Abstract
Quinolone resistance has been largely related to the presence of specific point mutations in chromosomal targets, with an accessory role of impaired uptake and enhanced pump-out. Meanwhile the relevance of transferable mechanisms of resistance able to protect the target of pump-out or inactivate quinolones has been increasingly reported since 1998. Nevertheless, bacteria have other strategies and mechanisms allowing them to survive and even proliferate in the presence of quinolones, which might be qualified as resistance or resilience mechanisms. These include decreasing levels of quinolone target production, transient amoeba protection, benthonic lifestyle, nutrient-independent slow growth, activation of stringent response, inactivation or degradation of quinolones as well as apparently unrelated or forgotten chromosomal mutations. These mechanisms have been largely overlooked, either because of the use of classical approaches to antibiotic resistance determination or due to the low increase in final minimum inhibitory concentration levels. This article is devoted to a review of a series of these mechanisms.
1. Introduction
Quinolones are fully synthetic molecules which may be classified within four different main subgroups as regards the position of nitrogen atoms in the molecule [1] (Figure 1).
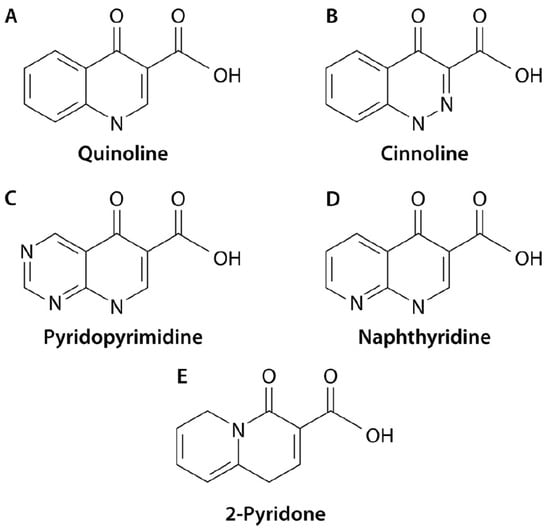
Figure 1.
Subclasses of quinolones. At present, 4 subclasses of quinolones (A) quinolines; (B) cinnolines; (C) Pyridopyrimidine; (D) Naphthyridine have been described; atom numeration is described following the quinolines (A) structure, with position 1 risen in the nitrogen atom. The figure also illustrates the structure of 2-Pyridons (E), because they are structurally related, but at present, none have been introduced in human or veterinary practice. Reproduced from reference [1], with permission from the American Society of Microbiology.
These antimicrobial agents are able to inhibit DNA synthesis, transcription and cell division through their interaction with Type II Topoisomerases (i.e., DNA Gyrase and Topoisomerase IV) [2,3]. Thus, quinolones bind to the complex DNA-Topoisomerase blocking the replication forks [4], with these findings leading to bacterial kill through different events [3].
Despite the presence of previous reports [5,6,7], the official history of quinolones as antimicrobial agents is considered to have started in 1962 with the description of nalidixic acid [8]. Although several studies reported the use of nalidixic acid for the treatment of infections such as diarrhea or dysentery [9,10,11], nalidixic acid as well as ancient quinolone therapeutic uses were almost limited to fighting urinary tract infections [12]. In the following years, the family of quinolones grew exponentially, and with the incorporation of a fluorine atom in position 6, their uses expanded from human urinary infections to a great variety of uses, including those in human therapeutics, such as dermic, gastrointestinal, ocular, osteoarticular, respiratory, skin and soft-tissue and systemic infections, among others [13,14,15,16,17,18,19,20,21]. In the 2000s, a series of quinolones lacking a fluorine atom at position 6 were synthesized, with several of them, such as ozenoxacin and nemonoxacin introduced in clinical practice [22,23]. Quinolones have also been used in the treatment of sick animals or for prophylactic purposes [24,25,26,27], in addition to use as livestock growth promoters, despite the potential side effects on cartilage [28]. Furthermore, apart from therapeutic uses related to bacterial pathogens, the use of quinolones against fungi, viruses and parasites has also been explored [29,30,31,32].
These expansive uses have led to an increasing isolation of quinolone-resistant microorganisms, resulting in a worldwide epidemic of quinolone resistance from the mid-1990s onward [1]. Subsequently, quinolone resistance levels, selection, mechanisms, and dispersion pathways have been largely and increasingly studied since the 1960s [21,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47].
In the late 1970s, the implication of GyrA in the development of quinolone resistance was observed [48]. The relevant role of punctual mutations in quinolone targets encoding genes (gyrA and gyrB, encoding DNA-Gyrase, and parC and parE, encoding Topoisomerase IV), able to avoid or limit the ability of quinolones to interact with their targets, was clearly established in the 1980s and 1990s and was subsequently considered a universal pathway of the development of quinolone resistance in both Gram-negative or Gram-positive bacteria, including particular microorganisms, such as Mycobacterium spp. [21,34,46,49,50,51,52,53,54,55,56]. Furthermore, it was described that several microorganisms presenting wild-type-specific amino acids in critical positions of GyrA and/or ParC showed natural resistance to quinolones [57,58]. In parallel, the role of efflux pumps and impaired permeability was also demonstrated, being considered as responsible for the basal minimal inhibitory concentration (MIC) levels of quinolones and an accessory mechanism of the development of quinolone resistance, when specific mutations lead to down-expression of porins or overexpression of efflux pumps [45,55,59,60,61,62]. The role of alterations in quinolone uptake has been considered as the most relevant route of quinolone resistance acquisition only in specific microorganisms, such as Stenotrophomonas maltophilia [63,64], while target mutations seem to play a residual role [64,65,66].
Despite the publication of previous unconfirmed or misinterpreted studies [1,37,67,68,69,70,71], the first undoubtable transferable mechanism of quinolone resistance (TMQR) was described in 1998 [39], with different TMQRs being reported in the following years and classified into three main generic mechanisms, quinolone target protection (qnr), quinolone inactivation/modification (aac(6′)Ib-cr and crpP) and quinolone pump-out (qepA, oqxAB and others), overall accounting for more than 12 different genes and hundreds of allelic variants [1,72,73,74,75,76]. Of note, the role of CrpP as a mechanism of quinolone resistance has recently been questioned, and it has been proposed to not consider it among TMQRs [77].
These well-established and largely studied mechanisms of quinolone resistance are referred to as “canonical mechanisms” in the present text. The vast majority of studies on quinolone resistance mechanisms, as well as specific thematic reviews, are focused on what was mentioned above [1,42,45,46,49,55,60], while the approaches on acquisition and dispersion of quinolone resistance mostly describe the selection of target mutations as well as the role of mobile (or mobilizable) genetic elements, such as plasmids, transposons, genomic islands, and integrons [1,44,76,78,79,80,81,82]. Nonetheless, in addition to these well-established mechanisms of resistance to quinolones, several less frequent mechanisms as well as non-classical routes of acquisition of quinolone resistance or quinolone tolerance have been reported in the literature, but they seem to have fallen by the wayside and are not considered (Table 1). The present review is focused on these less-studied mechanisms.

Table 1.
Main unconsidered mechanisms of resistance/resilience to quinolones.
From early studies focused on the analysis of the mechanisms of resistance to quinolones, the presence of unexplained quinolone-resistant bacteria or microorganisms with discordances between quinolone resistance levels and reported mechanisms of quinolone resistance has been frequent. While the acquisition of new knowledge has allowed sequential identification of new mechanisms of quinolone resistance in these unexplained quinolone-resistant strains, which often become a frequent motif of further studies and are classified as new canonical mechanisms of quinolone resistance [1,55], a series of minority or mostly neglected quinolone resistance mechanisms are present in the literature. In addition, although they are not strictly resistance mechanisms, several pathways also drive to increased survival in the presence of quinolones.
In the next sections, a series of pathways (Table 1) leading to increased bacterial survival in the presence of quinolones are presented in alphabetical order; therefore, no conclusion about higher or lower relevance may be inferred for the order in which they are exposed within the text.
2. Altered Production of Quinolone Targets
While target overexpression has been considered a mechanism of resistance to different antibacterial agents because the quantity of the target surpasses the inhibitory capacity of antibiotics, with overexpression of β-lactam inhibitor-sensitive TEM-1 leading to intermediate-to-full resistance to β-lactam plus β-lactam inhibitor combinations being a classic example [83,84,85], quinolones show an opposite phenomenon: lower levels of quinolone targets lead to higher levels of resistance. This phenomenon was first described in an in vitro selected quinolone-resistant Staphylococcus aureus in 2003 [86]. Using premafloxacin, Ince et al. selected an in vitro mutant exhibiting a 4-fold increase in the MIC levels of premafloxacin (from 0.004–0.008 mg/L to 0.016–0.032 mg/L) and ciprofloxacin (from 0.125–0.25 mg/L to 1 mg/L), with no alterations in either the MICs of ethidium bromide (suggesting no overexpression of efflux pumps) or amino acid substitution in GyrA, GrlA (ParC), GyrB or GrlB (ParE), and with a single nucleotide change G→A in the grlBA operon promoter at position -13, just downstream of a presumptive Shine–Dalgarno sequence [86]. The role of this alteration in the development of quinolone resistance was demonstrated by introducing the mutation in a wild-type S. aureus, confirming the increase in ciprofloxacin and premafloxacin MIC levels [86]. When the authors determined the physiological effects of this alteration, they observed a 3-fold reduction in the overall expression levels of grlA and grlB [86]. Nevertheless, no apparent effect on bacterial growth was observed, suggesting the presence of compensatory mutations within the chromosome [86].
The presence of alterations in the gyrA promoter region impacting sensitivity and supercoiling regulation and altering final expression levels has also been described [87], but no data on the effect on final quinolone levels are available in the literature. Of note, quinolones relax DNA by inhibiting supercoiling [88], which, in turn, results in increased expression of gyrA [89].
In relation to the above, potential alterations in the level of expression of genes encoding quinolone targets have been proposed as one of the mechanisms involved in the increased levels of resistance of the so-called “Small Colony Variants” (SCVs) [90] (see Section 8: Unconsidered Chromosomal Mutations).
While to the best of my knowledge no description has been made, it could be hypothesized that mutations at the initial codon of genes encoding quinolone targets leading to modifications from ATG to, for instance, GTG or TTG, may also affect transcription efficiency, as these initial codons are less efficient [91], thereby resulting in lower protein levels and subsequent similar effects on the final quinolone susceptibility levels.
3. Amoeba Protection
Free-living amoebae are potential pathogenic environmental microorganisms which live in water or soil environments predating bacteria communities [92]. Nonetheless, two singularities have been observed in the amoeba/bacteria relationships. These are the presence of a series of endosymbiotic bacteria, which in several cases seem to be unable to live in an amoeba-independent form [92], as well as the ability of several predated bacteria (including relevant pathogens, such as Legionella pneumophila or Pseudomonas aeruginosa) to survive and proliferate inside amoebae according to transient adaptations [93,94]. Thus, the amoeba environment might act both as an additional barrier for quinolones (as for other antimicrobial agents) to access bacteria, because of the need of antibacterial agents to cross the amoeba cell membrane, as well as directly influencing the bacterial transcriptome. Furthermore, the ability of bacteria to exchange genetic material inside amoebae has been demonstrated [95].
