
Transcriptomic and miRNA Signatures of ChAdOx1 nCoV-19 Vaccine Response Using Machine Learning

 ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Data
2.2. Feature Ranking Algorithms
2.2.1. Least Absolute Shrinkage and Selection Operator
2.2.2. Monte Carlo Feature Selection
2.2.3. Minimum Redundancy Maximum Relevance
2.2.4. CatBoost
2.2.5. XGBoost
2.2.6. AdaBoost
2.2.7. Random Forest
2.2.8. ExtraTrees
2.2.9. LightGBM
2.2.10. Ridge Regression
2.3. Incremental Feature Selection
2.4. Synthetic Minority Over-Sampling Technique
2.5. Classification Algorithms
2.5.1. Decision Tree
2.5.2. K-Nearest Neighbors
2.5.3. Support Vector Machine
2.5.4. Nearest Centroid Classifier
2.5.5. Stochastic Gradient Descent Classifier
2.5.6. Naïve Bayes Classifier
2.5.7. Quadratic Discriminant Analysis Classifier
2.6. Performance Evaluation
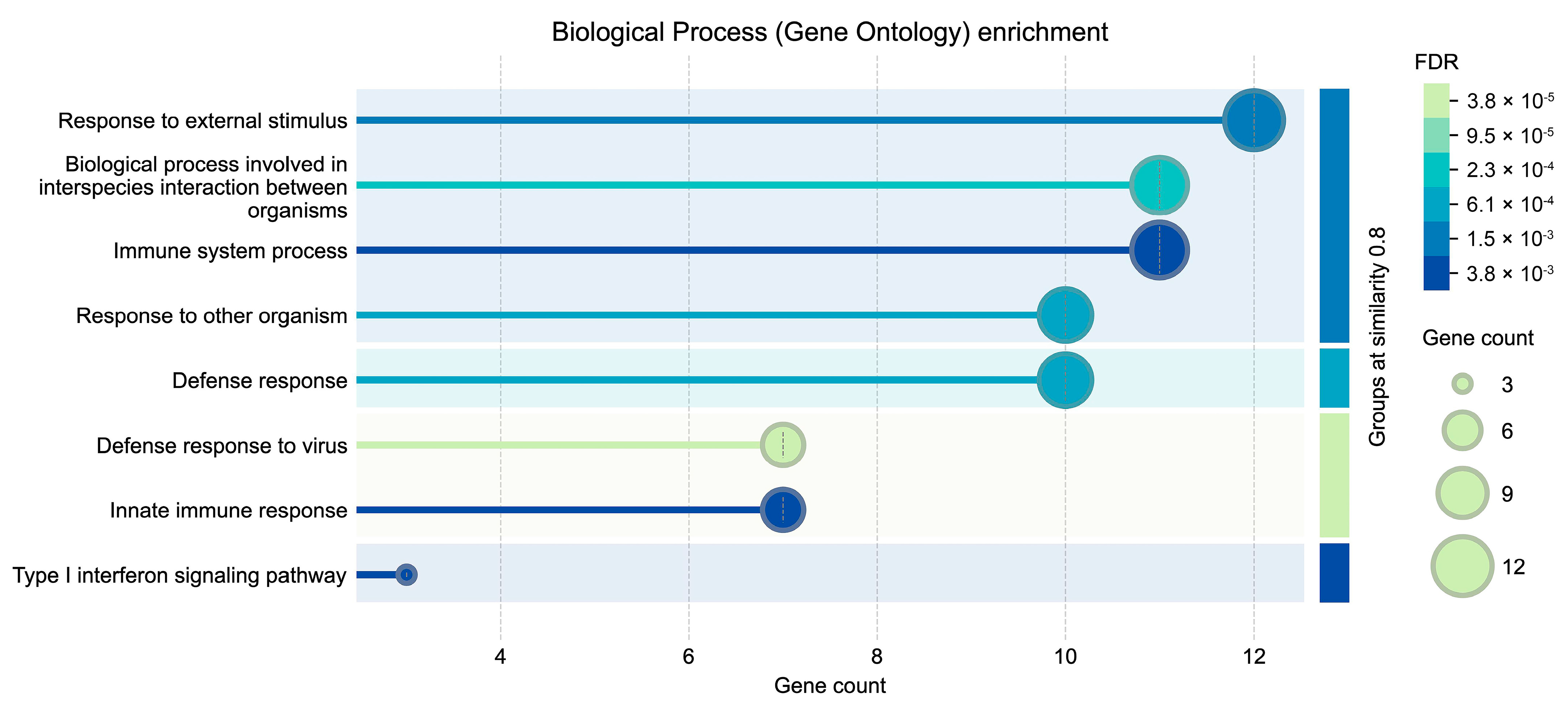
2.7. Protein–Protein Interaction Network Prediction and GO Enrichment Analysis (Biological Process)
2.8. Outline of the Analysis Procedure
3. Results
3.1. Feature Ranking Results
3.2. Results of IFS with Different Classification Algorithms
3.3. Intersection of Essential Features Identified by Different Feature Ranking Algorithms
3.4. Classification Rules Created by Decision Tree
4. Discussion
4.1. Essential Genes Associated with ChAdOx1 nCoV-19 Vaccine Effect Identified by Multiple Feature Ranking Algorithms
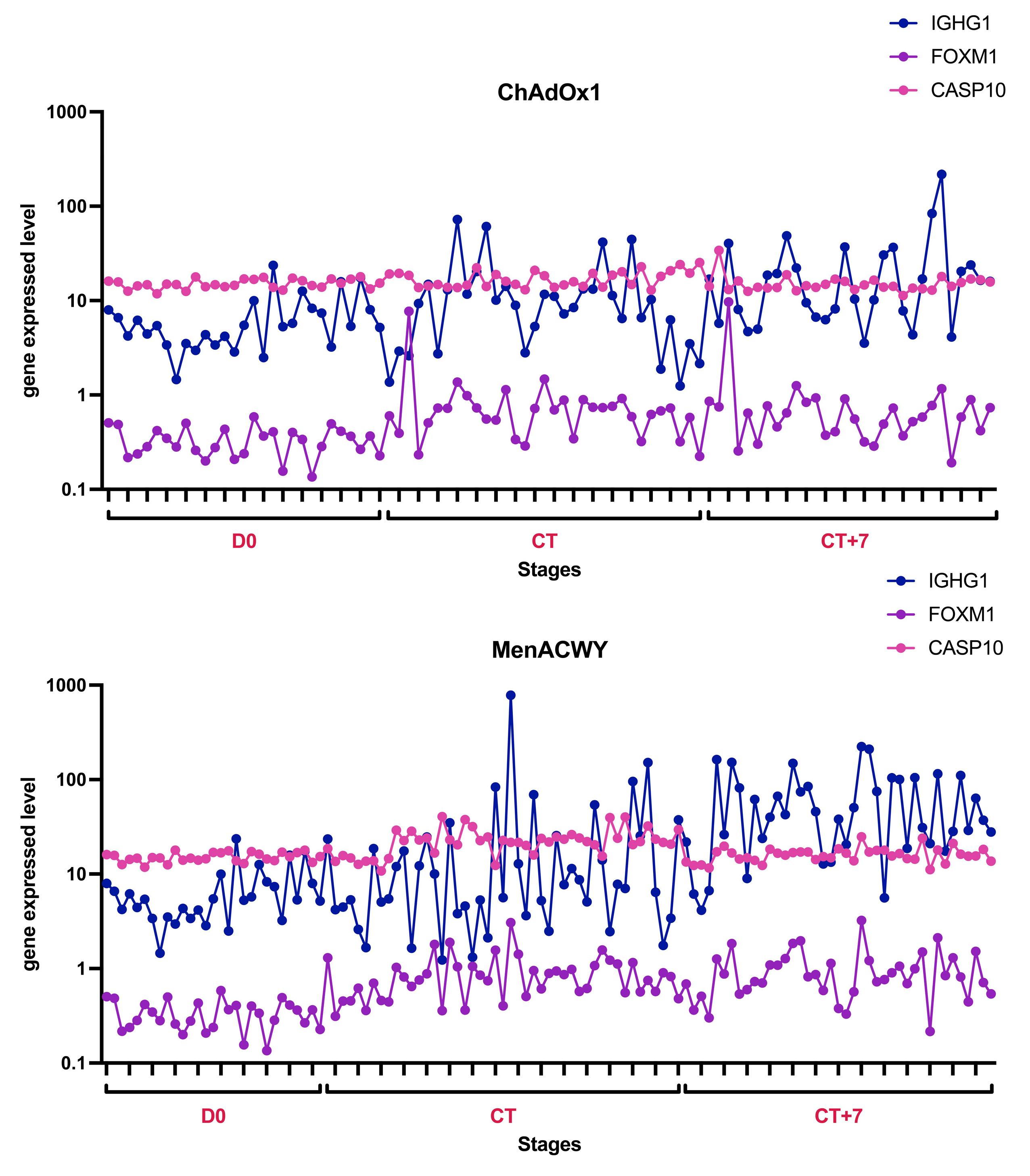
4.1.1. Role of IGHG1
4.1.2. Role of FOXM1
4.1.3. Role of CASP10
4.2. Analysis of Decision Rules to Identify Changes in Gene Expression Resulting from COVID-19 Vaccination
4.2.1. Rule 0: Distinguishing Between the Pre-Vaccination Group and the ChAdOx1-Onset Group
4.2.2. Rule 1: Identifying the MenACWY-Onset Group
4.2.3. Rule 2: Identifying the ChAdOx1-7D Group
4.2.4. Rule 3: Identifying the MenACWY-7D Group
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| COVID-19 | Coronavirus Disease 2019 |
| SARS-CoV-2 | Severe Acute Respiratory Syndrome Coronavirus 2 |
| IFS | Incremental Feature Selection |
| DT | Decision Tree |
| LASSO | Least Absolute Shrinkage and Selection Operator |
| MCFS | Monte Carlo Feature Selection |
| mRMR | Minimum Redundancy Maximum Relevance |
| CatBoost | Categorical Boosting |
| XGBoost | Extreme Gradient Boosting |
| AdaBoost | Adaptive Boosting |
| RF | Random Forest |
| ExtraTrees | Extreme Randomized Tree |
| LightGBM | Light Gradient Boosting Machine |
| SMOTE | Synthetic Minority Over-sampling Technique |
| KNN | K-Nearest Neighbors |
| SVM | Support Vector Machine |
| SGD | Stochastic Gradient Descent |
| QDA | Quadratic Discriminant Analysis |
| LDA | Linear Discriminant Analysis |
| ACC | Accuracy |
| MCC | Matthews Correlation Coefficient |
| PPI | Protein–Protein Interaction |
| SD | Standard Deviation |
References
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Zhou, X.; Huang, K.; Chen, L.; Guo, W.; Feng, K.; Huang, T.; Cai, Y.-D. Identification of key genes associated with persistent immune changes and secondary immune activation responses induced by influenza vaccination after COVID-19 recovery by machine learning methods. Comput. Biol. Med. 2024, 169, 107883. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Gao, Q.; Zhou, X.; Chen, L.; Guo, W.; Feng, K.; Hu, J.; Huang, T.; Cai, Y.-D. Identification of gene and protein signatures associated with long-term effects of COVID-19 on the immune system after patient recovery by analyzing single-cell multi-omics data using a machine learning approach. Vaccine 2024, 42, 126253. [Google Scholar] [CrossRef] [PubMed]
- Folegatti, P.M.; Ewer, K.J.; Aley, P.K.; Angus, B.; Becker, S.; Belij-Rammerstorfer, S.; Bellamy, D.; Bibi, S.; Bittaye, M.; Clutterbuck, E.A.; et al. Safety and immunogenicity of the chadox1 nCoV-19 vaccine against SARS-CoV-2: A preliminary report of a phase 1/2, single-blind, randomised controlled trial. Lancet 2020, 396, 467–478. [Google Scholar] [CrossRef]
- Ewer, K.J.; Barrett, J.R.; Belij-Rammerstorfer, S.; Sharpe, H.; Makinson, R.; Morter, R.; Flaxman, A.; Wright, D.; Bellamy, D.; Bittaye, M.; et al. T cell and antibody responses induced by a single dose of chadox1 nCoV-19 (azd1222) vaccine in a phase 1/2 clinical trial. Nat. Med. 2021, 27, 270–278. [Google Scholar] [CrossRef]
