bukowska-Ośko, I.; Kucia, M.; Popiel, M.; Wietrak, E.; Kramkowski, K.; Wrzosek, K.; Kaczyńska, K. Mitochondrial oxidative stress-A causative factor and therapeutic target in many diseases. Int. J. Mol. Sci. 2021, 22, 13384. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Kokkinopoulou, I.; Moutsatsou, P. Mitochondrial glucocorticoid receptors and their actions. Int. J. Mol. Sci. 2021, 22, 6054. [Google Scholar] [CrossRef] [PubMed]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef]
- Zhao, R.-Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Cadenas, E.; Davies, K.J.A. Mitochondrial free radical generation, oxidative stress, and aging11This article is dedicated to the memory of our dear friend, colleague, and mentor Lars Ernster (1920–1998), in gratitude for all he gave to us. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.L.; Norhaizan, M.E.; Liew, W.-P.-P. Nutrients and oxidative stress: Friend or foe? Oxid. Med. Cell. Longev. 2018, 2018, 9719584. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.J.; Igamberdiev, A.U. Reactive nitrogen species in mitochondria and their implications in plant energy status and hypoxic stress tolerance. Front. Plant Sci. 2016, 7, 369. [Google Scholar] [CrossRef] [PubMed]
- Görlach, A.; Dimova, E.Y.; Petry, A.; Martínez-Ruiz, A.; Hernansanz-Agustín, P.; Rolo, A.P.; Palmeira, C.M.; Kietzmann, T. Reactive oxygen species, nutrition, hypoxia and diseases: Problems solved? Redox Biol. 2015, 6, 372–385. [Google Scholar] [CrossRef]
- Vermot, A.; Petit-Härtlein, I.; Smith, S.M.E.; Fieschi, F. NADPH oxidases (NOX): An overview from discovery, molecular mechanisms to physiology and pathology. Antioxidants 2021, 10, 890. [Google Scholar] [CrossRef]
- Raad, H.; Derkawi, R.A.; Tlili, A.; Belambri, S.A.; Dang, P.M.-C.; El-Benna, J. Phosphorylation of gp91phox/NOX2 in human neutrophils. Methods Mol. Biol. 2019, 1982, 341–352. [Google Scholar]
- Mohanty, P. Glucose challenge stimulates reactive oxygen species (ROS) generation by leucocytes. J. Clin. Endocrinol. Metab. 2000, 85, 2970–2973. [Google Scholar] [CrossRef]
- Mohanty, P.; Ghanim, H.; Hamouda, W.; Aljada, A.; Garg, R.; Dandona, P. Both lipid and protein intakes stimulate increased generation of reactive oxygen species by polymorphonuclear leukocytes and mononuclear cells. Am. J. Clin. Nutr. 2002, 75, 767–772. [Google Scholar] [CrossRef]
- Dandona, P. Rapid Communication: Inhibitory effect of a two day fast on reactive oxygen species (ROS) generation by leucocytes and plasma ortho-tyrosine and meta-tyrosine concentrations. J. Clin. Endocrinol. Metab. 2001, 86, 2899–2902. [Google Scholar] [CrossRef]
- Ghemrawi, R.; Battaglia-Hsu, S.-F.; Arnold, C. Endoplasmic reticulum stress in metabolic disorders. Cells 2018, 7, 63. [Google Scholar] [CrossRef]
- Tu, B.P.; Weissman, J.S. Oxidative protein folding in eukaryotes: Mechanisms and consequences: Mechanisms and consequences. J. Cell Biol. 2004, 164, 341–346. [Google Scholar] [CrossRef]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef] [PubMed]
- Bachar, E.; Ariav, Y.; Ketzinel-Gilad, M.; Cerasi, E.; Kaiser, N.; Leibowitz, G. Glucose amplifies fatty acid-induced endoplasmic reticulum stress in pancreatic beta-cells via activation of mTORC1. PLoS ONE 2009, 4, e4954. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.; Liu, Y.; Zhang, L.; Huang, S.; Ding, H.-F.; Dong, Z. mTOR contributes to ER stress and associated apoptosis in renal tubular cells. Am. J. Physiol. Renal Physiol. 2015, 308, F267–F274. [Google Scholar] [CrossRef]
- Arruda, A.P.; Pers, B.M.; Parlakgül, G.; Güney, E.; Inouye, K.; Hotamisligil, G.S. Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nat. Med. 2014, 20, 1427–1435. [Google Scholar] [CrossRef]
- Pereda, A.E.; Curti, S.; Hoge, G.; Cachope, R.; Flores, C.E.; Rash, J.E. Gap junction-mediated electrical transmission: Regulatory mechanisms and plasticity. Biochim. Biophys. Acta 2013, 1828, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Bravo, R.; Gutierrez, T.; Paredes, F.; Gatica, D.; Rodriguez, A.E.; Pedrozo, Z.; Chiong, M.; Parra, V.; Quest, A.F.G.; Rothermel, B.A.; et al. Endoplasmic reticulum: ER stress regulates mitochondrial bioenergetics. Int. J. Biochem. Cell Biol. 2012, 44, 16–20. [Google Scholar] [CrossRef]
- Rieusset, J. The role of endoplasmic reticulum-mitochondria contact sites in the control of glucose homeostasis: An update. Cell Death Dis. 2018, 9, 388. [Google Scholar] [CrossRef]
- Tang, Q.; Liu, Q.; Li, Y.; Mo, L.; He, J. CRELD2, endoplasmic reticulum stress, and human diseases. Front. Endocrinol. 2023, 14, 1117414. [Google Scholar] [CrossRef]
