
The plk1 Gene Regulatory Network Modeling Identifies Three Circuits for plk1-mediated Genomic Instability Leading to Neoplastic Transformation

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Retrieving Genomic Instability-Associated Biological Processes Using PathwayStudio
2.2. Delimiting the Core of plk1- Associated Genomic Instability Events
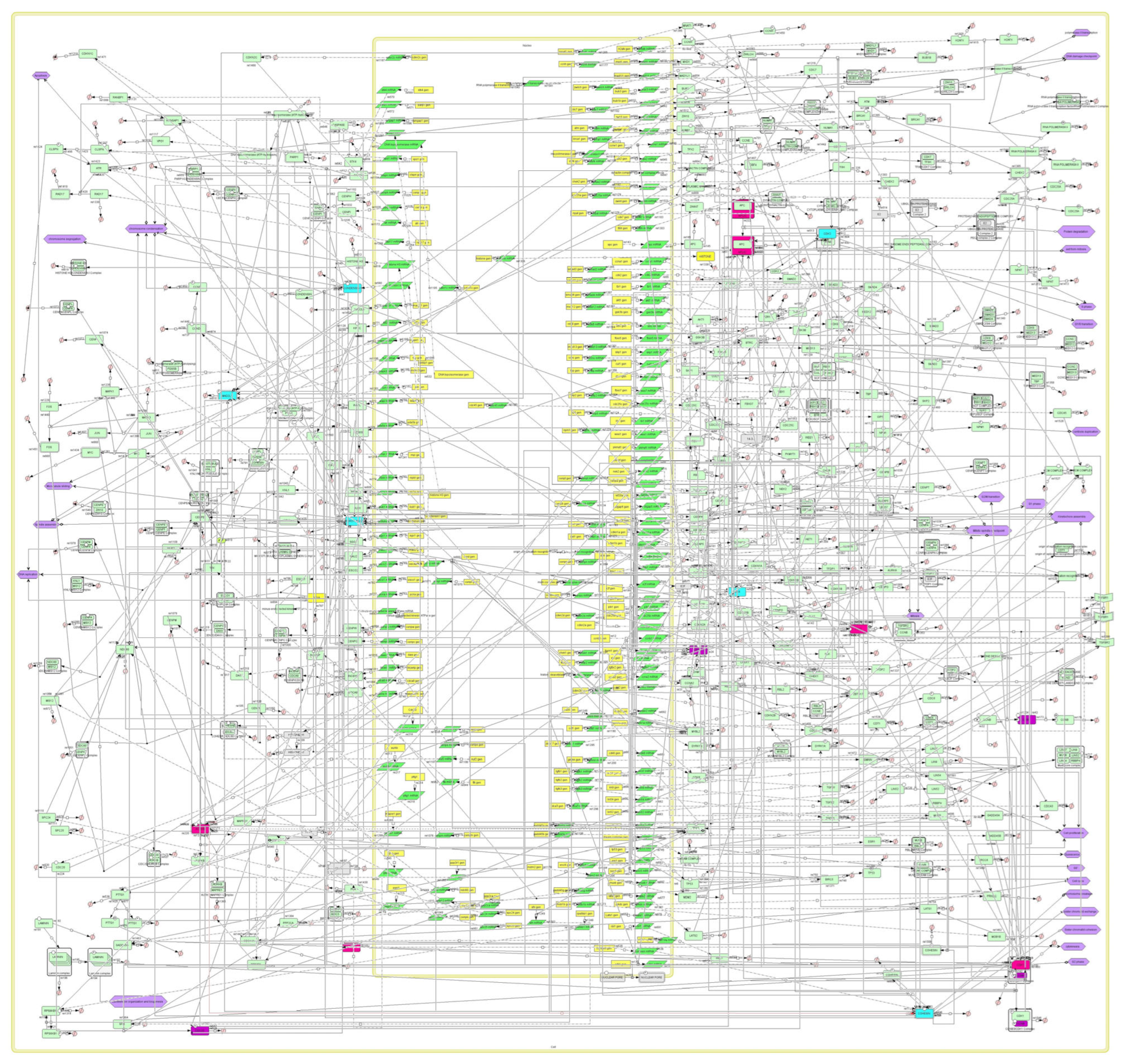
2.3. Construction of the plk1 Core Genomic Instability Network
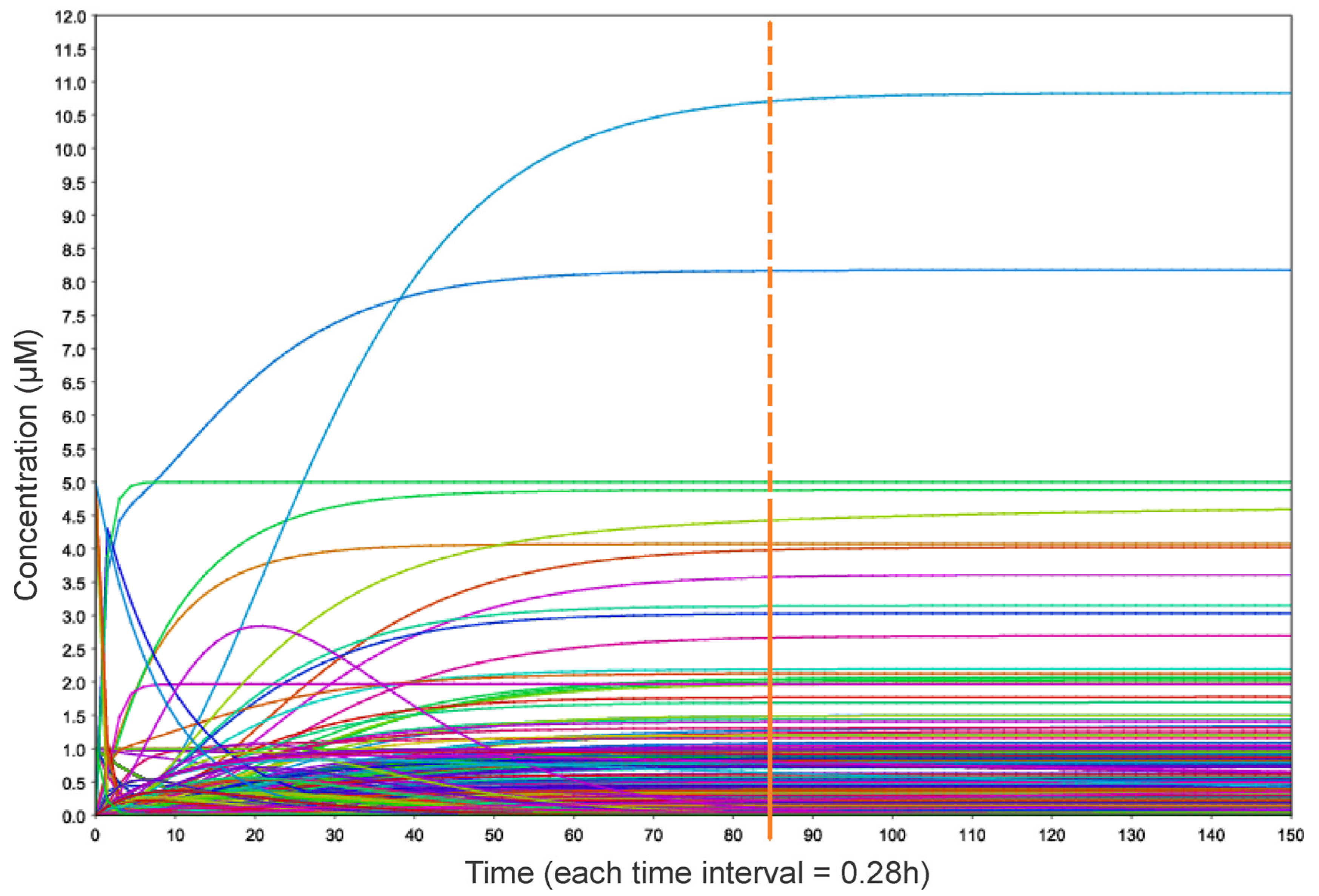
2.4. Mathematical Modeling and Generation of Relevant Simulation Scenarios
2.5. Transcriptomic Profiling and Survival Analysis
2.6. Cell Culture and Quantitative Real-Time Polymerase Chain Reaction
3. Results
3.1. Identifying and Integrating the Key Components of plk1-Mediated Genomic Instability Events in Cancer
3.2. Building the plk1 Core Genomic Instability Network
3.3. A Model for Genomic Instability Research
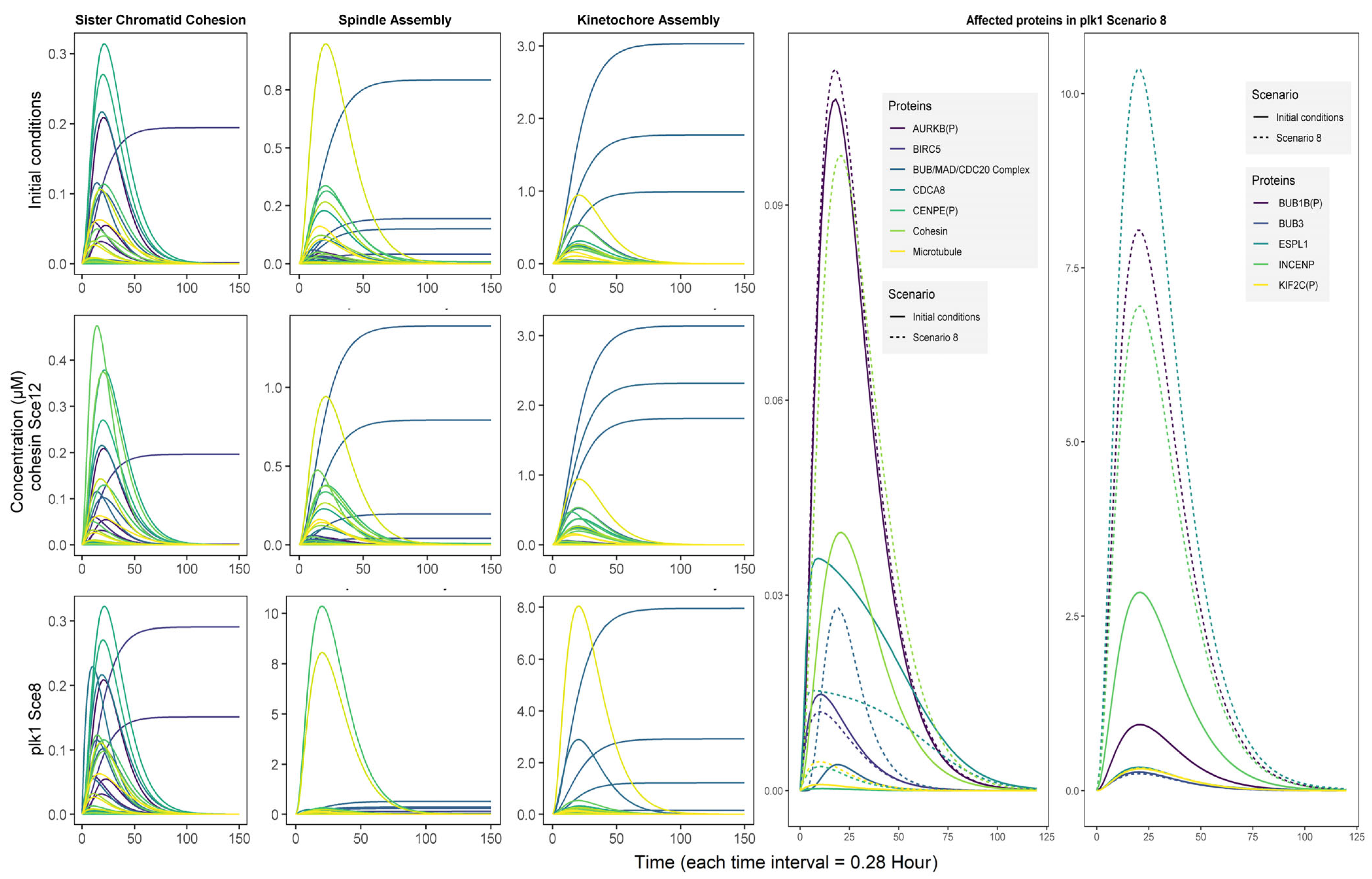
3.4. Simulation Scenarios Highlight a Synergy Between PLK1 and COHESIN Regulation
3.5. The Role of plk1 in Genomic Instability
3.5.1. First Circuit: Chromosomal Passenger Complex
3.5.2. Second Circuit: Mitotic Checkpoint Complex
3.5.3. Third Circuit: Anaphase/Cyclosome Promoter Complex
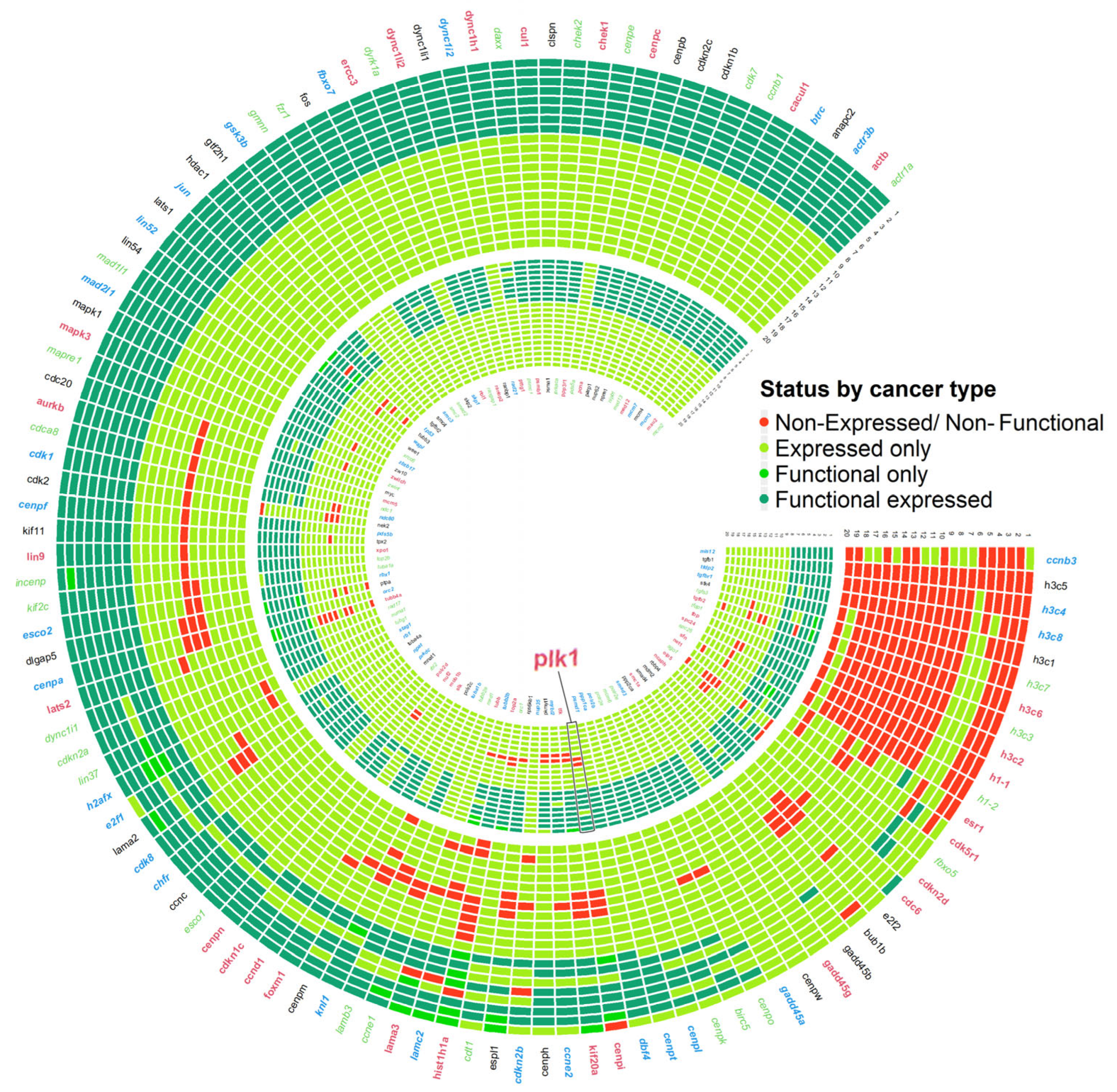
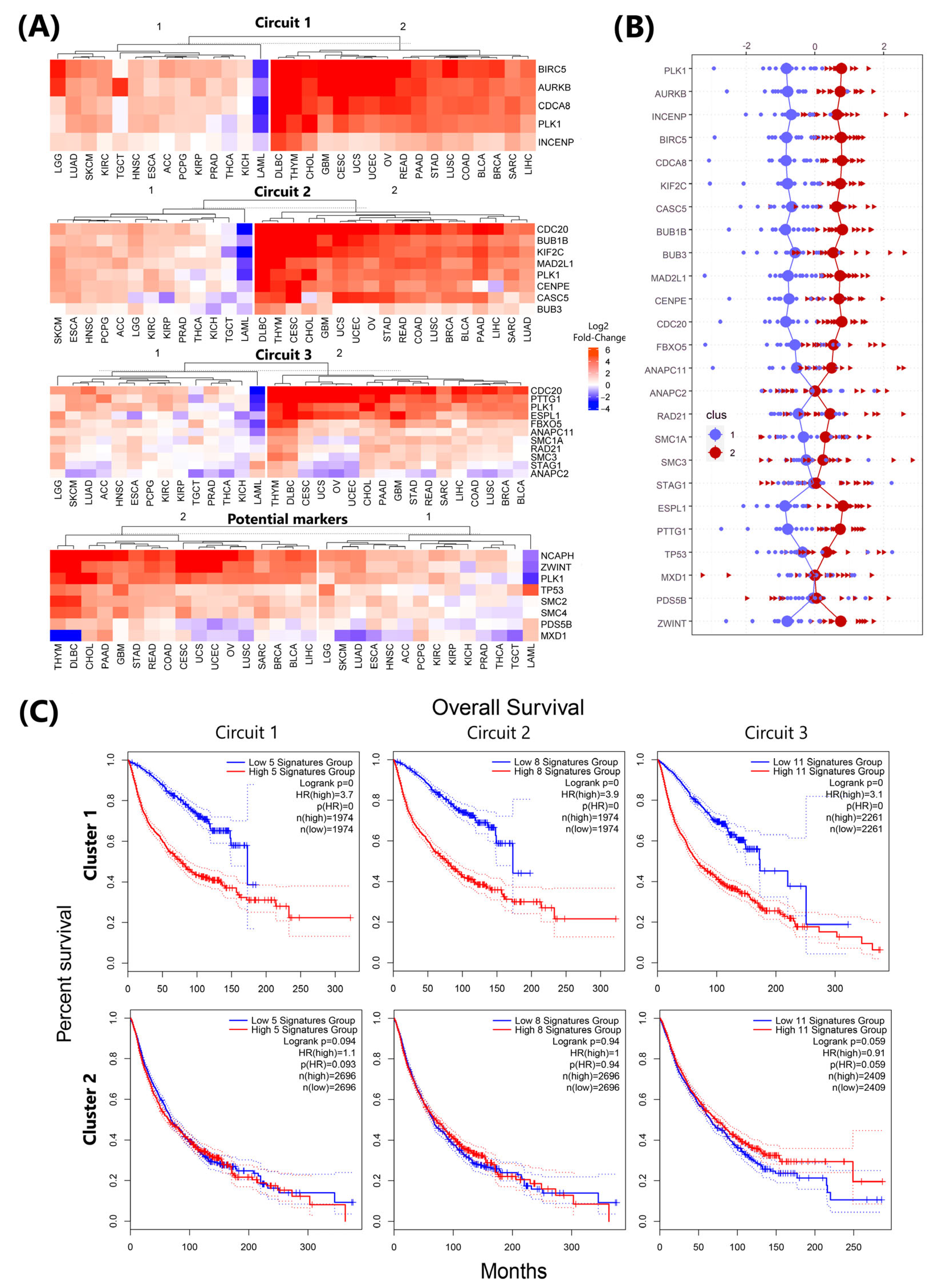
3.6. Circuit-Specific Deregulation and Prognostic Patterns in plk1-Driven Genomic Instability
3.7. Real Time PCR Analysis of Key Genes Predicted to Be Associated with Genome Instability Events
4. Discussion
4.1. Model Limitations and Assumptions
4.2. Deregulation Processes Leading to Genomic Instability
4.3. plk1: Oncogene and Tumor Suppressor
4.4. Model-Derived Hypotheses and Opportunities for Experimental Validation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Langie, S.A.S.; Koppen, G.; Desaulniers, D.; Al-Mulla, F.; Al-Temaimi, R.; Amedei, A.; Azqueta, A.; Bisson, W.H.; Brown, D.; Brunborg, G.; et al. Causes of Genome Instability: The Effect of Low Dose Chemical Exposures in Modern Society. Carcinogenesis 2015, 36, S61–S88. [Google Scholar] [CrossRef]
- Yao, Y.; Dai, W. Genomic Instability and Cancer. J. Carcinog. Mutagen. 2014, 5, 1000165. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, S.F.; Cantley, L.C. The Multifaceted Role of Chromosomal Instability in Cancer and Its Microenvironment. Cell 2018, 174, 1347. [Google Scholar] [CrossRef]
- Ippolito, M.R.; Martis, V.; Martin, S.; Tijhuis, A.E.; Hong, C.; Wardenaar, R.; Dumont, M.; Zerbib, J.; Spierings, D.C.J.; Fachinetti, D.; et al. Gene Copy-Number Changes and Chromosomal Instability Induced by Aneuploidy Confer Resistance to Chemotherapy. Dev. Cell 2021, 56, 2440–2454.e6. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.E.; MacAuley, M.J.; Vizeacoumar, F.S.; Abuhussein, O.; Freywald, A.; Vizeacoumar, F.J. The CINs of Polo-Like Kinase 1 in Cancer. Cancers 2020, 12, 2953. [Google Scholar] [CrossRef] [PubMed]