- Ramasamy, M.N.; Minassian, A.M.; Ewer, K.J.; Flaxman, A.L.; Folegatti, P.M.; Owens, D.R.; Voysey, M.; Aley, P.K.; Angus, B.; Babbage, G.; et al. Safety and immunogenicity of chadox1 nCoV-19 vaccine administered in a prime-boost regimen in young and old adults (cov002): A single-blind, randomised, controlled, phase 2/3 trial. Lancet 2021, 396, 1979–1993. [Google Scholar] [CrossRef]
- van Doremalen, N.; Lambe, T.; Spencer, A.; Belij-Rammerstorfer, S.; Purushotham, J.N.; Port, J.R.; Avanzato, V.A.; Bushmaker, T.; Flaxman, A.; Ulaszewska, M.; et al. Chadox1 nCoV-19 vaccine prevents SARS-CoV-2 pneumonia in rhesus macaques. Nature 2020, 586, 578–582. [Google Scholar] [CrossRef]
- Li, J.; Ren, J.; Liao, H.; Guo, W.; Feng, K.; Huang, T.; Cai, Y.D. Identification of dynamic gene expression profiles during sequential vaccination with chadox1/bnt162b2 using machine learning methods. Front. Microbiol. 2023, 14, 1138674. [Google Scholar] [CrossRef]
- Barros-Martins, J.; Hammerschmidt, S.I.; Cossmann, A.; Odak, I.; Stankov, M.V.; Morillas Ramos, G.; Dopfer-Jablonka, A.; Heidemann, A.; Ritter, C.; Friedrichsen, M. Immune responses against SARS-CoV-2 variants after heterologous and homologous chadox1 nCoV-19/bnt162b2 vaccination. Nat. Med. 2021, 27, 1525–1529. [Google Scholar] [CrossRef]
- Li, J.; Huang, F.; Ma, Q.; Guo, W.; Feng, K.; Huang, T.; Cai, Y.D. Identification of genes related to immune enhancement caused by heterologous chadox1-bnt162b2 vaccines in lymphocytes at single-cell resolution with machine learning methods. Front. Immunol. 2023, 14, 1131051. [Google Scholar] [CrossRef]
- Drury, R.E.; Camara, S.; Chelysheva, I.; Bibi, S.; Sanders, K.; Felle, S.; Emary, K.; Phillips, D.; Voysey, M.; Ferreira, D.M.; et al. Multi-omics analysis reveals COVID-19 vaccine induced attenuation of inflammatory responses during breakthrough disease. Nat. Commun. 2024, 15, 3402. [Google Scholar] [CrossRef] [PubMed]
- Santorsola, M.; Lescai, F. The promise of explainable deep learning for omics data analysis: Adding new discovery tools to AI. New Biotechnol. 2023, 77, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Maniaci, A.; Fakhry, N.; Chiesa-Estomba, C.; Lechien, J.R.; Lavalle, S. Synergizing chatgpt and general ai for enhanced medical diagnostic processes in head and neck imaging. Eur. Arch. Oto-Rhino-Laryngol. 2024, 281, 3297–3298. [Google Scholar] [CrossRef]
- Varadi, M.; Bordin, N.; Orengo, C.; Velankar, S. The opportunities and challenges posed by the new generation of deep learning-based protein structure predictors. Curr. Opin. Struct. Biol. 2023, 79, 102543. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.A.; Setiono, R. Incremental feature selection. Appl. Intell. 1998, 9, 217–230. [Google Scholar] [CrossRef]
- Safavian, S.R.; Landgrebe, D. A survey of decision tree classifier methodology. IEEE Trans. Syst. Man Cybern. 1991, 21, 660–674. [Google Scholar] [CrossRef]
- Tibshirani, R. Regression shrinkage and selection via the lasso. J. R. Stat. Soc. Ser. B (Methodol.) 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Draminski, M.; Rada-Iglesias, A.; Enroth, S.; Wadelius, C.; Koronacki, J.; Komorowski, J. Monte carlo feature selection for supervised classification. Bioinformatics 2008, 24, 110–117. [Google Scholar] [CrossRef]
- Peng, H.; Long, F.; Ding, C. Feature selection based on mutual information: Criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans. Pattern Anal. Mach. Intell. 2005, 27, 1226–1238. [Google Scholar] [CrossRef]
- Dorogush, A.V.; Ershov, V.; Gulin, A. Catboost: Gradient boosting with categorical features support. arXiv 2018, arXiv:1810.11363. [Google Scholar]
- Chen, T.; Guestrin, C. Xgboost: A scalable tree boosting system. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; Association for Computing Machinery: New York, NY, USA, 2016; pp. 785–794. [Google Scholar]
- Freund, Y.; Schapire, R.E. A decision-theoretic generalization of on-line learning and an application to boosting. J. Comput. Syst. Sci. 1997, 55, 119–139. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Geurts, P.; Ernst, D.; Wehenkel, L. Extremely randomized trees. Mach. Learn. 2006, 63, 3–42. [Google Scholar] [CrossRef]