In addition to the effects on quinolone resistance, the presence of microorganisms inside amoebae may have additional effects on human (and animal) health, acting as a reservoir of pathogens and becoming a neglected route for bacterial infections [96].
It is necessary to highlight that, in the present text, due to the scarcity of data on amoebae, a series of assumptions based on macrophages data have been formed.
3.1. Barrier Effect?
While quinolones may penetrate inside eukaryotic cells, the presence of quinolones within amoeba cytoplasm may be affected by the need to cross the border membrane and the presence of amoeba efflux pumps, with possible interspecific and/or intraspecific differences. While this finding seems intuitive, no article referring to the accumulation of quinolones inside amoebae has been found in the literature. This lack of data may be related to the good intracellular penetration of quinolones, which has been observed in macrophages, among other eukaryotic cell types [97,98], and with the observed anti-amoeba activity of fluoroquinolones [99]. Nonetheless, the presence of eukaryotic efflux pumps able to pump out quinolones has been highlighted [100]. Furthermore, the contribution of eukaryotic efflux pumps to intracellular bacteria survival in the presence of fluoroquinolones has been described [101]. In this regard, while no data on quinolone efflux in amoebae have been found in the literature, the abovementioned quinolone extrusion mediated by efflux pumps in eukaryotic cells suggests that the presence of similar mechanisms in amoebae is highly probable [102]. Thus, in the absence of specific data, a barrier effect, which might be amoeba-species- or amoeba-strain-dependent and affect quinolones in a selective manner, cannot be discarded.
A special situation is amoeba encystment. Encystment is a defensive strategy in which the amoeba remains inactive, in a resting form, allowing amoeba survival in special adverse situations, such as the presence of toxins, including several produced by microorganisms such as P. aeruginosa, starvation, dehydration or osmotic stress, among others [103,104]. Amoeba cysts possess an external layer, which varies from species to species and has been considered as a reason to explain the higher resistance to common treatments exhibited by amoeba cysts in comparison with trophozoites [103]. Of note, it has been observed that different amoeba-resident bacteria, such as Mycobacterium avium, may surpass the cyst period, thereby also remaining under the protective umbrella of the cyst layer during the cyst period [105]. While ciprofloxacin has shown activity against amoeba cysts [99], it is likely that the cyst layer contributes to hindering its access to the amoeba cytoplasm, especially in environments with low quinolone concentrations.
3.2. Effects on Bacterial Transcriptome
In 1995, when analyzing the effect of L. pneumophila grown inside amoebae and macrophages on the final bacterial MIC and its survival levels in the presence of antibiotics, Barker et al. observed that in L. pneumophila released from amoeba, the MIC of ciprofloxacin did not increase with respect to in vitro cultured L. pneumophila (MIC = 0.06 µg/mL). Nevertheless, when exposed to 1 µg/mL of ciprofloxacin, survival rates increased 1000-fold [106]. Of note, after 48 h of in vitro growth, the survival ability of amoeba-grown L. pneumophila was lost [106]. No analysis of the mechanisms of ciprofloxacin survival acquired was performed, but the authors observed deep morphological changes including motility affectation [106]. Although no analysis of quinolone targets was performed, all these findings strongly suggest a transient adaptative response and not a true quinolone-resistant mutant selection. Similarly, studies analyzing the intracellular activity of ancient quinolones against Francisella noatunensis subsp. noatunensis or Burkholderia mallei have shown the inability of flumequine to inhibit F. noatunensis replication, despite high flumequine intake within macrophages [98], and the inability of flumequine to completely clear B. mallei irrespective of patient improvement [97].
All these findings agree with the described effect of an intracellular lifestyle resulting in an altered bacterial transcriptome, which may lead to a different morphology, a modified duplication time or an altered surface [107,108]. Of note, different studies have shown that the altered expression levels of genes apparently unrelated to the development of quinolone resistance may result in increased MIC levels to quinolones with a few genes increasing levels of quinolone efflux or impairing quinolone intake (see Section 8: Unconsidered Chromosomal Mutations).
Furthermore, the expression of specific genes seems to be relevant for the ability of bacteria to survive and multiply within amoebae [109]. Among these genes, it has been observed that the lack of functionality of cmeB impairs the survival and replication of amoeba-internalized Campylobacter jejuni [109]. CmeABC is an RND efflux pump present in the Campylobacter spp. genome, which is able to efficiently extrude quinolones, among other antibacterial agents, with reports showing that cmeABC overexpression promotes the selection of fluoroquinolone-resistant C. jejuni isolates [110,111]. Therefore, amoeba-fed resistance as well as active efflux of fluoroquinolones are co-selected characteristics of Campylobacter.
4. Bacterial Benthonic Lifestyle
Since the dawn of microbiology, antimicrobial susceptibility has been established in metabolically active microorganisms with a planktonic lifestyle. When susceptibility/resistance to any antimicrobial agent is reported in a clinical or veterinarian setting, this involves these planktonic microorganisms. Nonetheless, other scenarios may be present, including benthonic communities, almost imperceptible grown or real quiescent states (see Section 6.1: Sporulation and Quiescence).
Bacteria may organize sessile communities imbibed within a matrix conformed by exopolysaccharides, eDNA, proteins and other compounds which vary among species; furthermore, intraspecies variations in matrix composition have been observed, suggesting clonal/strain specificity [112,113]. This matrix hinders the action of several antibacterial agents by blocking their access to bacteria. This finding has been directly associated with enhanced tolerability/resistance to a series of antibacterial agents including tobramycin and vancomycin, among others [114,115,116]. These communities are the so-called biofilms, which are of special concern with respect to medical device contamination and chronic or prosthesis infections. Thus, while the surface-fixed and non-motile nature of biofilms often underlies prothesis- or device-related infections [117,118,119], biofilms are not immutable structures, and different stages of biofilm maturity and different pathways of bacterial release have been described [116,120,121], with this release of bacteria mainly contributing to systemic dissemination of infections as well as infection chronicity. In recent years, there has been increasing attention to bacterial benthonic communities, with a continuous growth of basic studies focused on the characterization of these communities. However, only a scarce number of studies have been focused on the impact of bacterial biofilms on patient outcomes and their influence on antibiotic treatment failure, strongly suggesting a chronic forgotten and disconsideration of their real relevance in clinical settings [117].
Regarding quinolones, minimal biofilm inhibitory concentrations (MBICs) and minimal biofilm eradication concentrations (MBECs) may exceed planktonic MICs and minimal bactericidal concentrations (MBCs), respectively, by >256-fold [122,123]. The routes by which bacterial biofilms enhance resistance to quinolones (or other unrelated antibacterial agents) are diverse and multifactorial and are mainly socially and intimately interrelated [114,124]. Furthermore, in the case of quinolones, the levels of resistance conferred by these mechanisms are additive to those related to the presence of the abovementioned canonical chromosomal and transferable mechanisms of resistance.
4.1. Biofilm Access
Classically, as mentioned above, the main reason for the enhanced levels of resistance to members of this antibacterial agent family has been considered to be impaired access of quinolones to biofilm, related to the difficulty in passing through the imbibing matrix. Notwithstanding, this issue is controversial. Thus, while in several studies the extracellular matrix does not seem to significantly affect the access of quinolones, such as ciprofloxacin, which are able to penetrate the biofilm and achieve internal biofilm concentrations that are higher than the usual planktonic MICs [125], other studies have described that the penetration of ciprofloxacin within biofilm was hindered [126]. In this scenario, it has been suggested that these differences might be related to factors including bacterial species, growth conditions and biofilm thickness [114].
In this sense, the final levels of quinolone resistance/tolerance in bacterial biofilm communities are related to the specific composition of the imbibing matrix. Thus, the presence of the Psl exopolysaccharide in the matrix of P. aeruginosa biofilms has been related to enhanced ciprofloxacin resistance [127]. To the contrary, the presence of Pel, another exopolysaccharide, which may be present in the P. aeruginosa biofilm matrix, plays no role in ciprofloxacin resistance/tolerance [128]. The mechanisms by which Psl affects ciprofloxacin activity remain unclear. Despite the charge-neutral nature of Psl, the eradication of biofilms when cationic antibacterial agents (i.e., polymyxin B, and tobramycin) were combined with NaCl allowed Billings et al. to propose, as a partial explanation for this phenomenon, that the Psl matrix can sequestrate these positively charged antibacterial agents by electrostatic interactions. However, this explanation was not extended to ciprofloxacin (negatively charged) as biofilms were not disaggregate when ciprofloxacin was combined with NaCl [127].
4.2. Altered Bacterial Metabolic Activity
The metabolic activity of bacteria living in benthonic communities is different from that of bacteria in planktonic status. Thus, reduced metabolic activity and differences in gene expression have been described [129,130].
A reduction in metabolic activity has been described as being a quinolone resistance/tolerance mechanism [131]. These antimicrobial agents need DNA Gyrase and Topoisomerase IV, which are responsible for DNA relaxing and duplication, to be active to exert their action (see Section 6.1: Sporulation and Quiescence).
The decrease in metabolic activity of bacteria imbibed within biofilms is related to different factors including oxygen concentrations and nutrient availability, which are greater or lesser according to whether they are in located in external biofilm surfaces or the most internal strata [132]. In biofilms, the most extreme reduction in metabolic activity is that related to bacterial quiescent status, with most dormant cells being deep within the biofilm (see Section 6.1: Sporulation and Quiescence). Of note, lower oxygen concentrations have also been related to lower levels of formation of reactive oxygen species (ROS), hypothesizing that lower ROS levels affect the bactericidal activity of quinolones [133].
In these scenarios, different bacterial systems are activated, including the so-called stringent response, or toxin/antitoxin modules (T/A) (see Section 6: Stringent Response and Toxin/Antitoxin Systems) [134].
In addition, differences in the transcriptomes of bacteria living in planktonic and benthonic communities have been described [129]. This finding results in differences in bacterial functionalities, which might also lead to alterations in bacterial resistance to antibiotics. In fact, different studies on quinolone-resistant mutants have reported the selection of apparently unrelated mutations as well as modifications in the transcriptome leading to increases in the final MICs to quinolones, despite the absence of established mechanisms of quinolone resistance (see Section 8: Unconsidered Chromosomal Mutations).