- Oakes, S.A.; Papa, F.R. The role of endoplasmic reticulum stress in human pathology. Annu. Rev. Pathol. 2015, 10, 173–194. [Google Scholar] [CrossRef]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.D.; Kaufman, R.J. The endoplasmic reticulum and the unfolded protein response. Semin. Cell Dev. Biol. 2007, 18, 716–731. [Google Scholar] [CrossRef] [PubMed]
- Mony, V.K.; Benjamin, S.; O’Rourke, E.J. A lysosome-centered view of nutrient homeostasis. Autophagy 2016, 12, 619–631. [Google Scholar] [CrossRef]
- Li, F.; Zhang, H. Lysosomal acid lipase in lipid metabolism and beyond. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 850–856. [Google Scholar] [CrossRef]
- Antonenkov, V.D.; Grunau, S.; Ohlmeier, S.; Hiltunen, J.K. Peroxisomes are oxidative organelles. Antioxid. Redox Signal. 2010, 13, 525–537. [Google Scholar] [CrossRef]
- Kleiboeker, B.; Lodhi, I.J. Peroxisomal regulation of energy homeostasis: Effect on obesity and related metabolic disorders. Mol. Metab. 2022, 65, 101577. [Google Scholar] [CrossRef]
- Schrader, M.; Fahimi, H.D. Peroxisomes and oxidative stress. Biochim. Biophys. Acta 2006, 1763, 1755–1766. [Google Scholar] [CrossRef]
- Braz, G.R.F.; Silva SCde, A.; Pedroza AAda, S.; de Lemos, M.D.; de Lima, F.A.; da Silva, A.I.; Lagranha, C.J. Fluoxetine administration in juvenile overfed rats improves hypothalamic mitochondrial respiration and REDOX status and induces mitochondrial biogenesis transcriptional expression. Eur. J. Pharmacol. 2020, 881, 173200. [Google Scholar] [CrossRef] [PubMed]
- Lalrinzuali, S.; Khushboo, M.; Dinata, R.; Bhanushree, B.; Nisa, N.; Bidanchi, R.M.; Laskar, S.-A.; Manikandan, B.; Abinash, G.; Buragohain, P.; et al. Long-term consumption of fermented pork fat-based diets differing in calorie, fat content, and fatty acid levels mediates oxidative stress, inflammation, redox imbalance, germ cell apoptosis, disruption of steroidogenesis, and testicular dysfunction in Wistar rats. Environ. Sci. Pollut. Res. Int. 2023, 30, 52446–52471. [Google Scholar]
- Ghaddar, B.; Bringart, M.; Lefebvre d’Hellencourt, C.; Meilhac, O.; Diotel, N. Deleterious effects of overfeeding on brain homeostasis and plasticity in adult zebrafish. Zebrafish 2021, 18, 190–206. [Google Scholar] [CrossRef]
- Boden, G.; Homko, C.; Barrero, C.A.; Stein, T.P.; Chen, X.; Cheung, P.; Fecchio, C.; Koller, S.; Merali, S. Excessive caloric intake acutely causes oxidative stress, GLUT4 carbonylation, and insulin resistance in healthy men. Sci. Transl. Med. 2015, 7, 304re7. [Google Scholar] [CrossRef] [PubMed]
- Toledo, F.G.S.; Johannsen, D.L.; Covington, J.D.; Bajpeyi, S.; Goodpaster, B.; Conley, K.E.; Ravussin, E. Impact of prolonged overfeeding on skeletal muscle mitochondria in healthy individuals. Diabetologia 2018, 61, 466–475. [Google Scholar]
- Russo, L.; Babboni, S.; Andreassi, M.G.; Daher, J.; Canale, P.; Del Turco, S.; Basta, G. Treating metabolic dysregulation and senescence by caloric restriction: Killing two birds with one stone? Antioxidants 2025, 14, 99. [Google Scholar] [CrossRef] [PubMed]
- Abad-Jiménez, Z.; López-Domènech, S.; Pelechá, M.; Perea-Galera, L.; Rovira-Llopis, S.; Bañuls, C.; Blas-García, A.; Apostolova, N.; Morillas, C.; Víctor, V.M.; et al. Calorie restriction modulates mitochondrial dynamics and autophagy in leukocytes of patients with obesity. Free Radic. Biol. Med. 2024, 225, 677–686. [Google Scholar]
- Talebi, S.; Shab-Bidar, S.; Askari, G.; Mohammadi, H.; Moini, A.; Djafarian, K. Comparison of the impact of intermittent fasting diet alone or in conjunction with probiotic supplementation versus calorie-restricted diet on inflammatory, oxidative stress, and antioxidant capacity biomarkers in women with polycystic ovary syndrome: A randomized placebo-controlled trial. J. Res. Med. Sci. 2025, 30, 5. [Google Scholar] [PubMed]
- Čolak, E.; Žorić, L. Interrelation of oxidative stress and genetics in pathophysiology of obesity and obesity-related conditions. Genes 2025, 16, 489. [Google Scholar] [CrossRef]
- Krishnamurthy, H.K.; Rajavelu, I.; Pereira, M.; Jayaraman, V.; Krishna, K.; Wang, T.; Bei, K.; Rajasekaran, J.J. Inside the genome: Understanding genetic influences on oxidative stress. Front. Genet. 2024, 15, 1397352. [Google Scholar] [CrossRef]
- Phull, A.-R.; Arain, S.Q.; Majid, A.; Fatima, H.; Ahmed, M.; Kim, S.-J. Oxidative stress-mediated epigenetic remodeling, metastatic progression and cell signaling in cancer. Oncologie 2024, 26, 493–507. [Google Scholar] [CrossRef]