- Ke, G.; Meng, Q.; Finley, T.; Wang, T.; Chen, W.; Ma, W.; Ye, Q.; Liu, T.-Y. Lightgbm: A highly efficient gradient boosting decision tree. Adv. Neural Inf. Process. Syst. 2017, 30, 3146–3154. [Google Scholar]
- Hoerl, A.E.; Kennard, R.W. Ridge regression: Biased estimation for nonorthogonal problems. Technometrics 1970, 12, 55–67. [Google Scholar] [CrossRef]
- Kohavi, R. A study of cross-validation and bootstrap for accuracy estimation and model selection. In Proceedings of the International Joint Conference on Artificial Intelligence, Montreal, QC, Canada, 20–25 August 1995; Morgan Kaufmann Publishers Inc.: San Francisco, CA, USA, 1995; pp. 1137–1145. [Google Scholar]
- Chen, L.; Lu, Y.; Xu, J.; Zhou, B. Prediction of drug’s anatomical therapeutic chemical (atc) code by constructing biological profiles of atc codes. BMC Bioinform. 2025, 26, 86. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, S.; Zhou, B. Herb-disease association prediction model based on network consistency projection. Sci. Rep. 2025, 15, 3328. [Google Scholar] [CrossRef]
- Chen, L.; Zhu, W.; Chen, D. An end-to-end 3d graph neural network for predicting drug-target-disease associations. Curr. Bioinform. 2025. [Google Scholar] [CrossRef]
- Chawla, N.V.; Bowyer, K.W.; Hall, L.O.; Kegelmeyer, W.P. Smote: Synthetic minority over-sampling technique. J. Artif. Intell. Res. 2002, 16, 321–357. [Google Scholar] [CrossRef]
- Cover, T.; Hart, P. Nearest neighbor pattern classification. IEEE Trans. Inf. Theory 1967, 13, 21–27. [Google Scholar] [CrossRef]
- Cortes, C. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Levner, I. Feature selection and nearest centroid classification for protein mass spectrometry. BMC Bioinform. 2005, 6, 68. [Google Scholar] [CrossRef] [PubMed]
- Osho, O.; Hong, S. An overview: Stochastic gradient descent classifier, linear discriminant analysis, deep learning and naive bayes classifier approaches to network intrusion detection. Int. J. Eng. Tech. Res. 2021, 10, 294–308. [Google Scholar]
- Rish, I. An empirical study of the naive bayes classifier. In Proceedings of the IJCAI 2001 Workshop on Empirical Methods in Artificial Intelligence, Seattle, WA, USA, 4–6 August 2001; pp. 41–46. [Google Scholar]
- Bose, S.; Pal, A.; SahaRay, R.; Nayak, J. Generalized quadratic discriminant analysis. Pattern Recogn. 2015, 48, 2676–2684. [Google Scholar] [CrossRef]
- Powers, D.M. Evaluation: From precision, recall and f-measure to roc, informedness, markedness and correlation. arXiv 2020, arXiv:2010.16061. [Google Scholar]
- Liao, H.; Ma, Q.; Chen, L.; Guo, W.; Feng, K.; Bao, Y.; Zhang, Y.; Shen, W.; Huang, T.; Cai, Y.-D. Machine learning analysis of cd4+ t cell gene expression in diverse diseases: Insights from cancer, metabolic, respiratory, and digestive disorders. Cancer Genet. 2025, 290–291, 56–60. [Google Scholar] [CrossRef]
- Chen, L.; Li, J. PDTDAHN: Predicting drug-target-disease associations using a heterogeneous network. Curr. Bioinform. 2025, in press. [Google Scholar] [CrossRef]
- Chen, L.; Gu, J.; Zhou, B. PMiSLocMF: Predicting mirna subcellular localizations by incorporating multi-source features of mirnas. Brief. Bioinform. 2024, 25, bbae386. [Google Scholar] [CrossRef]
- Matthews, B. Comparison of the predicted and observed secondary structure of t4 phage lysozyme. Biochim. Biophys. Acta (BBA) Protein Struct. 1975, 405, 442–451. [Google Scholar] [CrossRef]
- Gorodkin, J. Comparing two k-category assignments by a k-category correlation coefficient. Comput. Biol. Chem. 2004, 28, 367–374. [Google Scholar] [CrossRef]