- Zitouni, S.; Nabais, C.; Jana, S.C.; Guerrero, A.; Bettencourt-Dias, M. Polo-like Kinases: Structural Variations Lead to Multiple Functions. Nat. Rev. Mol. Cell Biol. 2014, 15, 433–452. [Google Scholar] [CrossRef]
- Tavassoly, I.; Goldfarb, J.; Iyengar, R. Systems Biology Primer: The Basic Methods and Approaches. Essays Biochem. 2018, 62, 487–500. [Google Scholar] [CrossRef]
- Abroudi, A.; Samarasinghe, S.; Kulasiri, D. A Comprehensive Complex Systems Approach to the Study and Analysis of Mammalian Cell Cycle Control System in the Presence of DNA Damage Stress. J. Theor. Biol. 2017, 429, 204–228. [Google Scholar] [CrossRef]
- Gauthier, J.H.; Pohl, P.I. A General Framework for Modeling Growth and Division of Mammalian Cells. BMC Syst. Biol. 2011, 5, 3. [Google Scholar] [CrossRef]
- Alberghina, L.; Coccetti, P.; Orlandi, I. Systems Biology of the Cell Cycle of Saccharomyces Cerevisiae: From Network Mining to System-Level Properties. Biotechnol. Adv. 2009, 27, 960–978. [Google Scholar] [CrossRef]
- Verghese, J.; Abrams, J.; Wang, Y.; Morano, K.A. Biology of the Heat Shock Response and Protein Chaperones: Budding Yeast (Saccharomyces Cerevisiae) as a Model System. Microbiol. Mol. Biol. Rev. 2012, 76, 115. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, K.; Tashima, Y.; Hamada, H.; Eguchi, Y.; Okamoto, M. Mathematical Modeling and Sensitivity Analysis of G1/S Phase in the Cell Cycle Including the DNA-Damage Signal Transduction Pathway. BioSystems 2008, 94, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, K.; Hamada, H.; Eguchi, Y.; Okamoto, M. Mathematical Modeling of Cell Cycle Regulation in Response to DNA Damage: Exploring Mechanisms of Cell-Fate Determination. Biosystems 2011, 103, 384–391. [Google Scholar] [CrossRef]
- Li, L.; Zhang, X.; Tian, T.; Pang, L. Mathematical Modelling the Pathway of Genomic Instability in Lung Cancer. Sci. Rep. 2019, 9, 14136. [Google Scholar] [CrossRef]
- Jaiswal, H.; Benada, J.; Müllers, E.; Akopyan, K.; Burdova, K.; Koolmeister, T.; Helleday, T.; Medema, R.H.; Macurek, L.; Lindqvist, A. ATM /Wip1 Activities at Chromatin Control Plk1 Re-activation to Determine G2 Checkpoint Duration. EMBO J. 2017, 36, 2161–2176. [Google Scholar] [CrossRef] [PubMed]
- Terhune, S.S.; Jung, Y.; Cataldo, K.M.; Dash, R.K. Network Mechanisms and Dysfunction within an Integrated Computational Model of Progression through Mitosis in the Human Cell Cycle. PLoS Comput. Biol. 2020, 16, e1007733. [Google Scholar] [CrossRef]