- Huang, M.; Wu, Q.; Jiang, Z.-H. Epigenetic alterations under oxidative stress in stem cells. Oxid. Med. Cell Longev. 2022, 2022, 6439097. [Google Scholar] [CrossRef]
- Dubois-Deruy, E.; Peugnet, V.; Turkieh, A.; Pinet, F. Oxidative stress in cardiovascular diseases. Antioxidants 2020, 9, 864. [Google Scholar] [CrossRef]
- Sinenko, S.A.; Starkova, T.Y.; Kuzmin, A.A.; Tomilin, A.N. Physiological signaling functions of reactive oxygen species in stem cells: From flies to man. Front. Cell Dev. Biol. 2021, 9, 714370. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y. Roles of oxidative stress and inflammation in vascular endothelial dysfunction-related disease. Antioxidants 2022, 11, 1958. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Xia, N.; Li, H. Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef]
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef]
- Tran, N.; Garcia, T.; Aniqa, M.; Ali, S.; Ally, A.; Nauli, S.M. Endothelial nitric oxide synthase (eNOS) and the cardiovascular system: In physiology and in disease states. Am. J. Biomed. Sci. Res. 2022, 15, 153. [Google Scholar]
- Janaszak-Jasiecka, A.; Płoska, A.; Wierońska, J.M.; Dobrucki, L.W.; Kalinowski, L. Endothelial dysfunction due to eNOS uncoupling: Molecular mechanisms as potential therapeutic targets. Cell. Mol. Biol. Lett. 2023, 28, 21. [Google Scholar] [CrossRef]
- Libby, P. The changing landscape of atherosclerosis. Nature 2021, 592, 524–533. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Nikiforov, N.G.; Markin, A.M.; Kashirskikh, D.A.; Myasoedova, V.A.; Gerasimova, E.V.; Orekhov, A.N. Overview of OxLDL and its impact on cardiovascular health: Focus on atherosclerosis. Front. Pharmacol. 2021, 11, 613780. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chang, J.; Chen, V.; Beg, M.A.; Huang, W.; Vick, L.; Wang, Y.; Zhang, H.; Yttre, E.; Gupta, A.; et al. Oxidized LDL stimulates PKM2-mediated mtROS production and phagocytosis. J. Lipid Res. 2025, 66, 100809. [Google Scholar] [CrossRef]
- Donia, T.; Khamis, A. Management of oxidative stress and inflammation in cardiovascular diseases: Mechanisms and challenges. Environ. Sci. Pollut. Res. Int. 2021, 28, 34121–34153. [Google Scholar] [CrossRef]
- Li, S.; Li, Q.; Zhou, Q.; Li, S.; Wang, S.; Yao, Q.; Ouyang, C.; Liu, C.; Li, M. Attenuating atherosclerosis through inhibition of the NF-κB/NLRP3/IL-1β pathway-mediated pyroptosis in vascular smooth muscle cells (VSMCs). Cardiovasc. Ther. 2024, 2024, 1506083. [Google Scholar] [CrossRef] [PubMed]
- Samah, N.; Ugusman, A.; Hamid, A.A.; Sulaiman, N.; Aminuddin, A. Role of matrix metalloproteinase-2 in the development of atherosclerosis among patients with coronary artery disease. Mediators Inflamm. 2023, 2023, 9715114. [Google Scholar] [CrossRef]
- Nagareddy, P.R.; Rajput, P.S.; Vasudevan, H.; McClure, B.; Kumar, U.; Macleod, K.M.; McNeill, J.H. Inhibition of matrix metalloproteinase-2 improves endothelial function and prevents hypertension in insulin-resistant rats: MMP-2 impairs endothelial function. Br. J. Pharmacol. 2012, 165, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Momi, S.; Falcinelli, E.; Petito, E.; Ciarrocca Taranta, G.; Ossoli, A.; Gresele, P. Matrix metalloproteinase-2 on activated platelets triggers endothelial PAR-1 initiating atherosclerosis. Eur. Heart J. 2022, 43, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.-H.; Kim, Y.; Kim, M.; Jang, J.; Lee, S. Emerging roles of vascular cell adhesion molecule-1 (VCAM-1) in immunological disorders and cancer. Int. J. Mol. Sci. 2018, 19, 1057. [Google Scholar] [CrossRef]
- Sun, S.-L.; Liu, L.-M. Urotensin II: An inflammatory cytokine. J. Endocrinol. 2019, 240, R107–R117. [Google Scholar] [CrossRef]
- Simunovic, M.; Jukic, A.; Paradzik, M.; Supe-Domic, D.; Stanisic, L.; Degoricija, M.; Hillestad, A.H.; Skrabic, V.; Bozic, J. The role of urotensin-II in obesity and metabolic syndrome in pediatric population. Children 2022, 9, 204. [Google Scholar] [CrossRef]
- Mihovilovic, A.; Dogas, Z.; Martinovic, D.; Tokic, D.; Puizina Mladinic, E.; Kumric, M.; Ivkovic, N.; Vilovic, M.; Bozic, J. Serum urotensin II levels are elevated in patients with obstructive sleep apnea. Biomolecules 2023, 13, 914. [Google Scholar] [CrossRef]
- Coats, A.; Jain, S. Protective effects of nebivolol from oxidative stress to prevent hypertension-related target organ damage. J. Hum. Hypertens. 2017, 31, 376–381. [Google Scholar] [CrossRef]
- Rodrigo, R.; González, J.; Paoletto, F. The role of oxidative stress in the pathophysiology of hypertension. Hypertens. Res. 2011, 34, 431–440. [Google Scholar] [CrossRef]