- Ren, J.; Chen, L.; Guo, W.; Feng, K.; Huang, T.; Cai, Y.-D. Patterns of gene expression profiles associated with colorectal cancer in colorectal mucosa by using machine learning methods. Comb. Chem. High Throughput Screen. 2024, 27, 2921–2934. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Gao, Q.; Zhou, X.; Chen, L.; Guo, W.; Feng, K.; Huang, T.; Cai, Y.-D. Identification of key gene expression associated with quality of life after recovery from COVID-19. Med. Biol. Eng. Comput. 2024, 62, 1031–1048. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Ma, Q.; Chen, L.; Feng, K.; Guo, W.; Huang, T.; Cai, Y.-D. Recognizing SARS-CoV-2 infection of nasopharyngeal tissue at the single-cell level by machine learning method. Mol. Immunol. 2025, 177, 44–61. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Bao, Y.; Wang, S.; Yu, H.; Guo, W.; Feng, K.; Huang, T.; Cai, Y.-D. Investigating the unique transcriptional mirna-mrna regulatory network of alk-positive lung adenocarcinoma using machine learning methods. Curr. Bioinform. 2025. [Google Scholar] [CrossRef]
- Li, X.; Ni, R.; Chen, J.; Liu, Z.; Xiao, M.; Jiang, F.; Lu, C. The presence of ighg1 in human pancreatic carcinomas is associated with immune evasion mechanisms. Pancreas 2011, 40, 753–761. [Google Scholar] [CrossRef]
- Zhang, M.; Li, Z.; Li, J.; Huang, T.; Peng, G.; Tang, W.; Yi, G.; Zhang, L.; Song, Y.; Liu, T.; et al. Revisiting the pig ighc gene locus in different breeds uncovers nine distinct ighg genes. J. Immunol. 2020, 205, 2137–2145. [Google Scholar] [CrossRef]
- Bashirova, A.A.; Zheng, W.; Akdag, M.; Augusto, D.G.; Vince, N.; Dong, K.L.; O’HUigin, C.; Carrington, M. Population-specific diversity of the immunoglobulin constant heavy g chain (ighg) genes. Genes Immun. 2021, 22, 327–334. [Google Scholar] [CrossRef]
- Wang, G.; Li, H.; Pan, J.; Yan, T.; Zhou, H.; Han, X.; Su, L.; Hou, L.; Xue, X. Upregulated expression of cancer-derived immunoglobulin g is associated with progression in glioma. Front. Oncol. 2021, 11, 758856. [Google Scholar] [CrossRef]
- Chu, J.; Li, Y.; Deng, Z.; Zhang, Z.; Xie, Q.; Zhang, H.; Zhong, W.; Pan, B. Ighg1 regulates prostate cancer growth via the mek/erk/c-myc pathway. BioMed Res. Int. 2019, 2019, 7201562. [Google Scholar] [CrossRef]
- Reeves, E.; James, E. Antigen processing and immune regulation in the response to tumours. Immunology 2017, 150, 16–24. [Google Scholar] [CrossRef]
- Trailin, A.; Mrazova, P.; Hruba, P.; Voska, L.; Sticova, E.; Slavcev, A.; Novotny, M.; Kocik, M.; Viklicky, O. Chronic active antibody-mediated rejection is associated with the upregulation of interstitial but not glomerular transcripts. Front. Immunol. 2021, 12, 729558. [Google Scholar] [CrossRef] [PubMed]
- Blanco, I.K.; Heuer, S.; Catalina, M.; Robl, R.; Madamanchi, S.; Lipsky, P.; Grammer, A. Tissue infiltration of plasma cells/plasmablasts in patients with active systemic lupus erthematosus (sle). J. Immunol. 2017, 198, 55.8. [Google Scholar] [CrossRef]
- Carlsson, P.; Mahlapuu, M. Forkhead transcription factors: Key players in development and metabolism. Dev. Biol. 2002, 250, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Wierstra, I.; Alves, J. Foxm1, a typical proliferation-associated transcription factor. Biol. Chem. 2007, 388, 1257–1274. [Google Scholar] [CrossRef]
- Kalin, T.V.; Ustiyan, V.; Kalinichenko, V.V. Multiple faces of foxm1 transcription factor: Lessons from transgenic mouse models. Cell Cycle 2011, 10, 396–405. [Google Scholar] [CrossRef]
- Costa, R.H.; Kalinichenko, V.V.; Major, M.L.; Raychaudhuri, P. New and unexpected: Forkhead meets arf. Curr. Opin. Genet. Dev. 2005, 15, 42–48. [Google Scholar] [CrossRef]
- Myatt, S.S.; Lam, E.W. The emerging roles of forkhead box (fox) proteins in cancer. Nat. Rev. Cancer 2007, 7, 847–859. [Google Scholar] [CrossRef]
- Song, X.; Fiati Kenston, S.S.; Zhao, J.; Yang, D.; Gu, Y. Roles of foxm1 in cell regulation and breast cancer targeting therapy. Med. Oncol. 2017, 34, 41. [Google Scholar] [CrossRef]
- Anders, L.; Ke, N.; Hydbring, P.; Choi, Y.J.; Widlund, H.R.; Chick, J.M.; Zhai, H.; Vidal, M.; Gygi, S.P.; Braun, P.; et al. A systematic screen for cdk4/6 substrates links foxm1 phosphorylation to senescence suppression in cancer cells. Cancer Cell 2011, 20, 620–634. [Google Scholar] [CrossRef]
- Balli, D.; Ren, X.; Chou, F.S.; Cross, E.; Zhang, Y.; Kalinichenko, V.V.; Kalin, T.V. Foxm1 transcription factor is required for macrophage migration during lung inflammation and tumor formation. Oncogene 2012, 31, 3875–3888. [Google Scholar] [CrossRef]