5. Nutrient-Independent Slow Growth
In 1996, it was shown that the presence of the plasmid pKM101 (belonging to the IncN incompatibility group) used in the de Ames test confers both a slow growth pattern and, in the presence of ciprofloxacin, a parallel increase in the survival of Escherichia coli growing on minimal media [135]. This slow-growth phenotype was related to a region of 2.2 kb including the korB, traL, korA and traM genes. The authors further hypothesized the role of the region surrounded by the korB and korA genes. The increased survival may be related to the mode of action of quinolones, which require the presence of active biological processes involving Topoisomerase activity [136,137]. Nonetheless, no data of the exact reasons for the slow growth associated with this plasmid have been provided, and to our knowledge, no further studies have been carried out.
6. Stringent Response and Toxin/Antitoxin Systems
Stringent response and the so-called toxin/antitoxin systems are bacterial systems involved in bacterial stress response [138,139].
Stringent response is a bacterial mechanism involved in a series of adaptations in response to different stress situations, such as nutrient or iron deprivation, oxidative stress, or others [140,141]. Meanwhile, toxin/antitoxin systems encode both a toxin able to interfere with different metabolic pathways leading to cell death or arresting cell growth and an antitoxin that inhibits the action of the toxin component [139]. To date, up to 8 T/A systems have been described, classified based on the mechanism of action of antitoxin component [141].
Different proposals about how these metabolic routes drive towards dormant and quiescent bacteria have been made, including interdependent and independent routes of stringent response and T/A systems (Figure 2). Thus, while several authors propose that stringent response and T/A systems are two independent routes, it has also been proposed that the action of stringent response results in increased production of the Lon protein which, in turn, proceeds with the degradation of the antitoxin, allowing the toxin to exert its action, or the inverse scenario in which the inactivation of the antitoxin starts a series of metabolic processes leading to the overproduction of RelA, resulting in the activation of stringent response [138,142].
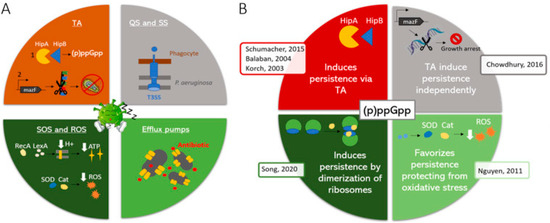
Figure 2.
Proposed model of independent action of stringent and toxin/antitoxin systems. TA: Toxin/antitoxin system; QS: Quorum sensing; SS: Secretion systems; ROS. Reactive Oxygen Species; T3SS: Type 3 secretion systems. (A) Molecular mechanisms underlying bacterial persistence. (B) Different models explaining the involvement of (p)ppGpp in persistence and representative publications (see reference [135]). (B) red: T/A systems are activated by (p)ppGpp. grey: T/A systems induce quiescence independently; light green: (p)ppGpp protects against oxidative stress, favoring the development of persister cells; dark green: (p)ppGpp induces dimerization of ribosomes, which subsequently lead to persistence. References presents in the figure may be found at [138]. Reproduced from reference [138], with the permission of Elsevier.
Stringent response is related to the action of a series of genes, including the relA, spoT and dksA genes [140,141]. The induction of stringent response results in the synthesis of the alarmones ppGpp or pppGpp (collectively referred to as (p)ppGpp) [143]. The impact of this mechanism on survival has been highlighted in studies in which mutants with impaired (p)ppGpp synthesis showed decreased survival in the presence of quinolones and lower MBC/MIC compared with parental isolates, while those with impaired hydrolysis of (p)ppGpp showed an opposite scenario [144].
It has been confirmed that a series of toxins from T/A systems, such as ParE toxin (not to be confounded with ParE, a subunit of Topoisomerase IV) or TisB from ParDE or TisAB-IstR-1, are released during ciprofloxacin exposure, favoring bacterial survival [145,146]. In the case of TisB, this finding is directly related to the presence of a Lex-box in its promoter region, which is activated by the SOS system that, in turn, is activated by DNA lesions related to the action of the quinolone [145]. On the other hand, increased levels of Lon have been claimed as the reason for the release of ParE [147]. Of note, there is currently controversy about the real role of T/A systems in the development of dormant cells [148].
6.1. Sporulation and Quiescence
The abovementioned mechanisms led to the development of dormant bacteria (Figure 3). Sporulation and quiescence are conceptually related phenomena leading to long-term inactive or almost inactive microorganisms. Two notes: bacteria may remain viable for years in spore or quiescent states [149], and it should be considered that antibiotic susceptibility is restored when they are reactivated.
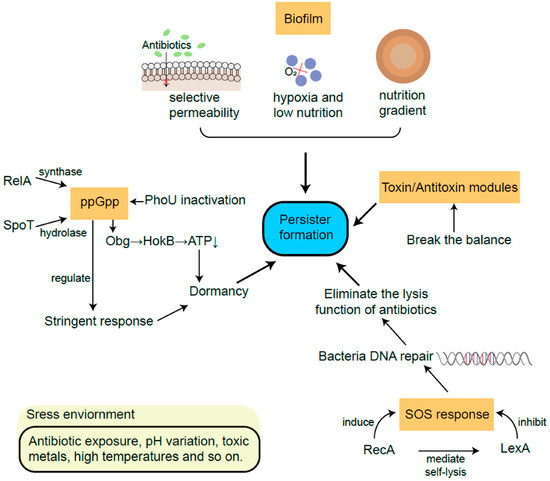
Figure 3.
Models of development of persister cells. Note that while all pathways are represented as independent routes, different interactions among them may be present. Reproduced from reference [142], published under a Creative Commons license.
Sporulation is a bacterial mechanism of bacterial survival in extreme conditions in which the bacteria suffer a series of morphological alterations and enter into a truly dormant state, in which no metabolic process takes place until environmental conditions are more favorable [149]. Meanwhile, quiescence is another bacterial response to environmental stress. Quiescent microorganisms are in a viable non-replicating state, but in contrast to spores, quiescent bacteria display a basal metabolic process with no morphological alterations [149]. As mentioned previously, the quiescent state is also present within biofilm communities in bacteria living in the most internal biofilm layers [114].
DNA Gyrase and Topoisomerase IV, both classified as Type II Topoisomerases, are actively involved in different DNA processes, with the introduction of negative supercoiling and DNA decatenation of replication products as the most relevant and well established [55,150]. The quinolones need Topoisomerase activity to interact with its targets in order to block the DNA replication forks, generating cleaved DNA-Type II Topoisomerase complexes, and subsequently inducing rapid bacterial death through different protein synthesis-dependent or -independent pathways, with the SOS system possibly being involved in slow bacterial death [136,137].
In agreement, a series of studies have reported negative relations between bacteriostatic ribosomal-targeting antibacterial agents and different quinolones, ranging from indifference tending to antagonism, to declared antagonism leading to suppressive effects [131,136,151]. A related and paradoxical effect has been described in which bacterial survival at high nalidixic acid concentrations was higher than when the concentrations did not reach these values. This was explained by the bactericidal action of nalidixic acid being modified to bacteriostatic action, inducing a metabolic block of bacterial activity leading to a bacterial quiescent state [131]. Crumplin et al. [131] also showed that after exposure to >200 mg/L of nalidixic acid, the surviving E. coli K16 did not show higher nalidixic acid MIC than those at baseline. In accordance with this scenario, full bacterial dormancy in the sporulate and the almost inactive quiescent states allow bacteria to remain unaffected by the presence of quinolones, displaying higher resistance levels and increasing bacterial survival in the presence of these antimicrobial agents and, in summary, escaping the action of quinolones irrespective of their MIC to quinolones when reactivated.
7. Quinolone Inactivation/Modification
While not included in the present study because both AAC(6′)Ib-cr and CrpP (despite the aforementioned controversy) qualify as canonical mechanisms of quinolone resistance, the presence of numerous variants, and thus possible different spectra, affecting different quinolones or level of activity should be highlighted [1,76].
In addition to the largely described ability of AAC(6′)Ib-cr to inactivate several quinolones through acetylation [73,152], and the recently described CrpP proposed to be able to phosphorylate several quinolones, such as ciprofloxacin [153], a series of poorly described bacterial mechanisms of inactivation of quinolones are present in different microorganisms [1].
7.1. Fungi
Thus, in the 1990s, the ability of several wood-rotting fungi, including Irpex lacteus, Gloeophyllum striatum or Phanerochaete chrysosporium, among others, to degrade enrofloxacin was first described [154]. Thereafter, the ability of other fungi, such as Clitocybe odora, Coriolopsis gallica, Cyathus stercoreus, Irpex lacteus, Xylaria longipes or others, to degrade or modify quinolones has been largely described [155,156,157,158]. Of note, the resulting metabolites may retain a degree of antibacterial activity, which varies between processed quinolones and fungi species [156].
The fungi degradation/modification routes of quinolones are diverse. Thus, working with Gloeophyllum striatum, Wetzstein et al. proposed four possible degradation routes: oxidative decarboxylation, defluorination, hydroxylation at C-8 or oxidation of the amino moiety (Figure 4) [158]. Data regarding the exact enzymes involved in these degradation/modification routes are scarce. Nevertheless, the role of enzymes such as cytochrome P450, or those possessing laccase-like and peroxidase-like activity, has been explored [155,156,159]; of note, the two latter types of enzymes are considered ligninolytic enzymes [160], in agreement with the apparent facility of wood-rooting fungi to degrade or modify quinolone. Along this line, a recent study analyzing the ability of Coriolopsis gallica to degrade levofloxacin suggested that enzymes with laccase-like and peroxidase-like activity may play a role due to their prevalence in fungi secretome [155]. Similarly, the chloroperoxidase of Leptoxyphium fumago (formerly Caldariomyces fumago) is also able to degrade norfloxacin [161]. On the other side, other authors have proposed that several peroxidases, such as for instance manganese peroxidases, play no role in the detoxification of quinolones, limiting this role to cytochrome P450 and laccases [159,162]. Regarding this latter issue, as mentioned previously, it should be considered that different amino acid sequences may result in different activity.
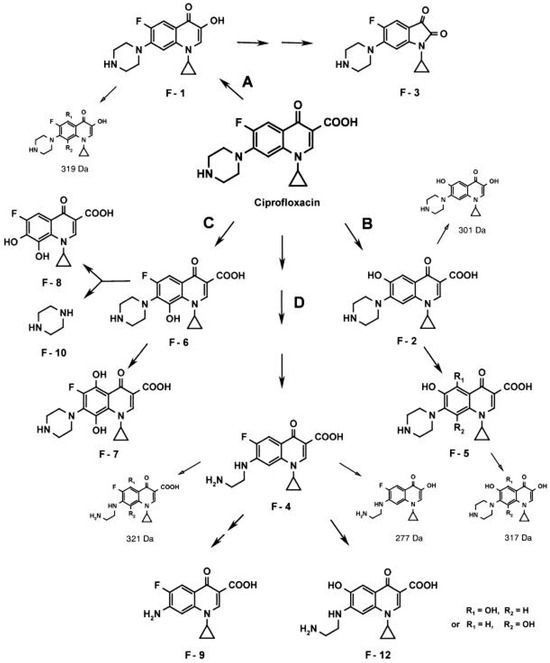
Figure 4.