- Shariq, O.A.; McKenzie, T.J. Obesity-related hypertension: A review of pathophysiology, management, and the role of metabolic surgery. Gland. Surg. 2020, 9, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Lambert, E.A.; Esler, M.D.; Schlaich, M.P.; Dixon, J.; Eikelis, N.; Lambert, G.W. Obesity-associated organ damage and sympathetic nervous activity: A target for treatment? Hypertension 2019, 73, 1150–1159. [Google Scholar] [CrossRef]
- Shu, L.; Xiao, L.; Hu, B.; Yu, Q.; Dai, D.; Chen, J.; Wang, J.; Xi, Z.; Zhang, J.; Bao, M. Carotid baroreceptor stimulation attenuates obesity-related hypertension through sympathetic-driven IL- 22 restoration of intestinal homeostasis. Eur. J. Med. Res. 2025, 30, 291. [Google Scholar] [CrossRef]
- Muntzel, M.S.; Al-Naimi, O.A.S.; Barclay, A.; Ajasin, D. Cafeteria diet increases fat mass and chronically elevates lumbar sympathetic nerve activity in rats. Hypertension 2012, 60, 1498–1502. [Google Scholar] [CrossRef] [PubMed]
- Bentley-Lewis, R.; Adler, G.K.; Perlstein, T.; Seely, E.W.; Hopkins, P.N.; Williams, G.H.; Garg, R. Body mass index predicts aldosterone production in normotensive adults on a high-salt diet. J. Clin. Endocrinol. Metab. 2007, 92, 4472–4475. [Google Scholar] [CrossRef] [PubMed]
- Engeli, S.; Sharma, A.M. The renin-angiotensin system and natriuretic peptides in obesity-associated hypertension. J. Mol. Med. 2001, 79, 21–29. [Google Scholar] [CrossRef]
- Cabandugama, P.K.; Gardner, M.J.; Sowers, J.R. The renin angiotensin aldosterone system in obesity and hypertension. Med. Clin. N. Am. 2017, 101, 129–137. [Google Scholar] [CrossRef]
- Ehrhart-Bornstein, M.; Lamounier-Zepter, V.; Schraven, A.; Langenbach, J.; Willenberg, H.S.; Barthel, A.; Hauner, H.; McCann, S.M.; Scherbaum, W.A.; Bornstein, S.R. Human adipocytes secrete mineralocorticoid-releasing factors. Proc. Natl. Acad. Sci. USA 2003, 100, 14211–14216. [Google Scholar] [CrossRef]
- Hall, J.E.; do Carmo, J.M.; da Silva, A.A.; Wang, Z.; Hall, M.E. Obesity-induced hypertension: Interaction of neurohumoral and renal mechanisms. Circ. Res. 2015, 116, 991–1006. [Google Scholar] [CrossRef]
- da Silva, A.A.; do Carmo, J.M.; Li, X.; Wang, Z.; Mouton, A.J.; Hall, J.E. Role of hyperinsulinemia and insulin resistance in hypertension: Metabolic syndrome revisited. Can. J. Cardiol. 2020, 36, 671–682. [Google Scholar] [CrossRef]
- Gruber, T.; Pan, C.; Contreras, R.E.; Wiedemann, T.; Morgan, D.A.; Skowronski, A.A.; Lefort, S.; Murat, C.D.B.; Le Thuc, O.; Legutko, B.; et al. Obesity-associated hyperleptinemia alters the gliovascular interface of the hypothalamus to promote hypertension. Cell Metab. 2021, 33, 1155–1170.e10. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Sathyapalan, T.; Atkin, S.L.; Sahebkar, A. Molecular mechanisms linking oxidative stress and diabetes mellitus. Oxid. Med. Cell. Longev. 2020, 2020, 8609213. [Google Scholar] [CrossRef]
- Ijsselmuiden, A.J.J.; Musters, R.J.P.; de Ruiter, G.; van Heerebeek, L.; Alderse-Baas, F.; van Schilfgaarde, M.; Leyte, A.; Tangelder, G.-J.; Laarman, G.J.; Paulus, W.J. Circulating white blood cells and platelets amplify oxidative stress in heart failure. Nat. Clin. Pract. Cardiovasc. Med. 2008, 5, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Copps, K.D.; White, M.F. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582. [Google Scholar] [CrossRef]
- Ho, C.K.; Sriram, G.; Dipple, K.M. Insulin sensitivity predictions in individuals with obesity and type II diabetes mellitus using mathematical model of the insulin signal transduction pathway. Mol. Genet. Metab. 2016, 119, 288–292. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.L.; Goldfine, I.D.; Maddux, B.A.; Grodsky, G.M. Are oxidative Stress−Activated signaling pathways mediators of insulin resistance and β-cell dysfunction? Diabetes 2003, 52, 1–8. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Farrokhi, F.R.; Butler, A.E.; Sahebkar, A. Insulin resistance: Review of the underlying molecular mechanisms. J. Cell. Physiol. 2019, 234, 8152–8161. [Google Scholar] [CrossRef] [PubMed]
- Elmarakby, A.A.; Sullivan, J.C. Relationship between oxidative stress and inflammatory cytokines in diabetic nephropathy: Relationship between oxidative stress and inflammatory cytokines. Cardiovasc. Ther. 2012, 30, 49–59. [Google Scholar] [CrossRef]
- Evans, J.L.; Maddux, B.A.; Goldfine, I.D. The molecular basis for oxidative stress-induced insulin resistance. Antioxid. Redox Signal. 2005, 7, 1040–1052. [Google Scholar] [CrossRef]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef]