- Gage, M.C.; Bécares, N.; Louie, R.; Waddington, K.E.; Zhang, Y.; Tittanegro, T.H.; Rodríguez-Lorenzo, S.; Jathanna, A.; Pourcet, B.; Pello, O.M.; et al. Disrupting lxrα phosphorylation promotes foxm1 expression and modulates atherosclerosis by inducing macrophage proliferation. Proc. Natl. Acad. Sci. USA 2018, 115, E6556–E6565. [Google Scholar] [CrossRef] [PubMed]
- Khongkow, P.; Karunarathna, U.; Khongkow, M.; Gong, C.; Gomes, A.R.; Yagüe, E.; Monteiro, L.J.; Kongsema, M.; Zona, S.; Man, E.P.; et al. Foxm1 targets nbs1 to regulate DNA damage-induced senescence and epirubicin resistance. Oncogene 2014, 33, 4144–4155. [Google Scholar] [CrossRef] [PubMed]
- Pabla, S.; Conroy, J.M.; Nesline, M.K.; Glenn, S.T.; Papanicolau-Sengos, A.; Burgher, B.; Hagen, J.; Giamo, V.; Andreas, J.; Lenzo, F.L.; et al. Proliferative potential and resistance to immune checkpoint blockade in lung cancer patients. J. Immunother. Cancer 2019, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Koh, Y.W.; Han, J.H.; Park, S.Y.; Yoon, D.H.; Suh, C.; Huh, J. Glut1 as a prognostic factor for classical hodgkin’s lymphoma: Correlation with pd-l1 and pd-l2 expression. J. Pathol. Transl. Med. 2017, 51, 152–158. [Google Scholar] [CrossRef]
- Petri, B.J.; Klinge, C.M. M6a readers, writers, erasers, and the m6a epitranscriptome in breast cancer. J. Mol. Endocrinol. 2023, 70, e220110. [Google Scholar] [CrossRef]
- Sawaya, A.P.; Stone, R.C.; Brooks, S.R.; Pastar, I.; Jozic, I.; Hasneen, K.; O’Neill, K.; Mehdizadeh, S.; Head, C.R.; Strbo, N. Deregulated immune cell recruitment orchestrated by foxm1 impairs human diabetic wound healing. Nat. Commun. 2020, 11, 4678. [Google Scholar] [CrossRef]
- Xu, Z.; Pei, C.; Cheng, H.; Song, K.; Yang, J.; Li, Y.; He, Y.; Liang, W.; Liu, B.; Tan, W. Comprehensive analysis of foxm1 immune infiltrates, m6a, glycolysis and cerna network in human hepatocellular carcinoma. Front. Immunol. 2023, 14, 1138524. [Google Scholar] [CrossRef]
- Cho, M.; Dho, S.H.; Shin, S.; Lee, Y.; Kim, Y.; Lee, J.; Yu, S.J.; Park, S.H.; Lee, K.-A.; Kim, L.K. Caspase-10 affects the pathogenesis of primary biliary cholangitis by regulating inflammatory cell death. J. Autoimmun. 2022, 133, 102940. [Google Scholar] [CrossRef]
- Wang, J.; Zheng, L.; Lobito, A.; Chan, F.K.; Dale, J.; Sneller, M.; Yao, X.; Puck, J.M.; Straus, S.E.; Lenardo, M.J. Inherited human caspase 10 mutations underlie defective lymphocyte and dendritic cell apoptosis in autoimmune lymphoproliferative syndrome type ii. Cell 1999, 98, 47–58. [Google Scholar] [CrossRef]
- Wang, J.; Chun, H.J.; Wong, W.; Spencer, D.M.; Lenardo, M.J. Caspase-10 is an initiator caspase in death receptor signaling. Proc. Natl. Acad. Sci. USA 2001, 98, 13884–13888. [Google Scholar] [CrossRef]
- Lai, C.; Lu, S.; Yang, Y.; You, X.; Xu, F.; Deng, X.; Lan, L.; Guo, Y.; Kuang, Z.; Luo, Y.; et al. Myeloid-driven immune suppression subverts neutralizing antibodies and t cell immunity in severe COVID-19. J. Med. Virol. 2025, 97, e70335. [Google Scholar] [CrossRef] [PubMed]
- Vanli, S.; Kurtoglu, F.; Alan, B.S.; Akcakavak, G.; Ozdemir, O. Investigation of the effects of theranekron and sorafenib treatments on carcinogenesis, apoptosis and biochemical profile in hepatocellular carcinoma in rats. Toxicol. Mech. Methods 2024, 34, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Shteinfer-Kuzmine, A.; Verma, A.; Bornshten, R.; Ben Chetrit, E.; Ben-Ya’acov, A.; Pahima, H.; Rubin, E.; Mograbi, Y.; Shteyer, E.; Shoshan-Barmatz, V. Elevated serum mtdna in COVID-19 patients is linked to SARS-CoV-2 envelope protein targeting mitochondrial vdac1, inducing apoptosis and mtdna release. Apoptosis Int. J. Program. Cell Death 2024, 29, 2025–2046. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Wong, C.K.; Li, P.; Xie, Y. A SARS-CoV protein, orf-6, induces caspase-3 mediated, er stress and jnk-dependent apoptosis. Biochim. Biophys. Acta 2008, 1780, 1383–1387. [Google Scholar] [CrossRef]
- Sonehara, K.; Uwamino, Y.; Saiki, R.; Takeshita, M.; Namba, S.; Uno, S.; Nakanishi, T.; Nishimura, T.; Naito, T.; Sato, G.; et al. Germline variants and mosaic chromosomal alterations affect COVID-19 vaccine immunogenicity. Cell Genom. 2025, 5, 100783. [Google Scholar] [CrossRef]