- Nikitin, A.; Egorov, S.; Daraselia, N.; Mazo, I. Pathway Studio—the Analysis and Navigation of Molecular Networks. Bioinformatics 2003, 19, 2155–2157. [Google Scholar] [CrossRef]
- Aguilera, A.; Gómez-González, B. Genome Instability: A Mechanistic View of Its Causes and Consequences. Nat. Rev. Genet. 2008, 9, 204–217. [Google Scholar] [CrossRef]
- Wei, W.; Cheng, Y.; Wang, B. Cancer and genome instability. In Genome Stability: From Virus to Human Application, 1st ed.; Rao, M.R.S., Ed.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 463–486. [Google Scholar] [CrossRef]
- Prüter, H.; Quimbaya, M. Diseño de Una Estrategia Para la Detección y Caracterización de Nuevos Genes Asociados al Proceso de Inestabilidad Genómica. Bachelor’s Thesis, Pontificia Universidad Javeriana Cali, Cali, Colombia, 2016. [Google Scholar]
- Putnam, C.D.; Kolodner, R.D. Pathways and Mechanisms That Prevent Genome Instability in Saccharomyces Cerevisiae. Genetics 2017, 206, 1187. [Google Scholar] [CrossRef]
- Shen, Z. Genomic Instability and Cancer: An Introduction. J. Mol. Cell Biol. 2011, 3, 1–3. [Google Scholar] [CrossRef]
- Paulsen, R.D.; Soni, D.V.; Wollman, R.; Hahn, A.T.; Yee, M.C.; Guan, A.; Hesley, J.A.; Miller, S.C.; Cromwell, E.F.; Solow-Cordero, D.E.; et al. A Genome-Wide SiRNA Screen Reveals Diverse Cellular Processes and Pathways That Mediate Genome Stability. Mol. Cell 2009, 35, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Safran, M.; Dalah, I.; Alexander, J.; Rosen, N.; Iny Stein, T.; Shmoish, M.; Nativ, N.; Bahir, I.; Doniger, T.; Krug, H.; et al. GeneCards Version 3: The Human Gene Integrator. Database 2010, 2010, baq020. [Google Scholar] [CrossRef] [PubMed]
- Funahashi, A.; Matsuoka, Y.; Jouraku, A.; Morohashi, M.; Kikuchi, N.; Kitano, H. CellDesigner 3.5: A Versatile Modeling Tool for Biochemical Networks. Proc. IEEE 2008, 96, 1254–1265. [Google Scholar] [CrossRef]
- Horn, F.; Jackson, R. General Mass Action Kinetics. Arch. Ration. Mech. Anal. 1972, 47, 81–116. [Google Scholar] [CrossRef]
- Le Novère, N.; Bornstein, B.; Broicher, A.; Courtot, M.; Donizelli, M.; Dharuri, H.; Li, L.; Sauro, H.; Schilstra, M.; Shapiro, B.; et al. BioModels Database: A Free, Centralized Database of Curated, Published, Quantitative Kinetic Models of Biochemical and Cellular Systems. Nucleic Acids Res. 2006, 34, D689–D691. [Google Scholar] [CrossRef]
- Schomburg, I.; Chang, A.; Ebeling, C.; Gremse, M.; Heldt, C.; Huhn, G.; Schomburg, D. BRENDA, the Enzyme Database: Updates and Major New Developments. Nucleic Acids Res. 2004, 32, D431–D433. [Google Scholar] [CrossRef]