Proposed network of metabolites generated from ciprofloxacin by the brown rot fungus Gloeophyllum striatum. (A) Oxidative decarboxylation. (B) Defluorination. (C) Hydroxylation at C-8. (D) Oxidation of the amino moiety. Trace metabolites, detected only by HPLC–MS are represented at a reduced size. Reproduced from reference [158], with the permission of Elsevier.
7.2. Bacteria
The ability of different bacteria to degrade or inactivate quinolones by non-canonical mechanisms of resistance has also been observed and may undoubtedly be considered as one of the factors underlying species-specific intrinsic levels of resistance to quinolones. Among the microorganisms with a demonstrated ability to degrade or inactivate quinolones, environmental bacteria, such as Labrys portucalensis, members of the genera Mycobacteria or Microbacterium, or extremofiles like members of the genus Thermus, among others, may be found [163,164,165,166]. Furthermore, the ability of relevant human pathogens to degrade or inactivate quinolones not related to canonical mechanisms of quinolones modification (i.e., AAC(6′)Ib-cr, CrpP) has also been reported, with descriptions of danofloxacin modifications by P. aeruginosa [167].
As with fungi, there are a variety of quinolone-modifying routes. For instance, Kim et al. described four different norfloxacin modification routes in Microbacterium: hydroxylation, oxidative defluorination, desethylation and N-acetylation [165]. In 2013, the same group described that norfloxacin N-acetylation in Microbacterium was related to the action of a glutamine synthetase encoded in the gene glnA (GenBank access: JX901058), confirming this finding by cloning and expressing this gene in E. coli, and observing both the ability of the enzyme cloned in E. coli to modify norfloxacin and its effect on ciprofloxacin susceptibility [168] (Figure 5).
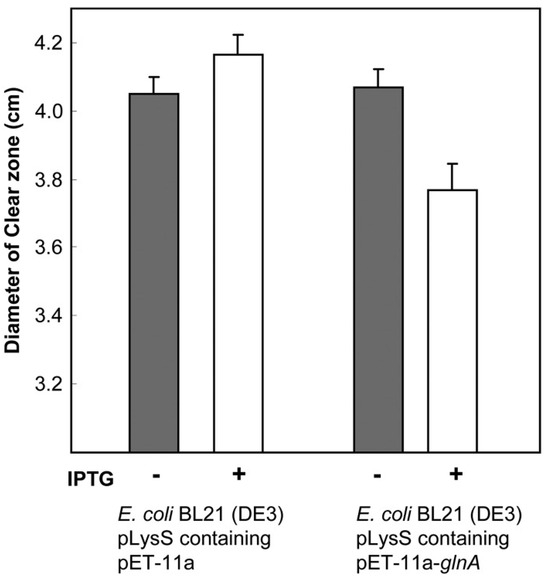
Figure 5.
Effect of glnA on the susceptibility levels to norfloxacin. Norfloxacin disk assay of non-induced (gray bars) and IPTG-induced (white bars) cultures of E. coli BL21(DE3) pLysS containing plasmids with or without the glnA insert. In the cells with the glnA insert, the reduced diameter of the clear zones of inhibition due to induction of glutamine synthetase was statistically significant (p < 0.05). Reproduced from reference [168] with the permission of Elsevier.
Nevertheless, the effect of glutamine synthetase on the final MIC of quinolones may also be influenced by a parallel effect on the production of OmpF related to the expression of glnA. Thus, an inverse relationship has been observed between the levels of expression of the glnA and ompF genes in Salmonella enterica, with lower levels of OmpF negatively affecting the intake of several quinolones, and thereby contributing to increasing the MIC levels to these agents [169].
SilA is another bacterial enzyme which has demonstrated its in vitro activity against different quinolones. Thus, Blanquez et al. cloned E. coli and purified SilA from the plant–pathogen Streptomyces ipomoeae, showing its ability to degrade ciprofloxacin and norfloxacin [170]. Of note, this enzyme also belongs to the laccases family [170]. Further in silico studies of the SilA protein have proposed that the amino acids His102, Val103 and Tyr108 are key in the modification of quinolones, with the first two being proposed to be involved in oxidative decarboxylation of the COOH radical at position 3, while Tyr108 is involved in the remotion of an oxygen for the carbonyl radical raised at position 4 [171]. A search in GenBank showed that SilA is present in a long series of microorganisms, with most having a high degree of identity with that of S. ipomoeae, including the critical amino acids proposed for anti-quinolone activity.
While almost all the abovementioned studies were focused on the biodegradation of quinolones in environmental or solid/liquid waste residues, it should be taken into account that all these quinolone-modifying mechanisms are at risk of mobilization to plasmids or other transferable genetic elements, as has been reported, for instance, with qepA, with the original source being proposed to be among Comamonadaceae [75], qnrB, which is indigenous of Citrobacter freundii [172], or oqxAB, chromosomally encoded in Klebsiella pneumoniae [173]. If this mobilization results in the lack of regulation, it will lead to a constitutive expression of the encoded protein and subsequently confer higher levels of resistance than in the original microorganism. As a general rule, all mechanisms of quinolone resistance are additive [1,55], and therefore, in a scenario in which silA, or any other gene encoding a protein with the potential of fully or partially inactivating quinolones, is mobilized to a plasmid—for instance by the action of insertion sequences—its exogenous acquisition by another microorganism should contribute to increasing its MIC levels to quinolones.
The fact that almost imperceptible amino acid alterations may expand or constrict the substrate profiles of enzymes able to introduce structural modifications, such as acetylation, phosphorylation or adenylation in antibacterial agents, as shown with AAC(6′)Ib [73], and the presence of a series of other fungi and bacterial chromosomal encoded proteins able, to a greater or lesser extent, to modify and degrade quinolones, suggest that new transferable quinolone-inactivating enzymes will be described in the forthcoming years.
8. Unconsidered Chromosomal Mutations
The selection of gyrA and/or parC mutations under quinolone exposure is a well-established phenomenon [44,50,80,81,174,175], with these mutations altering quinolone–DNA–DNA-Gyrase interactions [46,52,54]. Similarly, increased efflux levels as well as porin alterations resulting in decreased quinolone intake are also selected when microorganisms are exposed to quinolones, with both alterations resulting in lower cytoplasmic concentrations of quinolones and subsequent increased levels of antibiotic resistance [44,82,176]. While mutations at quinolone targets or either in regulator or constitutive genes related to efflux pumps or outer membrane proteins are clearly involved in the development of quinolone resistance, alterations in other genes outside the classical scope may induce subtle alterations. While these alterations produce slight changes in quinolone susceptibility levels, they do not modify the quinolone-susceptible status of microorganisms and remain unnoticed by classical methodologies, being considered as background noise or inter-strain MIC variations. However, these alterations can impact the ability of bacteria to develop full quinolone resistance. Of note, in multiple cases, these alterations induce modifications in different bacterial pathway regulation, which, over time, result in reduced quinolone uptake within the bacterial cytoplasm (Table 2).
Table 2.
Non-classical chromosomal mutations.
In this regard, when analyzing the exposure to ciprofloxacin of E. coli J53 carrying cloned qnr (designed below as J53-qnr) determinants (qnrA1, qnrA3, qnrB1 and qnrS1), Cesaro et al. showed that while no differences in the selection of mutants were observed, the mutant prevention concentration was ~10-fold higher than the original E. coli J53 [182]. Thus, the presence of qnr determinants facilitates the acquisition of quinolone resistance in the presence of higher antibiotic concentrations [182]. Surprisingly, while only 20% (65/329) of resistant isolates derived from J53-qnr presented mutation(s) in quinolone targets, this finding was observed in 79% (94/119) of quinolone-resistant mutants derived from E. coli J53, p < 0.0001 [182]. The authors suggested that Qnr blocks the access of quinolones to their targets, thereby favoring the selection of other mutations [182]. Other authors have observed similar phenomenon in E. coli, with Goto et al. also describing an increase in the mutant prevention concentrations as well as highlighting that parental strains acquire mutations in quinolone targets earlier than their derived strains with cloned qnr genes [183]. Vinué et al. expanded the scenario to other TMQRs, such as qepA and aac(6′)Ib-cr [177].
8.1. Toxic Metabolites Accumulation
These apparently unrelated mutations which are selected in this scenario were explored in different studies, with some probably being directly or indirectly involved in the regulation of efflux pump or porin expression levels, thereby resulting in the action of canonical mechanisms of resistance. For instance, alterations impairing cysH (cysteine biosynthesis), icdA (tricarboxylic acid cycle), metE (methionine synthesis) or purB (adenine biosynthesis) gene expression have been involved in metabolite accumulation-induced acrAB-tolC activation, increasing efflux and resistance levels to the tested quinolone, nalidixic acid [178]. While the metabolites related to these genes are different and belong to different metabolic processes, they lead to a common scenario, strongly suggesting the activation of efflux pumps to pump out toxic accumulations of intermediate products [178]. Of note, a possible association between purB mutations and quinolone resistance was suggested as early as 1970 [184]. On the other side, Vinué et al. also selected an in vitro ciprofloxacin-resistant E. coli mutant with an impaired icdA because of an IS10 insertion, but it was not correlated with increased levels of AcrAB-TolC [177].
8.2. RNA Polymerase
RNA polymerase subunits have also been involved in the development of low levels of quinolone resistance, with the rpoA mutation N294Y being observed in 4 S. enterica mutants [179]. While selected concomitantly with mutations in other genes, including well-established acrAB-TolC regulator genes, the cloning of the mutant rpoA results in final increases in the MICs of nalidixic acid (from 2 mg/L to 32 mg/L) and ciprofloxacin (from <0.015 mg/L to 0.015 mg/L) [179]. Regarding another subunit, the easy selection of rpoB mutations after ciprofloxacin exposure has been highlighted, with 20–30% of selected mutants carrying an rpoB alteration [180]. Thus, Pietsch et al. observed the presence of the amino acid substitutions H1244L, E1272G, A1277V, E1279G, Δ442–445 and duplication of the S455 (DuS445) in E. coli exposed to ciprofloxacin in vitro [180]. The authors cloned and analyzed three of the rpoB mutations (those leading to the changes in the amino acids A1277V, Δ442–445 and DuS445), observing increases from 0.015 mg/L to 0.023–0.045 mg/L in the MIC to ciprofloxacin [180]. Furthermore, the additive effect on the final MIC to ciprofloxacin was also observed when alterations in established quinolone resistance determinants (e.g., gyrA, gyrB, soxR, marR or acrR) were also present [180]. In addition, a very modest effect (maximum MIC of 48 mg/L) of these three amino acid changes in the MIC of rifampicin was also generated [180]. When the impact on the expression of other genes of the six rpoB mutations was determined, the authors observed that more than 100 genes were affected, including increased levels of mdtK expression, an efflux pump encoding gene, in all six cases, with further analysis suggesting a direct relationship between the presence of rpoB mutations and overexpression of mdtK [180]. Meanwhile, using qnrA1-carrying E. coli J53, Vinué et al. described that the rpoB mutation R1246C increased the MIC levels of nalidixic and ciprofloxacin from 8 mg/L to 64 mg/L and from 0.016 mg/L to 1,5 mg/L, respectively [177]. The rpoB mutant showed a significant decreased expression of ompF, thereby impacting quinolone intake within the bacterial cytoplasm [177]. Of note, while no effect on fitness was observed by Vinué et al., the opposite scenario was reported by Pietsch et al., a finding that might be related to the final alteration induced [177,180]. While not further explored, additional mutations in rpoB and rpoC have also been reported in ciprofloxacin-resistant E. coli mutants as well as in ciprofloxacin-resistant animal isolates [181,185].