- Houstis, N.; Rosen, E.D.; Lander, E.S. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 2006, 440, 944–948. [Google Scholar] [CrossRef] [PubMed]
- Hurrle, S.; Hsu, W.H. The etiology of oxidative stress in insulin resistance. Biomed. J. 2017, 40, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Moreira, P.I.; Sayre, L.M.; Zhu, X.; Nunomura, A.; Smith, M.A.; Perry, G. Detection and localization of markers of oxidative stress by in situ methods: Application in the study of Alzheimer disease. Methods Mol. Biol. 2010, 610, 419–434. [Google Scholar]
- Rahmanto, A.S.; Morgan, P.E.; Hawkins, C.L.; Davies, M.J. Cellular effects of peptide and protein hydroperoxides. Free Radic. Biol. Med. 2010, 48, 1071–1078. [Google Scholar] [CrossRef]
- Fazakerley, D.J.; Minard, A.Y.; Krycer, J.R.; Thomas, K.C.; Stöckli, J.; Harney, D.J.; Burchfield, J.G.; Maghzal, G.J.; Caldwell, S.T.; Hartley, R.C.; et al. Mitochondrial oxidative stress causes insulin resistance without disrupting oxidative phosphorylation. J. Biol. Chem. 2018, 293, 7315–7328. [Google Scholar] [CrossRef] [PubMed]
- Zalewska, E.; Kmieć, P.; Sworczak, K. Role of catestatin in the cardiovascular system and metabolic disorders. Front. Cardiovasc. Med. 2022, 9, 909480. [Google Scholar] [CrossRef]
- Bandyopadhyay, G.; Tang, K.; Webster, N.J.G.; van den Bogaart, G.; Mahata, S.K. Catestatin induces glycogenesis by stimulating the phosphoinositide 3-kinase-AKT pathway. Acta Physiol. 2022, 235, e13775. [Google Scholar] [CrossRef]
- Pankova, O.; Korzh, O. Plasma catestatin levels are related to metabolic parameters in patients with essential hypertension and type 2 diabetes mellitus. Heart Vessels 2024, 39, 144–159. [Google Scholar] [CrossRef]
- Simunovic, M.; Supe-Domic, D.; Karin, Z.; Degoricija, M.; Paradzik, M.; Bozic, J.; Unic, I.; Skrabic, V. Serum catestatin concentrations are decreased in obese children and adolescents. Pediatr. Diabetes 2019, 20, 549–555. [Google Scholar] [CrossRef]
- Paulus, W.J.; Tschöpe, C. A novel paradigm for heart failure with preserved ejection fraction. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef]
- Scarica, V.; Rinaldi, R.; Animati, F.M.; Manzato, M.; Montone, R.A. Coronary microvascular dysfunction: Pathophysiology, diagnosis, and therapeutic strategies across cardiovascular diseases. EXCLI J. 2025, 24, 454–478. [Google Scholar]
- Crea, F.; Montone, R.A.; Rinaldi, R. Pathophysiology of coronary microvascular dysfunction. Circ. J. 2022, 86, 1319–1328. [Google Scholar] [CrossRef] [PubMed]
- Bilak, J.M.; Alam, U.; Miller, C.A.; McCann, G.P.; Arnold, J.R.; Kanagala, P. Microvascular dysfunction in heart failure with preserved ejection fraction: Pathophysiology, assessment, prevalence and prognosis. Card. Fail. Rev. 2022, 8, e24. [Google Scholar] [CrossRef] [PubMed]
- Franssen, C.; Chen, S.; Unger, A.; Korkmaz, H.I.; De Keulenaer, G.W.; Tschöpe, C.; Leite-Moreira, A.F.; Musters, R.; Niessen, H.W.M.; Linke, W.A.; et al. Myocardial microvascular inflammatory endothelial activation in heart failure with preserved ejection fraction. JACC Heart Fail. 2016, 4, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Bond, R.M.; Ivy, K.; Crumbs, T.; Purewal, V.; Obang, S.; Sraow, D.I.S. Coronary microvascular dysfunction and its role in heart failure with preserved ejection fraction for future prevention and treatment. Am. J. Prev. Cardiol. 2025, 22, 100983. [Google Scholar] [CrossRef]
- Krüger, M.; Kötter, S.; Grützner, A.; Lang, P.; Andresen, C.; Redfield, M.M.; Butt, E.; dos Remedios, C.G.; Linke, W.A. Protein kinase G modulates human myocardial passive stiffness by phosphorylation of the titin springs. Circ. Res. 2009, 104, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Kötter, S.; Gout, L.; Von Frieling-Salewsky, M.; Müller, A.E.; Helling, S.; Marcus, K.; Dos Remedios, C.; Linke, W.A.; Krüger, M. Differential changes in titin domain phosphorylation increase myofilament stiffness in failing human hearts. Cardiovasc. Res. 2013, 99, 648–656. [Google Scholar] [CrossRef]
- Qi, Z.; Wu, D.; Yan, Z.; Wang, Q.; Li, Y.; Zhao, J.; Xu, F. Role of epicardial adipose tissue in heart failure with preserved ejection fraction: An emerging molecular mechanism and therapeutic potential. Obes. Rev. 2025, 26, e13912. [Google Scholar] [CrossRef]
- Iacobellis, G. Epicardial adipose tissue in contemporary cardiology. Nat. Rev. Cardiol. 2022, 19, 593–606. [Google Scholar] [CrossRef]