- Wittig, U.; Kania, R.; Golebiewski, M.; Rey, M.; Shi, L.; Jong, L.; Algaa, E.; Weidemann, A.; Sauer-Danzwith, H.; Mir, S.; et al. SABIO-RK—Database for Biochemical Reaction Kinetics. Nucleic Acids Res. 2012, 40, D790–D796. [Google Scholar] [CrossRef]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An Enhanced Web Server for Large-Scale Expression Profiling and Interactive Analysis. Nucleic Acids Res. 2019, 47, W556. [Google Scholar] [CrossRef]
- Wierstra, I. The Transcription Factor FOXM1 (Forkhead Box M1): Proliferation-Specific Expression, Transcription Factor Function, Target Genes, Mouse Models, and Normal Biological Roles. Adv. Cancer Res. 2013, 118, 97–398. [Google Scholar] [CrossRef]
- Zeng, J.; Zhang, Y.; Shang, Y.; Mai, J.; Shi, S.; Lu, M.; Bu, C.; Zhang, Z.; Zhang, Z.; Li, Y.; et al. CancerSCEM: A Database of Single-Cell Expression Map across Various Human Cancers. Nucleic Acids Res. 2022, 50, D1147–D1155. [Google Scholar] [CrossRef] [PubMed]
- Marzluff, W.F.; Wagner, E.J.; Duronio, R.J. Metabolism and Regulation of Canonical Histone MRNAs: Life without a Poly(A) Tail. Nat. Rev. Genet. 2008, 9, 843. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Huo, D.; Xie, H.; Wu, L.; Zhang, J.; Liu, L.; Jin, Q.; Chen, X. CHG: A Systematically Integrated Database of Cancer Hallmark Genes. Front. Genet. 2020, 11, 29. [Google Scholar] [CrossRef]
- Carter, S.L.; Eklund, A.C.; Kohane, I.S.; Harris, L.N.; Szallasi, Z. A Signature of Chromosomal Instability Inferred from Gene Expression Profiles Predicts Clinical Outcome in Multiple Human Cancers. Nat. Genet. 2006, 38, 1043–1048. [Google Scholar] [CrossRef]
- Cooper, G.M. The Eukaryotic Cell Cycle. In The Cell: A Molecular Approach, 2nd ed.; Sinauer Associates: Sunderland, MA, USA, 2000; Chapter 14. [Google Scholar]
- Haarhuis, J.H.I.; Elbatsh, A.M.O.; Rowland, B.D. Cohesin and Its Regulation: On the Logic of X-Shaped Chromosomes. Dev. Cell 2014, 31, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, M.; Lee, S.H. The Chromosomal Passenger Complex (CPC) as a Key Orchestrator of Orderly Mitotic Exit and Cytokinesis. Front. Cell Dev. Biol. 2015, 3, 14. [Google Scholar] [CrossRef]
- Marima, R.; Hull, R.; Penny, C.; Dlamini, Z. Mitotic Syndicates Aurora Kinase B (AURKB) and Mitotic Arrest Deficient 2 like 2 (MAD2L2) in Cohorts of DNA Damage Response (DDR) and Tumorigenesis. Mutat. Res./Rev. Mutat. Res. 2021, 787, 108376. [Google Scholar] [CrossRef]
- Bolton, M.A.; Lan, W.; Powers, S.E.; McCleland, M.L.; Kuang, J.; Stukenberg, P.T. Aurora B Kinase Exists in a Complex with Survivin and INCENP and Its Kinase Activity Is Stimulated by Survivin Binding and Phosphorylation. Mol. Biol. Cell 2002, 13, 3064–3077. [Google Scholar] [CrossRef]