8.3. Aminoacyl-tRNA Synthetase Genes
The presence of mutations in aminoacyl-tRNA synthetase aspS (D207A), leuS (L41H, D162N, S496P) and thrS (H244P, I582S) genes has also been involved in the development of low levels of resistance in E. coli exposed to ciprofloxacin [181]. Centering the study on leuS D162N, S496P amino acid substitutions, MIC studies suggest that alone these mutations play no role in the final MIC levels when introduced into strains carrying concomitant mutations in well-recognized quinolone targets, leading to a slight additive effect on final resistance levels [181]. The authors highlighted that the presence of these mutations influenced the final expression levels of around 200 genes and proposed the induction of a relA-related stringent response, in at least aspS and leuS mutants. This response leads to the overexpression of different genes, such as mdtK, encoding an efflux pump, acrZ, related to the efflux pump AcrAB-TolC or ydhIJK, a putative efflux pump [181]. Nevertheless, the scenario is highly complex with apparently contradictory data including the overexpression of acrA but the underexpression of acrB, or the contradictory overexpression of ompF (which theoretically leads to increased quinolone intake) [181]. The presence of a massive number of proteins expressed differently between quinolone-resistant and quinolone-susceptible microorganisms has also been described in other studies [186].
8.4. Mutations in Other Targets
While to the best of my knowledge no study on quinolones has been carried out, the deletion from amino acid 82 to 84 in the ribosomal protein L22 has been proposed to be related to an AcrAB-TolC overexpression [187]. Although further analysis is needed, if the impact of this deletion on AcrAB-TolC levels were definitively established, it would also result in modifications in the final cytoplasmatic quinolone levels and would thereby impact the final MIC to quinolones.
In addition, mutations in a series of other genes have been found when mutants were selected in the presence of quinolones, but the possible effect of these mutations on the final MIC levels remains unclear [177]. In this sense, the mutagenic power of quinolones must be considered [188,189].
Further studies on gene inactivation by the insertion of a transposon have shown that the inactivation of tens of genes (other than those classically considered) slightly increases or decreases ciprofloxacin susceptibility levels by 2-fold (in general) [190], and this might, therefore, underlie subtle differences in the basal levels of quinolone resistance. This finding demonstrates that point mutations in several of these genes cannot be discarded as actually additional contributors to the enhancement of quinolone resistance levels.
Vinué et al. suggested that the presence of the low levels of resistance conferred by a TMQR favors the development of a series of mutations, which, despite only having a low impact on the final susceptibility, altogether result in the development of full quinolone resistance [177]. While apparently unrelated, it has been shown that P. aeruginosa isolates carrying the exoU gene have an enhanced ability of acquiring high levels of resistance to quinolones and multiple mutations in gyrA and parC, while in a percentage of exoU- isolates, the acquisition of resistance to quinolones seems to be related to other pathways, including efflux pump overexpression, and the presence of isolates possessing high levels of resistance is significantly lower than among ExoU+ isolates [191,192,193]. This finding has been explained by the lower fitness cost of P. aeruginosa exoU+ related to the development of quinolone target mutations [191,192,193]. This fitness cost may better explain the abovementioned phenomenon observed by [177] Vinué et al. Thus, it can be hypothesized that these apparently unrelated mutations have no fitness cost, or the imposed fitness cost is lower than that related to quinolone target mutations. Isolates presenting a TMQR only need a slight increase in the MIC levels to become fully resistant to quinolones (i.e., survive and duplicate in the presence of quinolone concentrations achieved in their surrounding environment). Meanwhile, isolates which do not have this initial advantage need to become resistant with the presence of target mutations, irrespective of the fitness cost, and mutations in “non-canonical” genes may appear in a second phase.
Further analysis designed to obtain greater knowledge of the interactions of this constellation of punctual mutations and up- and downregulate genes and their derived effects is needed to better understand this phenomenon and clarify which genes play a role in the development of quinolone resistance and which do not.
8.5. Small Colony Variants
While different studies have not shown increased levels of quinolone resistance and, in fact, sometimes show slight increases in quinolone susceptibility [194,195], the SCVs have also been related to increased levels of quinolone resistance and enhanced persistence, even in the absence of target mutation [90,196,197]. SCVs are naturally occurring variants described in both Gram-negative and Gram-positive bacteria, such as Burkholderia pseudomallei, Coxiella burnetii, E. coli, P. aeruginosa or S. aureus, with an apparently unvaried morphology but exhibiting both a small size and a higher duplication time than their “normal” counterparts [90,194,196,198,199]. It has been proposed that these characteristics are related to the presence of a series of chromosomal mutations, leading to an altered electron-transport chain often resulting in menadione and/or hemin auxotrophy, or to the presence of thymidine auxotrophy [195,200,201,202,203], as well as other mutations, such as in the Pf1 prophage region [194]. Furthermore, the presence of genetic rearrangements as SCV inductors has also been described [204]. These alterations result in additional transcriptomic modifications [203], which might differ between different mutants presenting an SCV phenotype and thereby underlie the abovementioned differences related to the effect of quinolones on SCVs [205]. In this sense, alterations in the outer membrane and altered production of GyrA have been proposed as plausible explanations [90,197].
8.6. Atypical Amino Acid Substitutions in GyrA
Finally, while the classical quinolone resistance hot spot of the amino acid codon Ser83 of GyrA is affected [54], the rareness of the Ser83STOP (amber codon) mutation requires a brief comment [177,182]. Although this mutation should be lethal because of the essential role of the DNA-Gyrase, several microorganisms dispose of a “rescue system” for premature stops, the so-called tRNA suppressors, which recognize the STOP codon and insert an amino acid [206]. Regarding both of the abovementioned studies, Vinué et al. described the presence of an amber suppressor, while Cesaro et al. hypothesized the same option [177,182]. The presence of an amber suppressor may lead to the introduction of tryptophan, leucine or tyrosine, among other amino acids [206], with all three mentioned amino acids leading to quinolone resistance when raised in position 83 of GyrA [34,42,55,207]. Of note, the efficiency of these suppressors is lower than normal [206], and therefore, the presence of a lower amount of GyrA is predictable, which, as commented above, might also impact, by itself, the final quinolone resistance levels. Of note, a similar rescue system operates for ochre (TAA) and opal (TGA) terminators [206].
9. Unknown Mechanisms
The presence of undescribed mechanisms of quinolone resistance, either classified within canonical mechanisms, for instance as new transferable enzymes able to modify and inactivate one or more quinolones, or not, as in mutations in other apparently unrelated genes, cannot be ruled out, independently of the level of resistance conferred, or the extension and clinical relevance. In this sense, Chavez-Jacobo et al. noted that the original plasmid containing the crpP gene was able to confer higher levels of ciprofloxacin resistance than in vitro recombinant plasmids carrying crpP, thereby hypothesizing the presence of an additional undescribed plasmid-encoded mechanism of quinolone resistance [153].
10. Conclusions
While canonical mechanisms of quinolone resistance are undoubtedly the mechanisms most frequently described in quinolone-resistant microorganisms [55], a series of mechanisms and lifestyles may result in a low level of resistance to quinolones or in a quinolone escape route for microorganisms. Several of these mechanisms, as chromosomal mutations out of classical targets, may underlie the first steps towards the development of quinolone resistance and may thereby be selected in the presence of low concentrations of these agents. In this sense, the fitness cost of some of these mutations seems to be lower than that related to quinolone target mutations. This finding is of relevance in the microbial world beyond hospital settings, as natural environments in which quinolone residues arrive due to anthropogenic actions favor the initial selection of microbial populations with decreased susceptibility to quinolones, which, in turn, may facilitate the selection of full quinolone-resistant microorganisms. Meanwhile, at the clinical level, the possibility of the mobilization of these mechanisms, or at least of those involved in quinolone inactivation, is the most relevant risk.
A better understanding of these mechanisms may contribute to the fight against quinolone resistance and, by extension, to the struggle against antimicrobial resistance.