- Mazurek, T.; Zhang, L.; Zalewski, A.; Mannion, J.D.; Diehl, J.T.; Arafat, H.; Sarov-Blat, L.; O’Brien, S.; Keiper, E.A.; Johnson, A.G.; et al. Human epicardial adipose tissue is a source of inflammatory mediators. Circulation 2003, 108, 2460–2466. [Google Scholar] [CrossRef]
- Patel, V.B.; Mori, J.; McLean, B.A.; Basu, R.; Das, S.K.; Ramprasath, T.; Parajuli, N.; Penninger, J.M.; Grant, M.B.; Lopaschuk, G.D.; et al. ACE2 deficiency worsens epicardial adipose tissue inflammation and cardiac dysfunction in response to diet-induced obesity. Diabetes 2016, 65, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Couselo-Seijas, M.; Rodríguez-Mañero, M.; González-Juanatey, J.R.; Eiras, S. Updates on epicardial adipose tissue mechanisms on atrial fibrillation. Obes. Rev. 2021, 22, e13277. [Google Scholar] [CrossRef]
- Iacobellis, G.; Bianco, A.C. Epicardial adipose tissue: Emerging physiological, pathophysiological and clinical features. Trends Endocrinol. Metab. 2011, 22, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.G.; Renu, K.; Gopalakrishnan, A.V.; Jayaraj, R.; Dey, A.; Vellingiri, B.; Ganesan, R. Epicardial adipose tissue and cardiac lipotoxicity: A review. Life Sci. 2023, 328, 121913. [Google Scholar] [CrossRef]
- Badagliacca, R.; Vizza, C.D.; Lang, I.; Sadushi-Kolici, R.; Papa, S.; Manzi, G.; Filomena, D.; Ogawa, A.; Shimokawahara, H.; Matsubara, H. Pulmonary pressure recovery in idiopathic, hereditary and drug and toxin-induced pulmonary arterial hypertension: Determinants and clinical impact. Vasc. Pharmacol. 2022, 146, 107099. [Google Scholar] [CrossRef] [PubMed]
- Ltaief, Z.; Yerly, P.; Liaudet, L. Pulmonary hypertension in left heart diseases: Pathophysiology, hemodynamic assessment and therapeutic management. Int. J. Mol. Sci. 2023, 24, 9971. [Google Scholar] [CrossRef]
- Omote, K.; Borlaug, B.A. Left atrial myopathy in heart failure with preserved ejection fraction. Circ. J. 2023, 87, 1039–1046. [Google Scholar] [CrossRef]
- Schiattarella, G.G.; Altamirano, F.; Tong, D.; French, K.M.; Villalobos, E.; Kim, S.Y.; Luo, X.; Jiang, N.; May, H.I.; Wang, Z.V.; et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature 2019, 568, 351–356. [Google Scholar] [CrossRef]
- Yang, L.; Calay, E.S.; Fan, J.; Arduini, A.; Kunz, R.C.; Gygi, S.P.; Yalcin, A.; Fu, S.; Hotamisligil, G.S. METABOLISM. S-Nitrosylation links obesity-associated inflammation to endoplasmic reticulum dysfunction. Science 2015, 349, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Schiattarella, G.G.; Altamirano, F.; Tong, D.; Ferdous, A.; Piristine, H.; Dasgupta, S.; Wang, X.; French, K.M.; Villalobos, E.; Spurgin, S.B. Xbp1s–FoxO1 axis governs lipid accumulation and contractile performance in heart failure with preserved ejection fraction. Nat. Commun. 2021, 12, 1684. [Google Scholar] [CrossRef]
- Wang, Z.V.; Hill, J.A. Protein quality control and metabolism: Bidirectional control in the heart. Cell Metab. 2015, 21, 215–226. [Google Scholar] [CrossRef]
- Kitakata, H.; Endo, J.; Hashimoto, S.; Mizuno, E.; Moriyama, H.; Shirakawa, K.; Goto, S.; Katsumata, Y.; Fukuda, K.; Sano, M. Imeglimin prevents heart failure with preserved ejection fraction by recovering the impaired unfolded protein response in mice subjected to cardiometabolic stress. Biochem. Biophys. Res. Commun. 2021, 572, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Schiattarella, G.G.; Altamirano, F.; Kim, S.Y.; Smith, K.; Schisler, J.C. Mechanistic detail demonstrating that Xbp1s overexpression activates STUB1 to ubiquitinate and degrade FoxO1. Nat. Commun. 2021, 12, 1684. [Google Scholar] [CrossRef] [PubMed]
- Borlaug, B.A.; Jensen, M.D.; Kitzman, D.W.; Lam, C.S.P.; Obokata, M.; Rider, O.J. Obesity and heart failure with preserved ejection fraction: New insights and pathophysiological targets. Cardiovasc. Res. 2023, 118, 3434–3450. [Google Scholar] [CrossRef]
- Vanderpool, R.R.; Saul, M.; Nouraie, M.; Gladwin, M.T.; Simon, M.A. Association between hemodynamic markers of pulmonary hypertension and outcomes in heart failure with preserved ejection fraction. JAMA Cardiol. 2018, 3, 298. [Google Scholar] [CrossRef] [PubMed]
- Melenovsky, V.; Hwang, S.-J.; Lin, G.; Redfield, M.M.; Borlaug, B.A. Right heart dysfunction in heart failure with preserved ejection fraction. Eur. Heart J. 2014, 35, 3452–3462. [Google Scholar] [CrossRef]
- Obokata, M.; Reddy, Y.N.V.; Melenovsky, V.; Pislaru, S.; Borlaug, B.A. Deterioration in right ventricular structure and function over time in patients with heart failure and preserved ejection fraction. Eur. Heart J. 2019, 40, 689–697. [Google Scholar] [CrossRef]