- Kaisari, S.; Shomer, P.; Ziv, T.; Sitry-Shevah, D.; Miniowitz-Shemtov, S.; Teichner, A.; Hershko, A. Role of Polo-like Kinase 1 in the Regulation of the Action of P31 Comet in the Disassembly of Mitotic Checkpoint Complexes. Proc. Natl. Acad. Sci. USA 2019, 116, 11725–11730. [Google Scholar] [CrossRef]
- Rao, C.V.; Yamada, H.Y.; Yao, Y.; Dai, W. Enhanced Genomic Instabilities Caused by Deregulated Microtubule Dynamics and Chromosome Segregation: A Perspective from Genetic Studies in Mice. Carcinogenesis 2009, 30, 1469. [Google Scholar] [CrossRef]
- Tsou, M.-F.B.; Wang, W.-J.; George, K.A.; Uryu, K.; Stearns, T.; Jallepalli, P.V. Polo Kinase and Separase Regulate the Mitotic Licensing of Centriole Duplication in Human Cells. Dev. Cell 2009, 17, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Agircan, F.G.; Schiebel, E. Sensors at Centrosomes Reveal Determinants of Local Separase Activity. PLoS Genet. 2014, 10, e1004672. [Google Scholar] [CrossRef]
- McKenzie, L.; King, S.; Marcar, L.; Nicol, S.; Dias, S.S.; Schumm, K.; Robertson, P.; Bourdon, J.-C.; Perkins, N.; Fuller-Pace, F.; et al. P53-Dependent Repression of Polo-like Kinase-1 (PLK1). Cell Cycle 2010, 9, 4200–4212. [Google Scholar] [CrossRef] [PubMed]
- de Cárcer, G. The Mitotic Cancer Target Polo-Like Kinase 1: Oncogene or Tumor Suppressor? Genes 2019, 10, 208. [Google Scholar] [CrossRef]
- Li, J.; Hong, M.J.; Chow, J.P.H.; Man, W.Y.; Mak, J.P.Y.; Ma, H.T.; Poon, R.Y.C. Co-Inhibition of Polo-like Kinase 1 and Aurora Kinases Promotes Mitotic Catastrophe. Oncotarget 2015, 6, 9327. [Google Scholar] [CrossRef]
- Carmena, M.; Pinson, X.; Platani, M.; Salloum, Z.; Xu, Z.; Clark, A.; MacIsaac, F.; Ogawa, H.; Eggert, U.; Glover, D.M.; et al. The Chromosomal Passenger Complex Activates Polo Kinase at Centromeres. PLoS Biol. 2012, 10, e1001250. [Google Scholar] [CrossRef]
- Ando, K.; Ozaki, T.; Yamamoto, H.; Furuya, K.; Hosoda, M.; Hayashi, S.; Fukuzawa, M.; Nakagawara, A. Polo-like Kinase 1 (Plk1) Inhibits P53 Function by Physical Interaction and Phosphorylation *. J. Biol. Chem. 2004, 279, 25549–25561. [Google Scholar] [CrossRef]
- Gully, C.P.; Velazquez-Torres, G.; Shin, J.-H.; Fuentes-Mattei, E.; Wang, E.; Carlock, C.; Chen, J.; Rothenberg, D.; Adams, H.P.; Choi, H.H.; et al. Aurora B Kinase Phosphorylates and Instigates Degradation of P53. Proc. Natl. Acad. Sci. USA 2012, 109, E1513. [Google Scholar] [CrossRef] [PubMed]
- Putkey, F.R.; Cramer, T.; Morphew, M.K.; Silk, A.D.; Johnson, R.S.; McIntosh, J.R.; Cleveland, D.W. Unstable Kinetochore-Microtubule Capture and Chromosomal Instability Following Deletion of CENP-E. Dev. Cell 2002, 3, 351–365. [Google Scholar] [CrossRef]