Funding
This research was funded by the Universidad Científica del Sur.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
The author thanks Donna Pringle for language editing.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ruiz, J. Transferable mechanisms of quinolone resistance from 1998 onward. Clin. Microbiol. Rev. 2019, 32, e00007-19. [Google Scholar] [CrossRef]
- Correia, S.; Poeta, P.; Hébraud, M.; Capelo, J.L.; Igrejas, G. Mechanisms of quinolone action and resistance: Where do we stand? J. Med. Microbiol. 2017, 66, 551–559. [Google Scholar] [CrossRef]
- Hooper, D.C.; Jacoby, G.A. Topoisomerase Inhibitors: Fluoroquinolone mechanisms of action and resistance. Cold Spring Harb. Perspect. Med. 2016, 6, a025320. [Google Scholar] [CrossRef]
- Pohlhaus, J.R.; Kreuzer, K.N. Norfloxacin-induced DNA gyrase cleavage complexes block Escherichia coli replication forks, causing double-stranded breaks in vivo. Mol. Microbiol. 2005, 56, 1416–1429. [Google Scholar] [CrossRef]
- Barton, N.; Crowther, A.F.; Hepworth, W.; Richardson, N.D.; Driver, G.W. New Quinolones and Therapeutic Compositions Containing Them. British Patent 830832, 23 March 1960. [Google Scholar]
- Bisacchi, G.S. Origins of the quinolone class of antibacterials: An expanded “discovery story”. J. Med. Chem. 2015, 58, 4874–4882. [Google Scholar] [CrossRef]
- Hepworth, W. Manufacture of 1-Methyl-6-nitro-4-quinolone-3-carboxylic Acid. British Patent 820295, 16 September 1959. [Google Scholar]
- Lesher, G.Y.; Froelich, E.J.; Gruett, M.D.; Bailey, J.H.; Brundage, R.P. 1,8-Naphthyridine derivatives: A new class of chemotherapy agents. J. Med. Pharm. Chem. 1962, 5, 1063–1068. [Google Scholar] [CrossRef]
- Mandomando, I.M.; Macete, E.V.; Ruiz, J.; Sanz, S.; Abacassamo, F.; Vallés, X.; Sacarlal, J.; Vila, J.; Navia, M.M.; Alonso, P.L.; et al. Etiology of diarrhea in children younger than 5 years of age admitted in a rural hospital of southern Mozambique. Am. J. Trop. Med. Hyg. 2007, 76, 522–527. [Google Scholar] [CrossRef]
- Parry, H.E. Nalidixic acid for shigellosis. Lancet 1983, 322, 1206. [Google Scholar] [CrossRef]
- Sen, D.; Dutta, P.; Deb, B.C.; Pal, S.C. Nalidixic-acid resistant Shigella dysenteriae type 1 in eastern India. Lancet 1988, 332, 911. [Google Scholar] [CrossRef]
- Emmerson, A.M.; Jones, A.M. The quinolones: Decades of development and use. J. Antimicrob. Chemother. 2003, 51 (Suppl. S1), 13–20. [Google Scholar] [CrossRef]
- AlMahmoud, T.; Elhanan, M.; Elshamsy, M.H.; Alshamsi, H.N.; Abu-Zidan, F.M. Management of infective corneal ulcers in a high-income developing country. Medicine 2019, 98, e18243. [Google Scholar] [CrossRef]
- Alonso, D.; Muñoz, J.; Ruiz, J.; Carmona, F.; Nadal, A.; Gascón, J. Salmonella ovarian abscess following travel diarrhoea episode. Arch. Gynecol. Obstet. 2007, 276, 551–553. [Google Scholar] [CrossRef]
- Bassetti, M.; Cadeo, B.; Villa, G.; Sartor, A.; Cainero, V.; Causero, A. Current antibiotic management of prosthetic joint infections in Italy: The ‘Udine strategy’. J. Antimicrob. Chemother. 2014, 69 (Suppl. S1), i41–i45. [Google Scholar] [CrossRef]
- Gentry, L.O. Review of quinolones in the treatment of infections of the skin and skin structure. J. Antimicrob. Chemother. 1991, 28 (Suppl. SC), 97–110. [Google Scholar] [CrossRef]
- Koulenti, D.; Xu, E.; Mok, I.Y.S.; Song, A.; Karageorgopoulos, D.E.; Armaganidis, A.; Lipman, J.; Tsiodras, S. Novel antibiotics for multidrug-resistant gram-positive microorganisms. Microorganisms 2019, 7, 270. [Google Scholar] [CrossRef]
- Leonov, Y.; Schlaeffer, F.; Karpuch, J.; Bourvin, A.; Shemesh, Y.; Lewinson, G. Ciprofloxacin in the treatment of nosocomial multiply resistant Acinetobacter calcoaceticus bacteremia. Infection 1990, 18, 234–236. [Google Scholar] [CrossRef]
- Okubo, Y.; Michihata, N.; Morisaki, N.; Uda, K.; Miyairi, I.; Ogawa, Y.; Matsui, H.; Fushimi, K.; Yasunaga, H. Recent trends in practice patterns and impact of corticosteroid use on pediatric Mycoplasma pneumoniae-related respiratory infections. Respir. Investig. 2018, 56, 158–165. [Google Scholar] [CrossRef]
- Ruiz, J.; Marco, F.; Oliveira, I.; Vila, J.; Gascón, J. Trends in antimicrobial resistance in Campylobacter spp. causing traveler’s diarrhea. APMIS 2007, 115, 218–224. [Google Scholar] [CrossRef]
- Vila, J.; Ruiz, J.; Sanchez, F.; Navarro, F.; Mirelis, B.; Jiménez de Anta, M.T.; Prats, G. Increase in quinolone resistance in a Haemophilus influenzae strain isolated from a patient with recurrent respiratory infections treated with ofloxacin. Antimicrob. Agents Chemother. 1999, 43, 161–162. [Google Scholar] [CrossRef]
- Davino, G.; D’Alvano, T.; Esposito, S. The use of ozenoxacin in pediatric patients: Clinical evidence, efficacy and safety. Front. Pharmacol. 2020, 11, 559708. [Google Scholar] [CrossRef]
- Liapikou, A.; Cilloniz, C.; Palomeque, A.; Torres, T. Emerging antibiotics for community-acquired pneumonia. Expert Opin. Emerg. Drugs 2019, 24, 221–231. [Google Scholar] [CrossRef]
- Copeland, V.; McLaughlin, M.; Trifilio, S. Ciprofloxacin vs levofloxacin for prophylaxis during hematopoietic stem-cell transplantation. Clin. Transplant. 2018, 32, e13145. [Google Scholar] [CrossRef]
- Córdova-González, D.; Alfonseca-Silva, E.; Gutiérrez, L.; Tapia-Pérez, G.; Sumano, H. Intramammary preparation of enrofloxacin hydrochloride-dihydrate for bovine mastitis (biofilm-forming Staphylococcus aureus). J. Vet. Sci. 2024, 25, e6. [Google Scholar] [CrossRef]
- Nelson, J.M.; Chiller, T.M.; Powers, J.H.; Angulo, F.J. Fluoroquinolone-resistant Campylobacter species and the withdrawal of fluoroquinolones from use in poultry: A public health success story. Clin. Infect. Dis. 2007, 44, 977–980. [Google Scholar] [CrossRef]
- Trouchon, T.; Lefebvre, S. A review of enrofloxacin for veterinary use. Open J. Vet. Med. 2016, 6, 40–58. [Google Scholar] [CrossRef]
- World Organisation for Animal Health. OIE Annual Report on Antimicrobial Agents Intended for Use in Animals, 3rd ed.; World Organisation for Animal Health (OIE): Paris, France, 2018; pp. 1–129. Available online: https://www.oie.int/fileadmin/Home/eng/Our_scientific_expertise/docs/pdf/AMR/Annual_Report_AMR_3.pdf (accessed on 13 March 2024).
- Dalhoff, A. Antiviral, antifungal, and antiparasitic activities of fluoroquinolones optimized for treatment of bacterial infections: A puzzling paradox or a logical consequence of their mode of action? Eur. J. Clin. Microbiol. Infect. Dis. 2015, 34, 661–668. [Google Scholar] [CrossRef]
- Romero, I.C.; Saravia, N.G.; Walker, J. Selective action of fluoroquinolones against intracellular amastigotes of Leishmania (Viannia) panamensis in vitro. J. Parasitol. 2005, 91, 1474–1479. [Google Scholar] [CrossRef]
- Stergiopoulou, T.; Meletiadis, J.; Sein, T.; Papaioannidou, P.; Tsiouris, I.; Roilides, E.; Walsh, T.J. Comparative pharmacodynamic interaction analysis between ciprofloxacin, moxifloxacin and levofloxacin and antifungal agents against Candida albicans and Aspergillus fumigatus. J. Antimicrob. Chemother. 2009, 63, 343–348. [Google Scholar] [CrossRef]
- Yamaya, M.; Nishimura, H.; Hatachi, Y.; Yasuda, H.; Deng, X.; Sasaki, T.; Mizuta, K.; Kubo, H.; Nagatomi, R. Levofloxacin inhibits rhinovirus infection in primary cultures of human tracheal epithelial cells. Antimicrob. Agents Chemother. 2012, 56, 4052–4061. [Google Scholar] [CrossRef]
- Carroll, G. Neggram (nalidixic acid): A new antimicrobial chemotherapeutic agent. J. Urol. 1963, 90, 476–478. [Google Scholar] [CrossRef]
- Cullen, M.E.; Wyke, A.W.; Kuroda, R.; Fisher, L.M. Cloning and characterization of a DNA gyrase A gene from Escherichia coli that confers clinical resistance to 4-quinolones. Antimicrob. Agents Chemother. 1989, 33, 886–894. [Google Scholar] [CrossRef]
- Guibert, F.; Espinoza, K.; Taboada-Blanco, C.; Alonso, C.A.; Oporto, R.; Castillo, A.K.; Rojo-Bezares, B.; López, M.; Sáenz, Y.; Pons, M.J.; et al. Traditional marketed meats as a reservoir of multidrug-resistant Escherichia coli. Int. Microbiol. 2024; in press. [Google Scholar] [CrossRef]
- Hooper, D.C.; Wolfson, J.S.; Ng, E.Y.; Swartz, M.N. Mechanisms of action of and resistance to ciprofloxacin. Am. J. Med. 1987, 82, 12–20. [Google Scholar]
- Jonsson, M. Antibiotic resistance and R factors in gram-negative bacteria isolated in a hospital for infectious diseases. III. The effect of antibacterial treatment on the incidence of R factor-mediated antibiotic resistance. Scand. J. Infect. Dis. 1973, 5, 41–47. [Google Scholar] [CrossRef]
- Lishman, I.V.; Swinney, J. Studies of a new antibacterial agent, nalidixic acid. Br. J. Urol. 1963, 35, 116–121. [Google Scholar] [CrossRef]
- Martínez-Martínez, L.; Pascual, A.; Jacoby, G.A. Quinolone resistance from a transferable plasmid. Lancet 1998, 351, 797–799. [Google Scholar] [CrossRef]
- Medina, A.; Rivera, F.P.; Ochoa, T.J.; Riveros, M.; Pons, M.J.; Ruiz, J. Transferable mechanisms of quinolone resistance are more frequent among enterotoxigenic Escherichia coli isolates displaying low-level quinolone resistance. Trop. Biomed. 2023, 40, 183–187. [Google Scholar] [CrossRef]