- Reddy, Y.N.V.; Obokata, M.; Verbrugge, F.H.; Lin, G.; Borlaug, B.A. Atrial dysfunction in patients with heart failure with preserved ejection fraction and atrial fibrillation. J. Am. Coll. Cardiol. 2020, 76, 1051–1064. [Google Scholar] [CrossRef]
- Obokata, M.; Reddy, Y.N.V.; Pislaru, S.V.; Melenovsky, V.; Borlaug, B.A. Evidence supporting the existence of a distinct obese phenotype of heart failure with preserved ejection fraction. Circulation 2017, 136, 6–19. [Google Scholar] [CrossRef]
- Koepp, K.E.; Obokata, M.; Reddy, Y.N.V.; Olson, T.P.; Borlaug, B.A. Hemodynamic and functional impact of epicardial adipose tissue in heart failure with preserved ejection fraction. JACC Heart Fail. 2020, 8, 657–666. [Google Scholar] [CrossRef]
- Kumric, M.; Ticinovic Kurir, T.; Borovac, J.A.; Bozic, J. Role of novel biomarkers in diabetic cardiomyopathy. World J. Diabetes 2021, 12, 685–705. [Google Scholar] [CrossRef] [PubMed]
- Urlic, H.; Kumric, M.; Vrdoljak, J.; Martinovic, D.; Dujic, G.; Vilovic, M.; Ticinovic Kurir, T.; Bozic, J. Role of echocardiography in diabetic cardiomyopathy: From mechanisms to clinical practice. J. Cardiovasc. Dev. Dis. 2023, 10, 46. [Google Scholar] [CrossRef]
- Rubler, S.; Dlugash, J.; Yuceoglu, Y.Z.; Kumral, T.; Branwood, A.W.; Grishman, A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am. J. Cardiol. 1972, 30, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Seferović, P.M.; Paulus, W.J. Clinical diabetic cardiomyopathy: A two-faced disease with restrictive and dilated phenotypes. Eur. Heart J. 2015, 36, 1718–1727. [Google Scholar] [CrossRef]
- Packer, M. Differential pathophysiological mechanisms in heart failure with a reduced or preserved ejection fraction in diabetes. JACC Heart Fail. 2021, 9, 535–549. [Google Scholar] [CrossRef]
- American College of Cardiology. Focus on Heart Failure. Available online: https://www.acc.org/Latest-in-Cardiology/Articles/2025/06/01/01/Focus-on-Heart-Failure-HFpEF (accessed on 14 June 2025).
- Solomon, S.D.; McMurray, J.J.V.; Claggett, B.; de Boer, R.A.; DeMets, D.; Hernandez, A.F.; Inzucchi, S.E.; Kosiborod, M.N.; Lam, C.S.P.; Martinez, F.; et al. Dapagliflozin in heart failure with mildly reduced or preserved ejection fraction. N. Engl. J. Med. 2022, 387, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Böhm, M.; Brunner-La Rocca, H.-P.; Choi, D.-J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in heart failure with a preserved ejection fraction. N. Engl. J. Med. 2021, 385, 1451–1461. [Google Scholar] [CrossRef]
- Nassif, M.E.; Windsor, S.L.; Borlaug, B.A.; Kitzman, D.W.; Shah, S.J.; Tang, F.; Khariton, Y.; Malik, A.O.; Khumri, T.; Umpierrez, G.; et al. The SGLT2 inhibitor dapagliflozin in heart failure with preserved ejection fraction: A multicenter randomized trial. Nat. Med. 2021, 27, 1954–1960. [Google Scholar] [CrossRef]
- Voors, A.A.; Angermann, C.E.; Teerlink, J.R.; Collins, S.P.; Kosiborod, M.; Biegus, J.; Ferreira, J.P.; Nassif, M.E.; Psotka, M.A.; Tromp, J.; et al. The SGLT2 inhibitor empagliflozin in patients hospitalized for acute heart failure: A multinational randomized trial. Nat. Med. 2022, 28, 568–574. [Google Scholar] [CrossRef]
- Bhatt, D.L.; Szarek, M.; Steg, P.G.; Cannon, C.P.; Leiter, L.A.; McGuire, D.K.; Lewis, J.B.; Riddle, M.C.; Voors, A.A.; Metra, M.; et al. Sotagliflozin in patients with diabetes and recent worsening heart failure. N. Engl. J. Med. 2021, 384, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Kosiborod, M.N.; Abildstrøm, S.Z.; Borlaug, B.A.; Butler, J.; Rasmussen, S.; Davies, M.; Hovingh, G.K.; Kitzman, D.W.; Lindegaard, M.L.; Møller, D.V.; et al. Semaglutide in patients with heart failure with preserved ejection fraction and obesity. N. Engl. J. Med. 2023, 389, 1069–1084. [Google Scholar] [CrossRef] [PubMed]
- Deanfield, J.; Verma, S.; Scirica, B.M.; Kahn, S.E.; Emerson, S.S.; Ryan, D.; Lingvay, I.; Colhoun, H.M.; Plutzky, J.; Kosiborod, M.N.; et al. Semaglutide and cardiovascular outcomes in patients with obesity and prevalent heart failure: A prespecified analysis of the SELECT trial. Lancet 2024, 404, 773–786. [Google Scholar] [CrossRef]
- Lambers Heerspink, H.J.; de Zeeuw, D.; Wie, L.; Leslie, B.; List, J. Dapagliflozin a glucose-regulating drug with diuretic properties in subjects with type 2 diabetes. Diabetes Obes. Metab. 2013, 15, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Pabel, S.; Hamdani, N.; Luedde, M.; Sossalla, S. SGLT2 inhibitors and their mode of action in heart failure-has the mystery been unravelled? Curr. Heart Fail. Rep. 2021, 18, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.J.M.; Gandy, S.; McCrimmon, R.; Houston, J.G.; Struthers, A.D.; Lang, C.C. A randomized controlled trial of dapagliflozin on left ventricular hypertrophy in people with type two diabetes: The DAPA-LVH trial. Eur. Heart J. 2020, 41, 3421–3432. [Google Scholar] [CrossRef]