- Wagenbach, M.; Vicente, J.J.; Ovechkina, Y.; Domnitz, S.; Wordeman, L. Functional Characterization of MCAK/Kif2C Cancer Mutations Using High-Throughput Microscopic Analysis. Mol. Biol. Cell 2020, 31, 580–588. [Google Scholar] [CrossRef]
- Elowe, S.; Hümmer, S.; Uldschmid, A.; Li, X.; Nigg, E.A. Tension-Sensitive Plk1 Phosphorylation on BubR1 Regulates the Stability of Kinetochore–Microtubule Interactions. Genes. Dev. 2007, 21, 2205. [Google Scholar] [CrossRef]
- Zhang, L.; Shao, H.; Huang, Y.; Yan, F.; Chu, Y.; Hou, H.; Zhu, M.; Fu, C.; Aikhionbare, F.; Fang, G.; et al. PLK1 Phosphorylates Mitotic Centromere-Associated Kinesin and Promotes Its Depolymerase Activity. J. Biol. Chem. 2011, 286, 3033. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Li, B.; Yu, H. The Bub1–Plk1 Kinase Complex Promotes Spindle Checkpoint Signalling through Cdc20 Phosphorylation. Nat. Commun. 2016, 7, 10818. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.V.; Loktev, A.V.; Ban, K.H.; Jackson, P.K. Plk1 Regulates Activation of the Anaphase Promoting Complex by Phosphorylating and Triggering SCFbetaTrCP-Dependent Destruction of the APC Inhibitor Emi1. Mol. Biol. Cell 2004, 15, 5623–5634. [Google Scholar] [CrossRef] [PubMed]
- Reimann, J.D.R.; Freed, E.; Hsu, J.Y.; Kramer, E.R.; Peters, J.-M.; Jackson, P.K. Emi1 Is a Mitotic Regulator That Interacts with Cdc20 and Inhibits the Anaphase Promoting Complex. Cell 2001, 105, 645–655. [Google Scholar] [CrossRef]
- Mukherjee, S. El Emperador de Todos Los Males; Pons, H., Ed.; Scribner: New York, NY, USA, 2010; ISBN 8535920064. [Google Scholar]
- Gheghiani, L.; Wang, L.; Zhang, Y.; Moore, X.T.R.; Zhang, J.; Smith, S.C.; Tian, Y.; Wang, L.; Turner, K.; Jackson-Cook, C.K.; et al. PLK1 Induces Chromosomal Instability and Overrides Cell-Cycle Checkpoints to Drive Tumorigenesis. Cancer Res. 2021, 81, 1293–1307. [Google Scholar] [CrossRef]
- Jeong, S.B.; Im, J.H.; Yoon, J.-H.; Bui, Q.T.; Lim, S.C.; Song, J.M.; Shim, Y.; Yun, J.; Hong, J.; Kang, K.W. Essential Role of Polo-like Kinase 1 (Plk1) Oncogene in Tumor Growth and Metastasis of Tamoxifen-Resistant Breast Cancer. Mol. Cancer Ther. 2018, 17, 825–837. [Google Scholar] [CrossRef]
- de Cárcer, G.; Venkateswaran, S.V.; Salgueiro, L.; El Bakkali, A.; Somogyi, K.; Rowald, K.; Montañés, P.; Sanclemente, M.; Escobar, B.; de Martino, A.; et al. Plk1 Overexpression Induces Chromosomal Instability and Suppresses Tumor Development. Nat. Commun. 2018, 9, 3012. [Google Scholar] [CrossRef]
- Strebhardt, K.; Raab, M.; Sanhaji, M. The Role of PLK1 in Cancer Exhibiting Chromosomal Instability. Mol. Cell. Oncol. 2018, 5, e1485539. [Google Scholar] [CrossRef]