- Agidigbi, T.S.; Kwon, H.K.; Knight, J.R.; Zhao, D.; Lee, F.Y.; Oh, I. Transcriptomic identification of genes expressed in invasive s. Aureus diabetic foot ulcer infection. Front. Cell. Infect. Microbiol. 2023, 13, 1198115. [Google Scholar] [CrossRef]
- Wang, L.; Yang, B.; Jiang, H.; Yu, G.; Feng, M.; Lu, X.; Luo, Q.; Wu, H.; Zhang, S.; Liu, H. The molecular mechanism study of insulin in promoting wound healing under high-glucose conditions. J. Cell. Biochem. 2019, 120, 16244–16253. [Google Scholar] [CrossRef]
- Urban, C.F.; Ermert, D.; Schmid, M.; Abu-Abed, U.; Goosmann, C.; Nacken, W.; Brinkmann, V.; Jungblut, P.R.; Zychlinsky, A. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against candida albicans. PLoS Pathog. 2009, 5, e1000639. [Google Scholar] [CrossRef]
- Venkatesh, S.; Workman, J.L. Histone exchange, chromatin structure and the regulation of transcription. Nat. Rev. Mol. Cell Biol. 2015, 16, 178–189. [Google Scholar] [CrossRef]
- Frydman, G.H.; Tessier, S.N.; Wong, K.H.K.; Vanderburg, C.R.; Fox, J.G.; Toner, M.; Tompkins, R.G.; Irimia, D. Megakaryocytes contain extranuclear histones and may be a source of platelet-associated histones during sepsis. Sci. Rep. 2020, 10, 4621. [Google Scholar] [CrossRef]
- Winheim, E.; Rinke, L.; Lutz, K.; Reischer, A.; Leutbecher, A.; Wolfram, L.; Rausch, L.; Kranich, J.; Wratil, P.R.; Huber, J.E. Impaired function and delayed regeneration of dendritic cells in COVID-19. PLoS Pathog. 2021, 17, e1009742. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.-Y.R.; Perlman, S. Immune dysregulation and immunopathology induced by SARS-CoV-2 and related coronaviruses—are we our own worst enemy? Nat. Rev. Immunol. 2022, 22, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Calmarza, P.; Lapresta, C.; Martínez García, M.; Ochoa, J.; Sienes Bailo, P.; Acha Pérez, J.; Beltrán Audera, J.; González-Roca, E. Musculoskeletal pain and muscular weakness as the main symptoms of adult hypophosphatasia in a spanish cohort: Clinical characterization and identification of a new alpl gene variant. J. Bone Miner. Metab. 2023, 41, 654–665. [Google Scholar] [CrossRef]
- Rader, B.A. Alkaline phosphatase, an unconventional immune protein. Front. Immunol. 2017, 8, 897. [Google Scholar] [CrossRef] [PubMed]
- Shanmugham, L.N.; Petrarca, C.; Castellani, M.L.; Symeonidou, I.; Frydas, S.; Vecchiet, J.; Falasca, K.; Tetè, S.; Conti, P.; Salini, V. Il-1beta induces alkaline phosphatase in human phagocytes. Arch. Med. Res. 2007, 38, 39–44. [Google Scholar] [CrossRef]
- Li, H.; Zhao, Y.; Li, W.; Yang, J.; Wu, H. Critical role of neutrophil alkaline phosphatase in the antimicrobial function of neutrophils. Life Sci. 2016, 157, 152–157. [Google Scholar] [CrossRef]
- Meng, Y.; Ye, F.; Nie, P.; Zhao, Q.; An, L.; Wang, W.; Qu, S.; Shen, Z.; Cao, Z.; Zhang, X.; et al. Immunosuppressive CD10+ALPL+ neutrophils promote resistance to anti-PD-1 therapy in hcc by mediating irreversible exhaustion of T cells. J. Hepatol. 2023, 79, 1435–1449. [Google Scholar] [CrossRef]
- Hernández-Chirlaque, C.; Gámez-Belmonte, R.; Ocón, B.; Martínez-Moya, P.; Wirtz, S.; Sánchez de Medina, F.; Martínez-Augustin, O. Tissue non-specific alkaline phosphatase expression is needed for the full stimulation of t cells and t cell-dependent colitis. J. Crohns Colitis 2017, 11, 857–870. [Google Scholar]
- Shi, W.; Li, T.; Li, H.; Ren, J.; Lv, M.; Wang, Q.; He, Y.; Yu, Y.; Liu, L.; Jin, S.; et al. Bioinformatics approach to identify the hub gene associated with COVID-19 and idiopathic pulmonary fibrosis. IET Syst. Biol. 2023, 17, 336–351. [Google Scholar] [CrossRef]
- Cruz, P.D.; Wargowsky, R.; Gonzalez-Almada, A.; Sifontes, E.P.; Shaykhinurov, E.; Jaatinen, K.; Jepson, T.; Lafleur, J.E.; Yamane, D.; Perkins, J.; et al. Blood rna biomarkers identify bacterial and biofilm coinfections in COVID-19 intensive care patients. J. Intensive Care Med. 2024, 39, 1071–1082. [Google Scholar] [CrossRef]