- Pitout, J.D.; Wei, Y.; Church, D.L.; Gregson, D.B. Surveillance for plasmid-mediated quinolone resistance determinants in Enterobacteriaceae within the Calgary Health Region, Canada: The emergence of aac(6′)-Ib-cr. J. Antimicrob. Chemother. 2008, 61, 999–1002. [Google Scholar] [CrossRef]
- Pons, M.J.; Mosquito, S.G.; Gomes, C.; del Valle, L.J.; Ochoa, T.J.; Ruiz, J. Analysis of quinolone-resistance in commensal and diarrheagenic Escherichia coli isolates from infants in Lima, Peru. Trans. R. Soc. Trop. Med. Hyg. 2014, 108, 22–28. [Google Scholar] [CrossRef]
- Ricci, V.; Tzakas, P.; Buckley, A.; Piddock, L.J. Ciprofloxacin-resistant Salmonella enterica serovar Typhimurium strains are difficult to select in the absence of AcrB and TolC. Antimicrob. Agents Chemother. 2006, 50, 38–42. [Google Scholar] [CrossRef]
- Ruiz, E.; Sáenz, Y.; Zarazaga, M.; Rocha-Gracia, R.; Martínez-Martínez, L.; Arlet, G.; Torres, C. qnr, aac(6′)-Ib-cr and qepA genes in Escherichia coli and Klebsiella spp.: Genetic environments and plasmid and chromosomal location. J. Antimicrob. Chemother. 2012, 67, 886–897. [Google Scholar] [CrossRef]
- Tavío, M.M.; Vila, J.; Ruiz, J.; Ruiz, J.; Martin-Sanchez, A.M.; Jiménez de Anta, M.T. Mechanisms involved in the development of resistance to fluoroquinolones in Escherichia coli isolates. J. Antimicrob. Chemother. 1999, 44, 735–742. [Google Scholar] [CrossRef]
- Vila, J.; Ruiz, J.; Marco, F.; Barceló, A.; Goñi, P.; Giralt, E.; Jiménez de Anta, M.T. Association between double mutation in gyrA gene of ciprofloxacin-resistant clinical isolates of Escherichia coli and minimal inhibitory concentration. Antimicrob. Agents Chemother. 1994, 38, 2477–2479. [Google Scholar] [CrossRef]
- Vinué, L.; Hooper, D.C.; Jacoby, G.A. Chromosomal mutations that accompany qnr in clinical isolates of Escherichia coli. Int. J. Antimicrob. Agents 2018, 51, 479–483. [Google Scholar] [CrossRef]
- Gellert, M.; Mizuuchi, K.; O’Dea, M.H.; Itoh, T.; Tomizawa, J.I. Nalidixic acid resistance: A second genetic character involved in DNA gyrase activity. Proc. Natl. Acad. Sci. USA 1977, 74, 4772–4776. [Google Scholar] [CrossRef]
- Everett, M.J.; Jin, Y.F.; Ricci, V.; Piddock, L.J. Contributions of individual mechanisms to fluoroquinolone resistance in 36 Escherichia coli strains isolated from humans and animals. Antimicrob. Agents Chemother. 1996, 40, 2380–2386. [Google Scholar] [CrossRef]
- Guillemin, I.; Jarlier, V.; Cambau, E. Correlation between quinolone susceptibility patterns and sequences in the A and B subunits of DNA gyrase in Mycobacteria. Antimicrob. Agents Chemother. 1998, 42, 2084–2088. [Google Scholar] [CrossRef]
- Gomes, C.; Martínez-Puchol, S.; Ruiz-Roldán, L.; Pons, M.J.; del Valle Mendoza, J.; Ruiz, J. Development and characterisation of highly antibiotic resistant Bartonella bacilliformis mutants. Sci. Rep. 2016, 6, 33584. [Google Scholar] [CrossRef]
- Madurga, S.; Sánchez-Céspedes, J.; Belda, I.; Vila, J.; Giralt, E. Mechanism of binding of fluoroquinolones to the quinolone resistance-determining region of DNA gyrase: Towards an understanding of the molecular basis of quinolone resistance. ChemBioChem 2008, 9, 2081–2086. [Google Scholar] [CrossRef]
- Mendoza-Mujica, G.; Flores-Leon, D.; Ruiz, J. Molecular characterization of fluoroquinolone-resistant Bartonella bacilliformis. Pathogens 2021, 10, 876. [Google Scholar] [CrossRef]
- Palumbo, M.; Gatto, B.; Zagotto, G.; Palù, G. On the mechanism of action of quinolone drugs. Trends Microbiol. 1993, 1, 232–235. [Google Scholar] [CrossRef]
- Ruiz, J. Mechanisms of resistance to quinolones: Target alterations, decreased accumulation and DNA Gyrase protection. J. Antimicrob. Chemother. 2003, 51, 1109–1117. [Google Scholar] [CrossRef]
- Sierra, J.M.; Marco, F.; Ruiz, J.; Jiménez de Anta, M.T.; Vila, J. Correlation between the activity of different fluoroquinolones and the presence of mechanisms of quinolone resistance in epidemiologically related and unrelated strains of methicillin-susceptible and -resistant Staphylococcus aureus. Clin. Microbiol. Infect. 2002, 8, 781–790. [Google Scholar] [CrossRef]
- Han, X.Y.; Andrade, R.A. Brevundimonas diminuta infections and its resistance to fluoroquinolones. J. Antimicrob. Chemother. 2005, 55, 853–859. [Google Scholar] [CrossRef]
- del Valle, L.J.; Flores, L.; Vargas, M.; García-de-la-Guarda, R.; Quispe, R.L.; Ibañez, Z.B.; Alvarado, D.; Ramírez, P.; Ruiz, J. Bartonella bacilliformis, endemic pathogen of the Andean region, is intrinsically resistant to quinolones. Int. J. Infect. Dis. 2010, 14, e506–e510. [Google Scholar] [CrossRef][Green Version]
- Hirai, K.; Aoyama, H.; Suzue, S.; Irikura, T.; Iyobe, S.; Mitsuhashi, S. Isolation and characterization of norfloxacin-resistant mutants of Escherichia coli K-12. Antimicrob. Agents Chemother. 1986, 30, 248–253. [Google Scholar] [CrossRef]
- Palma, N.; Pons, M.J.; Gomes, C.; Mateu, J.; Riveros, M.; García, W.; Jacobs, J.; García, C.; Ochoa, T.J.; Ruiz, J. Resistance to quinolones, cephalosporins and macrolides in Escherichia coli causing bacteraemia in Peruvian children. J. Glob. Antimicrob. Resist. 2017, 11, 28–33. [Google Scholar] [CrossRef]
- Redgrave, L.S.; Sutton, S.B.; Webber, M.A.; Piddock, L.J. Fluoroquinolone resistance: Mechanisms, impact on bacteria, and role in evolutionary success. Trends Microbiol. 2014, 22, 438–445. [Google Scholar] [CrossRef]
- Sáenz, Y.; Ruiz, J.; Zarazaga, M.; Teixidó, M.; Torres, C.; Vila, J. Effect of the efflux pump inhibitor Phe-Arg-beta-naphthylamide on the MIC values of the quinolones, tetracycline and chloramphenicol, in Escherichia coli isolates of different origin. J. Antimicrob. Chemother. 2004, 53, 544–545. [Google Scholar] [CrossRef]
- García-León, G.; Ruiz de Alegría Puig, C.; García de la Fuente, C.; Martínez-Martínez, L.; Martínez, J.L.; Sánchez, M.B. High-level quinolone resistance is associated with the overexpression of smeVWX in Stenotrophomonas maltophilia clinical isolates. Clin. Microbiol. Infect. 2015, 21, 464–467. [Google Scholar] [CrossRef]
- Valdezate, S.; Vindel, A.; Saéz-Nieto, J.A.; Baquero, F.; Cantón, R. Preservation of topoisomerase genetic sequences during in vivo and in vitro development of high-level resistance to ciprofloxacin in isogenic Stenotrophomonas maltophilia strains. J. Antimicrob. Chemother. 2005, 56, 220–223. [Google Scholar] [CrossRef]
- Ribera, A.; Doménech-Sánchez, A.; Ruiz, J.; Benedi, V.J.; de Anta, M.T.J.; Vila, J. Mutations in gyrA and parC QRDRs are not relevant for quinolone resistance in epidemiological unrelated Stenotrophomonas maltophilia clinical isolates. Microb. Drug Resist. 2002, 8, 245–252. [Google Scholar] [CrossRef]
- Ribera, A.; Jurado, A.; Ruiz, J.; Marco, F.; Del Valle, O.; Mensa, J.; Chaves, J.; Hernández, G.; de Anta, M.J.; Vila, J. In vitro activity of clinafloxacin in comparison with other quinolones against Stenotrophomonas maltophilia clinical isolates in the presence and absence of reserpine. Diagn. Microbiol. Infect. Dis. 2002, 42, 123–128. [Google Scholar] [CrossRef]
- Babcock, G.F.; Berryhill, D.L.; Marsh, D.H. R-factors of Escherichia coli from dressed beef and humans. Appl. Microbiol. 1973, 25, 21–23. [Google Scholar] [CrossRef]
- Courvalin, P. Plasmid-mediated 4-quinolone resistance: A real or apparent absence? Antimicrob. Agents Chemother. 1990, 34, 681–684. [Google Scholar] [CrossRef]
- Lebek, G. Medizinische aspekte der infektiösen antibiotika-resistenz gram-negativer darmbakterien. Pathol. Microbiol. 1967, 30, 1015–1036. [Google Scholar] [CrossRef]
- Munshi, M.H.; Sack, D.A.; Haider, K.; Ahmed, Z.U.; Rahaman, M.M.; Morshed, M.G. Plasmid-mediated resistance to nalidixic acid in Shigella dysenteriae type 1. Lancet 1987, 330, 419–421. [Google Scholar] [CrossRef]
- Panhotra, B.R.; Desai, B.; Sharma, P.L. Nalidixic-acid-resistant Shigella dysenteriae I. Lancet 1985, 325, 763. [Google Scholar] [CrossRef]
- Pons, M.J.; Gomes, C.; Ruiz, J. QnrVC, a new transferable Qnr-like family. Enferm. Infecc. Microbiol. Clin. 2013, 31, 191–192. [Google Scholar] [CrossRef]
- Robicsek, A.; Strahilevitz, J.; Jacoby, G.A.; Macielag, M.; Abbanat, D.; Park, C.H.; Bush, K.; Hooper, D.C. Fluoroquinolone-modifying enzyme: A new adaptation of a common aminoglycoside acetyltransferase. Nat. Med. 2006, 12, 83–88. [Google Scholar] [CrossRef]
- Ruiz, J. Analysis of the presence of erroneous Qnr sequences in GenBank. J. Antimicrob. Chemother. 2018, 73, 1213–1216. [Google Scholar] [CrossRef]
- Ruiz, J. In silico analysis of transferable QepA variants and related chromosomal efflux pumps. Rev. Esp. Quimioterap. 2018, 31, 537–541. [Google Scholar]
- Ruiz, J. CrpP, a passenger or a hidden stowaway in the Pseudomonas aeruginosa genome? J. Antimicrob. Chemother. 2019, 74, 3397–3399. [Google Scholar] [CrossRef]
- Zubyk, H.L.; Wright, G.D. CrpP is not a fluoroquinolone-inactivating enzyme. Antimicrob. Agents Chemother. 2021, 65, e0077321. [Google Scholar] [CrossRef]