- Raab, M.; Sanhaji, M.; Matthess, Y.; Hörlin, A.; Lorenz, I.; Dötsch, C.; Habbe, N.; Waidmann, O.; Kurunci-Csacsko, E.; Firestein, R.; et al. PLK1 Has Tumor-Suppressive Potential in APC-Truncated Colon Cancer Cells. Nat. Commun. 2018, 9, 1106. [Google Scholar] [CrossRef]
- Lindqvist, A.; van Zon, W.; Rosenthal, C.K.; Wolthuis, R.M.F. Cyclin B1–Cdk1 Activation Continues after Centrosome Separation to Control Mitotic Progression. PLoS Biol. 2007, 5, e123. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, G.; Gao, Z.; Li, S.; Li, Z.; Bi, J.; Liu, X.; Li, Z.; Kong, C. Comprehensive Analysis of Differentially Expressed Genes Associated with PLK1 in Bladder Cancer. BMC Cancer 2017, 17, 861. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Initial Concentration Value (µM) Per Simulation Scenario (Sce) | |||||
|---|---|---|---|---|---|
| Gene | IC | Sce1 | Sce2 | Sce3 | Sce4 |
| cdk1 | 1.0 | 0.01 | 0.1 | 10 | 100 |
| Gene | IC | Sce5 | Sce6 | Sce7 | Sce8 |
| plk1 | 1.0 | 0.01 | 0.1 | 10 | 100 |
| Gene | IC | Sce9 | Sce10 | Sce11 | Sce12 |
| cohesin | 1.0 | 0.01 | 0.1 | 10 | 100 |
| Gene | IC | Sce13 | Sce14 | Sce15 | Sce16 |
| cdk1, plk1, cohesin | 1.0 | 0.01 | 0.1 | 10 | 100 |
| Category | Biological Process | Biological Species | Biological Interactions |
|---|---|---|---|
| Diseases | FOXM1 Signaling in Prostate Cancer | 13 | 17 |
| Cell processes | G2/M Phase Transition | 47 | 65 |
| Cell processes | Metaphase/Anaphase Phase Transition | 15 | 22 |
| Cell processes | Chromosome Condensation | 22 | 36 |
| Pathological processes | DREAM Complex and FOXM1/MYBL2 Promote Cell Cycle Progression in Cancer | 36 | 56 |
| Cell processes | Sister Chromatid Cohesion | 27 | 41 |
| Cell processes | Spindle Assembly | 29 | 45 |
| Cell processes | Kinetochore Assembly | 49 | 73 |
| Cell processes | Cell Cycle Overview | 140 | 213 |
| Total | 9 Biological processes | 378 | 568 |
| Tool/Platform | Data Consolidation (Curated or Integrative Sources) | Network Representation | Simulation Environment | Scenario Building (e.g., PLK1 Overexpression, Knockout) | Relevance to PLK1 Regulation/Genomic Instability | Limitations |
|---|---|---|---|---|---|---|
| PLK1 Model (this work) | High—Manual curation of PLK1-specific regulation from primary literature and reviews | Mechanistic, reaction-based (SBGN) | Yes—ODE simulation in CellDesigner | Yes—Edits to concentrations, rates, logic | Focused on PLK1 in the context of genomic instability | Requires parameter estimation; limited to encoded components |