- Gupta, R.K.; Nwachuku, E.L.; Zusman, B.E.; Jha, R.M.; Puccio, A.M. Drug repurposing for COVID-19 based on an integrative meta-analysis of SARS-CoV-2 induced gene signature in human airway epithelium. PLoS ONE 2021, 16, e0257784. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.N.; Wang, X.Y.; Cai, L.C.; Jian, W.G.; Zhang, C. Igll5 is correlated with tumor-infiltrating immune cells in clear cell renal cell carcinoma. FEBS Open Bio 2021, 11, 898–910. [Google Scholar] [CrossRef] [PubMed]
- Cornish, A.J.; Hoang, P.H.; Dobbins, S.E.; Law, P.J.; Chubb, D.; Orlando, G.; Houlston, R.S. Identification of recurrent noncoding mutations in b-cell lymphoma using capture hi-c. Blood Adv. 2019, 3, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Kasar, S.; Kim, J.; Improgo, R.; Tiao, G.; Polak, P.; Haradhvala, N.; Lawrence, M.S.; Kiezun, A.; Fernandes, S.M.; Bahl, S.; et al. Whole-genome sequencing reveals activation-induced cytidine deaminase signatures during indolent chronic lymphocytic leukaemia evolution. Nat. Commun. 2015, 6, 8866. [Google Scholar] [CrossRef]
- White, B.S.; Lanc, I.; O’Neal, J.; Gupta, H.; Fulton, R.S.; Schmidt, H.; Fronick, C.; Belter, E.A.; Fiala, M.; King, J.; et al. A multiple myeloma-specific capture sequencing platform discovers novel translocations and frequent, risk-associated point mutations in igll5. Blood Cancer J. 2018, 8, 35. [Google Scholar] [CrossRef]
- Hosoi, H.; Tabata, S.; Kosako, H.; Hori, Y.; Okamura, T.; Yamashita, Y.; Fujimoto, K.; Kajioka, D.; Suzuki, K.; Osato, M.; et al. Igll5 controlled by super-enhancer affects cell survival and myc expression in mature b-cell lymphoma. Leuk. Res. Rep. 2024, 21, 100451. [Google Scholar] [CrossRef]
- Guglielmi, P.; Davi, F. Expression of a novel type of immunoglobulin cλ transcripts in human mature b lymphocytes producing χ light chains. Eur. J. Immunol. 1991, 21, 501–508. [Google Scholar] [CrossRef]
- Post, N.; Eddy, D.; Huntley, C.; Van Schalkwyk, M.C.; Shrotri, M.; Leeman, D.; Rigby, S.; Williams, S.V.; Bermingham, W.H.; Kellam, P. Antibody response to SARS-CoV-2 infection in humans: A systematic review. PLoS ONE 2020, 15, e0244126. [Google Scholar] [CrossRef]
- Liu, X.; Wang, J.; Xu, X.; Liao, G.; Chen, Y.; Hu, C.H. Patterns of igg and igm antibody response in COVID-19 patients. Emerg. Microbes Infect. 2020, 9, 1269–1274. [Google Scholar] [CrossRef]
- Suthar, M.S.; Zimmerman, M.G.; Kauffman, R.C.; Mantus, G.; Linderman, S.L.; Hudson, W.H.; Vanderheiden, A.; Nyhoff, L.; Davis, C.W.; Adekunle, O.; et al. Rapid generation of neutralizing antibody responses in COVID-19 patients. Cell Rep. Med. 2020, 1, 100040. [Google Scholar] [CrossRef]
- Frasca, D.; Diaz, A.; Romero, M.; Mendez, N.V.; Landin, A.M.; Blomberg, B.B. Effects of age on h1n1-specific serum igg1 and igg3 levels evaluated during the 2011-2012 influenza vaccine season. Immun. Ageing 2013, 10, 14. [Google Scholar] [CrossRef]
- Moura, A.D.; da Costa, H.H.M.; Correa, V.A.; de S. Lima, A.K.; Lindoso, J.A.L.; De Gaspari, E.; Hong, M.A.; Cunha-Junior, J.P.; Prudencio, C.R. Assessment of avidity related to igg subclasses in SARS-CoV-2 brazilian infected patients. Sci. Rep. 2021, 11, 17642. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sub-Dataset | Sample Size | Features |
|---|---|---|
| Pre-vaccination group | 29 | 13,383 RNA-seq and 1662 small RNA-Seq |
| ChAdOx1-onset group | 33 | 13,383 RNA-seq and 1662 small RNA-Seq |
| MenACWY-onset group | 47 | 13,383 RNA-seq and 1662 small RNA-Seq |
| ChAdOx1-7D group | 30 | 13,383 RNA-seq and 1662 small RNA-Seq |
| MenACWY-7D group | 41 | 13,383 RNA-seq and 1662 small RNA-Seq |
| Feature List | Classification Algorithm | Number of Features | ACC | MCC | Macro F1 | Weigthed F1 |
|---|---|---|---|---|---|---|
| AdaBoost feature list | QDA | 80 | 0.753 | 0.716 | 0.774 | 0.774 |
| CatBoost feature list | LightGBM | 215 | 0.750 | 0.688 | 0.746 | 0.750 |
| ExtraTrees feature list | QDA | 580 | 0.736 | 0.708 | 0.748 | 0.748 |
| LASSO feature list | QDA | 65 | 0.719 | 0.674 | 0.735 | 0.735 |
| LightGBM feature list | LightGBM | 270 | 0.872 | 0.841 | 0.870 | 0.871 |
| MCFS feature list | LightGBM | 865 | 0.767 | 0.709 | 0.758 | 0.766 |
| mRMR feature list | QDA | 130 | 0.740 | 0.704 | 0.754 | 0.754 |
| RF feature list | RF | 70 | 0.750 | 0.690 | 0.748 | 0.750 |