- Kosiborod, M.N.; Petrie, M.C.; Borlaug, B.A.; Butler, J.; Davies, M.J.; Hovingh, G.K.; Kitzman, D.W.; Møller, D.V.; Treppendahl, M.B.; Verma, S.; et al. Semaglutide in patients with obesity-related heart failure and type 2 diabetes. N. Engl. J. Med. 2024, 390, 1394–1407. [Google Scholar] [CrossRef]
- Alsereidi, F.R.; Khashim, Z.; Marzook, H.; Gupta, A.; Al-Rawi, A.M.; Ramadan, M.M.; Saleh, M.A. Targeting inflammatory signaling pathways with SGLT2 inhibitors: Insights into cardiovascular health and cardiac cell improvement. Curr. Probl. Cardiol. 2024, 49, 102524. [Google Scholar] [CrossRef]
- Bendotti, G.; Montefusco, L.; Lunati, M.E.; Usuelli, V.; Pastore, I.; Lazzaroni, E.; Assi, E.; Seelam, A.J.; El Essawy, B.; Jang, J.; et al. The anti-inflammatory and immunological properties of GLP-1 receptor agonists. Pharmacol. Res. 2022, 182, 106320. [Google Scholar] [CrossRef]
- Wong, C.K.; McLean, B.A.; Baggio, L.L.; Koehler, J.A.; Hammoud, R.; Rittig, N.; Yabut, J.M.; Seeley, R.J.; Brown, T.J.; Drucker, D.J. Central glucagon-like peptide 1 receptor activation inhibits Toll-like receptor agonist-induced inflammation. Cell Metab. 2024, 36, 130–143.e5. [Google Scholar] [CrossRef]
- Yu, J.; Zhou, G.; Guo, Y.; Long, Y. Inflammation in heart failure: Mechanisms and therapeutic strategies. Cardiovasc. Innov. Appl. 2025, 10. [Google Scholar] [CrossRef]
- Lam, C.S.P.; Lund, L.H.; Shah, S.J.; Voors, A.A.; Erlinge, D.; Saraste, A.; Pirazzi, C.; Grove, E.L.; Barasa, A.; Schou, M.; et al. Myeloperoxidase inhibition in heart failure with preserved or mildly reduced ejection fraction: Satellite trial results. J. Card. Fail. 2024, 30, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Lund, L.H.; Lam, C.S.P.; Pizzato, P.E.; Gabrielsen, A.; Michaëlsson, E.; Nelander, K.; Ericsson, H.; Holden, J.; Folkvaljon, F.; Mattsson, A.; et al. Rationale and design of ENDEAVOR: A sequential phase 2b-3 randomized clinical trial to evaluate the effect of myeloperoxidase inhibition on symptoms and exercise capacity in heart failure with preserved or mildly reduced ejection fraction. Eur. J. Heart Fail. 2023, 25, 1696–1707. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.; Jha, R.K.; Keerti, A. Chronic kidney disease: Its relationship with obesity. Cureus 2022, 14, e30535. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Wang, Y.; Zhao, X.; Cui, H.; Han, M.; Ren, X.; Gang, X.; Wang, G. Obesity and chronic kidney disease. Am. J. Physiol. Endocrinol. Metab. 2023, 324, E24–E41. [Google Scholar] [CrossRef]
- Abasheva, D.; Ortiz, A.; Fernandez-Fernandez, B. GLP-1 receptor agonists in patients with chronic kidney disease and either overweight or obesity. Clin. Kidney J. 2024, 17 (Suppl. S2), 19–35. [Google Scholar] [CrossRef]
- Wheeler, D.C.; Stefansson, B.V.; Batiushin, M.; Bilchenko, O.; Cherney, D.Z.I.; Chertow, G.M.; Douthat, W.; Dwyer, J.P.; Escudero, E.; Pecoits-Filho, R.; et al. The dapagliflozin and prevention of adverse outcomes in chronic kidney disease (DAPA-CKD) trial: Baseline characteristics. Nephrol. Dial. Transplant. 2020, 35, 1700–1711. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Komic, L.; Kumric, M.; Komic, J.; Tomicic, M.; Kurir, T.T.; Grahovac, M.; Mornar, M.; Rusic, D.; Bukic, J.; Bozic, J. The Hidden Price of Plenty: Oxidative Stress and Calorie-Induced Cardiometabolic Dysfunction. Life 2025, 15, 1022. https://doi.org/10.3390/life15071022
Komic L, Kumric M, Komic J, Tomicic M, Kurir TT, Grahovac M, Mornar M, Rusic D, Bukic J, Bozic J. The Hidden Price of Plenty: Oxidative Stress and Calorie-Induced Cardiometabolic Dysfunction. Life. 2025; 15(7):1022. https://doi.org/10.3390/life15071022
Chicago/Turabian StyleKomic, Luka, Marko Kumric, Jelena Komic, Marion Tomicic, Tina Ticinovic Kurir, Marko Grahovac, Marin Mornar, Doris Rusic, Josipa Bukic, and Josko Bozic. 2025. "The Hidden Price of Plenty: Oxidative Stress and Calorie-Induced Cardiometabolic Dysfunction" Life 15, no. 7: 1022. https://doi.org/10.3390/life15071022
APA StyleKomic, L., Kumric, M., Komic, J., Tomicic, M., Kurir, T. T., Grahovac, M., Mornar, M., Rusic, D., Bukic, J., & Bozic, J. (2025). The Hidden Price of Plenty: Oxidative Stress and Calorie-Induced Cardiometabolic Dysfunction. Life, 15(7), 1022. https://doi.org/10.3390/life15071022