| Reactome Pathway Browser | High—Expert-curated, literature-based | Mechanistic, biochemical steps | No—Static visualization only | Limited—Overlay gene expression or mutations | Contains mitotic roles of PLK1; curated context for mitosis | No dynamic behavior or causal testing |
| Cytoscape + ReactomeFIViz | Medium—Uses imported pathways or user-generated data | Topological; interaction & influence networks | No native simulation | Partial—Node/edge manipulation, expression mapping | Visualizes PLK1’s neighborhood; combines pathway sources | Not mechanistic; lacks temporal modeling |
| Pathway Commons | Very high—Aggregates >20 DBs (Reactome, Panther, HPRD) | Interaction-based (BioPAX format) | No—Data portal only | No—Requires export to SBML or Cytoscape | Useful for mining extended PLK1 network | Static; not simulation-ready without processing |
| IPA (Ingenuity Pathway Analysis) | High—Curated and predicted interactions from multi-omics data | Causal and regulatory network inference | No—Statistical inference only | Limited—Predicts activation/inhibition based on input | Predicts PLK1 up/downstream effects from expression data | Proprietary; no kinetic or mechanistic modeling |
| GSEA (Gene Set Enrichment Analysis) | None—Depends on user-defined gene sets | Not network-based; gene list comparison | No—Statistical enrichment tool | No—Identifies gene set activity in datasets | Can evaluate whether PLK1 targets or affected genes are enriched | No network structure or dynamic context |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suescum-Holguín, J.F.; Clavijo-Buriticá, D.C.; Carrillo-Borda, E.F.; Quimbaya, M.A. The plk1 Gene Regulatory Network Modeling Identifies Three Circuits for plk1-mediated Genomic Instability Leading to Neoplastic Transformation. Life 2025, 15, 799. https://doi.org/10.3390/life15050799
Suescum-Holguín JF, Clavijo-Buriticá DC, Carrillo-Borda EF, Quimbaya MA. The plk1 Gene Regulatory Network Modeling Identifies Three Circuits for plk1-mediated Genomic Instability Leading to Neoplastic Transformation. Life. 2025; 15(5):799. https://doi.org/10.3390/life15050799
Chicago/Turabian StyleSuescum-Holguín, Jeison F., Diana Carolina Clavijo-Buriticá, Edward Fabian Carrillo-Borda, and Mauricio Alberto Quimbaya. 2025. "The plk1 Gene Regulatory Network Modeling Identifies Three Circuits for plk1-mediated Genomic Instability Leading to Neoplastic Transformation" Life 15, no. 5: 799. https://doi.org/10.3390/life15050799
APA StyleSuescum-Holguín, J. F., Clavijo-Buriticá, D. C., Carrillo-Borda, E. F., & Quimbaya, M. A. (2025). The plk1 Gene Regulatory Network Modeling Identifies Three Circuits for plk1-mediated Genomic Instability Leading to Neoplastic Transformation. Life, 15(5), 799. https://doi.org/10.3390/life15050799