| Ridge feature list | QDA | 1685 | 0.723 | 0.694 | 0.741 | 0.741 |
| XGBoost feature list | LightGBM | 440 | 0.844 | 0.805 | 0.843 | 0.843 |
| Feature List | Classification Algorithm | Number of Features | ACC | MCC | Macro F1 | Weigthed F1 |
|---|---|---|---|---|---|---|
| AdaBoost feature list $ | QDA | 80 | 0.753 | 0.716 | 0.774 | 0.774 |
| CatBoost feature list | LightGBM | 70 | 0.739 | 0.674 | 0.733 | 0.734 |
| ExtraTrees feature list | QDA | 50 | 0.698 | 0.675 | 0.714 | 0.714 |
| LASSO feature list $ | QDA | 65 | 0.719 | 0.674 | 0.735 | 0.735 |
| LightGBM feature list | LightGBM | 60 | 0.833 | 0.791 | 0.830 | 0.832 |
| MCFS feature list | LightGBM | 105 | 0.672 | 0.593 | 0.667 | 0.674 |
| mRMR feature list | QDA | 45 | 0.681 | 0.657 | 0.691 | 0.691 |
| RF feature list $ | RF | 70 | 0.750 | 0.690 | 0.748 | 0.750 |
| Ridge feature list | QDA | 60 | 0.698 | 0.661 | 0.711 | 0.711 |
| XGBoost feature list | LightGBM | 90 | 0.811 | 0.764 | 0.809 | 0.809 |
| Ensembl ID (Gene Symbol) | ENSG00000211896 (IGHG1) | ENSG00000111206 (FOXM1) | ENSG00000003400 (CASP10) |
|---|---|---|---|
| Description | Immunoglobulin heavy constant gamma 1 (G1m marker) | Forkhead box M1 | Caspase 10 |
| References | [48,49,50,51,52,53,54,55] | [56,57,58,59,60,61,62,63,64,65,66,67,68,69,70] | [71,72,73] |
| Corresponding algorithm | CatBoost, RF, XGBoost, MCFS, LightGBM | CatBoost, RF, XGBoost, MCFS, LightGBM | AsaBoost, XGBoost, MCFS, LightGBM |
| Immunological activity | IGHG1 is a subclass member of immunoglobulin G, which influences the interaction between the immune system and cancer cells, as well as the regulation of immune mechanisms, in certain cancers and immune-related diseases through modulating cancer cell immune evasion, suppressing immune cell function, and others. | FOXM1, as an important member of the forkhead transcription factors family, plays a crucial role in cell cycle, cell proliferation, and immune cell regulation. It can participate in immune regulation by up-regulating the expression of PD-L1 and is closely related to the function of immune cells. | Caspase-10 (CASP10), a member of the cysteine-aspartate protease family, is involved in apoptosis and cellular immunity, with dysfunction or mutations in CASP10 contributing to autoimmune diseases such as primary biliary cholangitis (PBC) and type IIA autoimmune lymphoproliferative syndrome (ALPS), suggesting its critical role in immune regulation. |
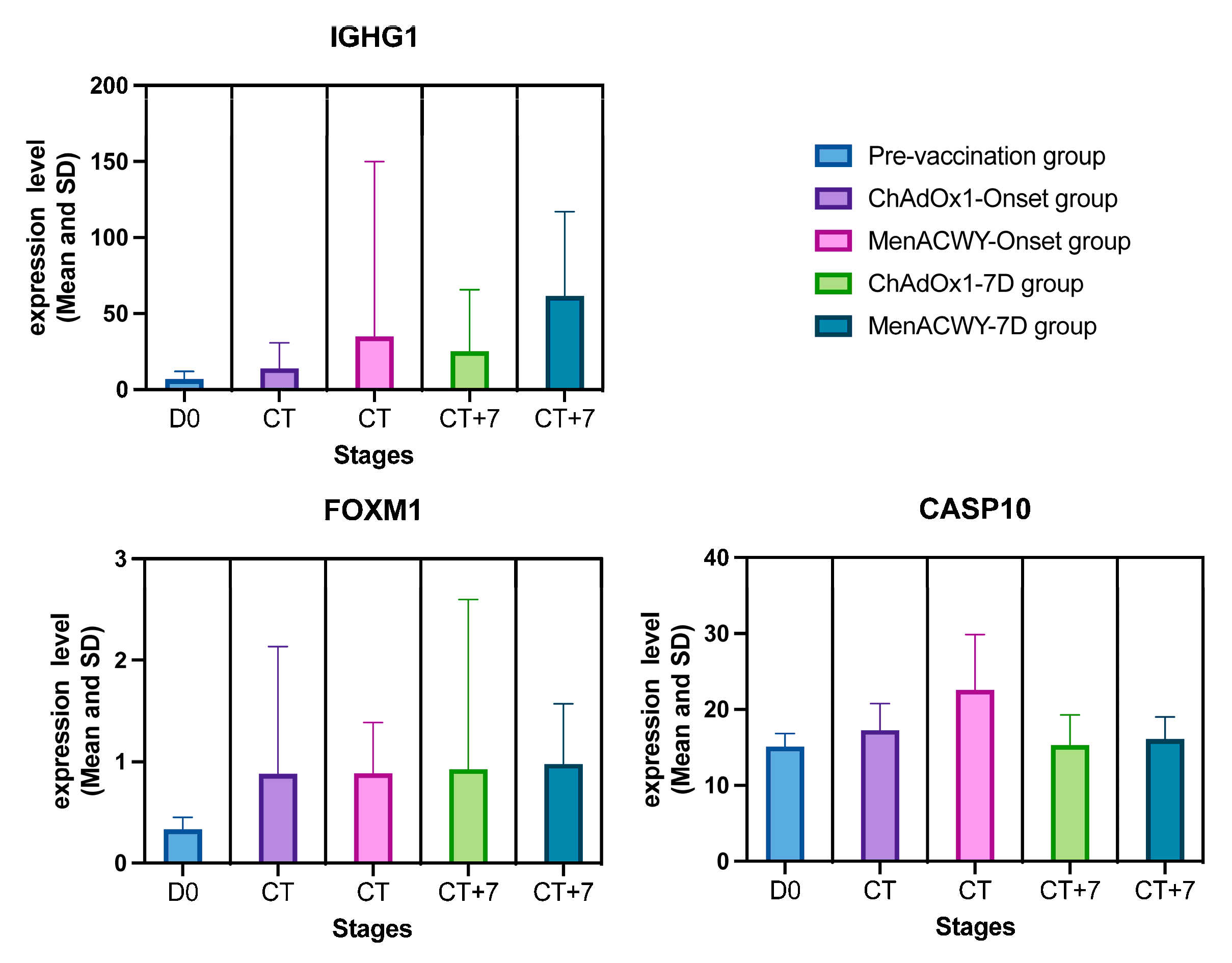
| Pre-vaccination group (mean ± SD) | 6.79 ± 4.97 | 0.33 ± 0.12 | 15.12 ± 1.70 |
| ChAdOx1-onset→7D group (mean ± SD) | 13.77 ± 16.76→25.31 ± 40.09 | 0.88 ± 1.26→0.92 ± 1.68 | 17.27 ± 3.49→15.32 ± 3.96 |
| MenACWY-onset→7D group (mean ± SD) | 34.77 ± 115.17→61.43 ± 55.69 | 0.88 ± 0.50→0.98 ± 0.59 | 22.63 ± 7.24→16.09 ± 2.93 |
| General tendency | Expression and volatility continued to rise over time. | The pre-vaccination group was significantly lower, and the ChAdOx1-/MenACWY-onset and ChAdOx1/-MenACWY-7D groups were similar. | The ChAdOx1-/MenACWY-onset group in-creased over time, while the ChAdOx1-/MenACWY-7D group remained basically unchanged. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, J.; Ma, Q.; Chen, L.; Guo, W.; Feng, K.; Huang, T.; Cai, Y.-D. Transcriptomic and miRNA Signatures of ChAdOx1 nCoV-19 Vaccine Response Using Machine Learning. Life 2025, 15, 981. https://doi.org/10.3390/life15060981
Lin J, Ma Q, Chen L, Guo W, Feng K, Huang T, Cai Y-D. Transcriptomic and miRNA Signatures of ChAdOx1 nCoV-19 Vaccine Response Using Machine Learning. Life. 2025; 15(6):981. https://doi.org/10.3390/life15060981
Chicago/Turabian StyleLin, Jinting, Qinglan Ma, Lei Chen, Wei Guo, Kaiyan Feng, Tao Huang, and Yu-Dong Cai. 2025. "Transcriptomic and miRNA Signatures of ChAdOx1 nCoV-19 Vaccine Response Using Machine Learning" Life 15, no. 6: 981. https://doi.org/10.3390/life15060981
APA StyleLin, J., Ma, Q., Chen, L., Guo, W., Feng, K., Huang, T., & Cai, Y.-D. (2025). Transcriptomic and miRNA Signatures of ChAdOx1 nCoV-19 Vaccine Response Using Machine Learning. Life, 15(6), 981. https://doi.org/10.3390/life15060981