- Pérez-Moreno, M.O.; Estepa, V.; Sáenz, Y.; Cortell-Ortolá, M.; Fort-Gallifa, I.; Ruiz, J.; Torres, C. Intrahospitalary dissemination of Klebsiella pneumoniae carrying blaDHA-1 and qnrB4 genes within a novel complex class 1 integron. Diagn. Microbiol. Infect. Dis. 2012, 73, 210–211. [Google Scholar] [CrossRef]
- Quiroga, M.P.; Arduino, S.M.; Merkier, A.K.; Quiroga, C.; Petroni, A.; Argentinian Integron Study Group; Roy, P.H.; Centrón, D. Distribution and functional identification of complex class 1 integrons. Infect. Genet. Evol. 2013, 19, 88–96. [Google Scholar] [CrossRef]
- Ruiz, J.; Jurado, A.; Garcia-Méndez, E.; Marco, F.; Aguilar, L.; Jiménez de Anta, M.T.; Vila, J. Frequency of selection of fluoroquinolone-resistant mutants of Neisseria gonorrhoeae exposed to gemifloxacin and four other quinolones. J. Antimicrob. Chemother. 2001, 48, 545–548. [Google Scholar] [CrossRef][Green Version]
- Ruiz, J.; Sierra, J.M.; Jiménez de Anta, M.T.; Vila, J. Characterization of sparfloxacin-resistant mutants of Staphylococcus aureus obtained in vitro. Int. J. Antimicrob. Agents 2001, 18, 107–112. [Google Scholar] [CrossRef]
- Soto, S.M.; Ruíz, J.; Mendoza, M.C.; Vila, J. In vitro fluoroquinolone-resistant mutants of Salmonella enterica serotype Enteritidis: Analysis of mechanisms involved in resistance. Int. J. Antimicrob. Agents 2003, 22, 537–540. [Google Scholar] [CrossRef]
- Ruíz, J.; Navia, M.M.; Marco, F.; Vila, J. Mecanismos de resistencia a betalactámicos y ácido nalidíxico en aislados clínicos de Salmonella enterica serotipo hadar y bsilla. Enferm. Infecc. Microbiol. Clin. 2004, 22, 251–256. [Google Scholar] [CrossRef]
- Stapleton, P.; Wu, P.J.; King, A.; Shannon, K.; French, G.; Philips, I. Incidence and mechanisms of resistance to the combination of amoxicillin and clavulanate in Escherichia coli. Antimicrob. Agents Chemother. 1995, 39, 2478–2483. [Google Scholar] [CrossRef]
- Wu, P.J.; Shannon, K.; Phillips, I. Effect of hyperproduction of TEM-1 β-lactamase on in vitro susceptibility of Escherichia coli to β-lactam antibiotics. Antimicrob. Agents Chemother. 1994, 38, 494–498. [Google Scholar] [CrossRef]
- Ince, D.; Hooper, D.C. Quinolone resistance due to reduced target enzyme expression. J. Bacteriol. 2003, 185, 6883–6892. [Google Scholar] [CrossRef]
- Straney, R.; Krah, R.; Menzel, R. Mutations in the -10 TATAAT sequence of the gyrA promoter affect both promoter strength and sensitivity to DNA supercoiling. J. Bacteriol. 1994, 176, 5999–6006. [Google Scholar] [CrossRef]
- Aleixandre, V.; Herrera, G.; Urios, A.; Blanco, M. Effects of ciprofloxacin on plasmid DNA supercoiling of Escherichia coli topoisomerase I and gyrase mutants. Antimicrob. Agents Chemother. 1991, 35, 20–23. [Google Scholar] [CrossRef]
- Dages, S.; Dages, K.; Zhi, X.; Leng, F. Inhibition of the gyrA promoter by transcription-coupled DNA supercoiling in Escherichia coli. Sci. Rep. 2018, 8, 14759. [Google Scholar] [CrossRef]
- Pan, X.S.; Hamlyn, P.J.; Talens-Visconti, R.; Alovero, F.L.; Manzo, R.H.; Fisher, L.M. Small-colony mutants of Staphylococcus aureus allow selection of gyrase-mediated resistance to dual-target fluoroquinolones. Antimicrob. Agents Chemother. 2002, 46, 2498–2506. [Google Scholar] [CrossRef]
- Hecht, A.; Glasgow, J.; Jaschke, P.R.; Bawazer, L.A.; Munson, M.S.; Cochran, J.R.; Endy, D.; Salit, M. Measurements of translation initiation from all 64 codons in E. coli. Nucleic Acids Res. 2017, 45, 3615–3626. [Google Scholar] [CrossRef]
- Samba-Louaka, A.; Delafont, V.; Rodier, M.H.; Cateau, E.; Héchard, Y. Free-living amoebae and squatters in the wild: Ecological and molecular features. FEMS Microbiol. Rev. 2019, 43, 415–434. [Google Scholar] [CrossRef]
- Cirillo, J.D.; Falkow, S.; Tompkins, L.S. Growth of Legionella pneumophila in Acanthamoeba castellanii enhances invasion. Infect. Immun. 1994, 62, 3254–3261. [Google Scholar] [CrossRef]
- Molmeret, M.; Horn, M.; Wagner, M.; Santic, M.; Abu Kwaik, Y. Amoebae as training grounds for intracellular bacterial pathogens. Appl. Environ. Microbiol. 2005, 71, 20–28. [Google Scholar] [CrossRef]
- Saisongkorh, W.; Robert, C.; La Scola, B.; Raoult, D.; Rolain, J.M. Evidence of transfer by conjugation of type IV secretion system genes between Bartonella species and Rhizobium radiobacter in amoeba. PLoS ONE 2010, 5, e12666. [Google Scholar] [CrossRef]
- Maschio, V.J.; Corção, G.; Rott, M.B. identification of Pseudomonas spp. as amoeba-resistant microorganisms in isolates of Acanthamoeba. Rev. Inst. Med. Trop. Sao Paulo 2015, 57, 81–83. [Google Scholar] [CrossRef]
- Judy, B.M.; Whitlock, G.C.; Torres, A.G.; Estes, D.M. Comparison of the in vitro and in vivo susceptibilities of Burkholderia mallei to ceftazidime and levofloxacin. BMC Microbiol. 2009, 9, 88. [Google Scholar] [CrossRef]
- Kaldestad, M.; Haugland, G.T.; Rønneseth, A.; Wergeland, H.I.; Samuelsen, O.B. Antibiotic uptake by cultured Atlantic cod leucocytes and effect on intracellular Francisella noatunensis subsp. noatunensis replication. Dis. Aquat. Organ. 2014, 108, 11–21. [Google Scholar] [CrossRef][Green Version]
- Ortillés, A.; Belloc, J.; Rubio, E.; Fernández, M.T.; Benito, M.; Cristóbal, J.A.; Calvo, B.; Goñi, P. In-vitro development of an effective treatment for Acanthamoeba keratitis. Int. J. Antimicrob. Agents 2017, 50, 325–333. [Google Scholar] [CrossRef]
- Michot, J.M.; Seral, C.; Van Bambeke, F.; Mingeot-Leclercq, M.P.; Tulkens, P.M. Influence of efflux transporters on the accumulation and efflux of four quinolones (ciprofloxacin, levofloxacin, garenoxacin, and moxifloxacin) in J774 macrophages. Antimicrob. Agents Chemother. 2005, 49, 2429–2437. [Google Scholar] [CrossRef]
- Lismond, A.; Tulkens, P.M.; Mingeot-Leclercq, M.P.; Courvalin, P.; Van Bambeke, F. Cooperation between prokaryotic (Lde) and eukaryotic (MRP) efflux transporters in J774 macrophages infected with Listeria monocytogenes: Studies with ciprofloxacin and moxifloxacin. Antimicrob. Agents Chemother. 2008, 52, 3040–3046. [Google Scholar] [CrossRef]
- Behera, H.S.; Satpathy, G. Characterisation and expression analysis of trophozoite and cyst proteins of Acanthamoeba spp. isolated from Acanthamoeba keratitis (AK) patient. Mol. Biochem. Parasitol. 2016, 205, 29–34. [Google Scholar] [CrossRef]
- Fouque, E.; Trouilhé, M.C.; Thomas, V.; Hartemann, P.; Rodier, M.H.; Héchard, Y. Cellular, biochemical, and molecular changes during encystment of free-living amoebae. Eukaryot. Cell 2012, 11, 382–387. [Google Scholar] [CrossRef]
- Lee, X.; Reimmann, C.; Greub, G.; Sufrin, J.; Croxatto, A. The Pseudomonas aeruginosa toxin L-2-amino-4-methoxy-trans-3-butenoic acid inhibits growth and induces encystment in Acanthamoeba castellanii. Microbes Infect. 2012, 14, 268–272. [Google Scholar] [CrossRef]
- Maal-Bared, R.; Dixon, B.; Axelsson-Olsson, D. Fate of internalized Campylobacter jejuni and Mycobacterium avium from encysted and excysted Acanthamoeba polyphaga. Exp. Parasitol. 2019, 199, 104–110. [Google Scholar] [CrossRef]
- Barker, J.; Scaife, H.; Brown, M.R. Intraphagocytic growth induces an antibiotic-resistant phenotype of Legionella pneumophila. Antimicrob. Agents Chemother. 1995, 39, 2684–2688. [Google Scholar] [CrossRef]
- Dubois, V.; Pawlik, A.; Bories, A.; Le Moigne, V.; Sismeiro, O.; Legendre, R.; Varet, H.; Rodríguez-Ordóñez, M.D.P.; Gaillard, J.L.; Coppée, J.Y.; et al. Mycobacterium abscessus virulence traits unraveled by transcriptomic profiling in amoeba and macrophages. PLoS Pathog. 2019, 15, e1008069. [Google Scholar] [CrossRef]
- Winiecka-Krusnell, J.; Linder, E. Bacterial infections of free-living amoebae. Res. Microbiol. 2001, 152, 613–619. [Google Scholar] [CrossRef]
- Vieira, A.; Ramesh, A.; Seddon, A.M.; Karlyshev, A.V. CmeABC Multidrug efflux pump contributes to antibiotic resistance and promotes Campylobacter jejuni survival and multiplication in Acanthamoeba polyphaga. Appl. Environ. Microbiol. 2017, 83, e01600-17. [Google Scholar] [CrossRef]
- Grinnage-Pulley, T.; Zhang, Q. Genetic basis and functional consequences of differential expression of the CmeABC efflux pump in Campylobacter jejuni isolates. PLoS ONE 2015, 10, 0131534. [Google Scholar] [CrossRef]
- Yan, M.; Sahin, O.; Lin, J.; Zhang, Q. Role of the CmeABC efflux pump in the emergence of fluoroquinolone-resistant Campylobacter under selection pressure. J. Antimicrob. Chemother. 2006, 58, 1154–1159. [Google Scholar] [CrossRef]
- Colvin, K.M.; Irie, Y.; Tart, C.S.; Urbano, R.; Whitney, J.C.; Ryder, C.; Howell, P.L.; Wozniak, D.J.; Parsek, M.R. The Pel and Psl polysaccharides provide Pseudomonas aeruginosa structural redundancy within the biofilm matrix. Environ. Microbiol. 2012, 14, 1913–1928. [Google Scholar] [CrossRef]
- Oliveira, F.; Lima, C.A.; Brás, S.; França, Â.; Cerca, N. Evidence for inter- and intraspecies biofilm formation variability among a small group of coagulase-negative staphylococci. FEMS Microbiol. Lett. 2015, 362, fnv175. [Google Scholar] [CrossRef]
- Hall, C.W.; Mah, T.F. Molecular mechanisms of biofilm-based antibiotic resistance and tolerance in pathogenic bacteria. FEMS Microbiol. Rev. 2017, 41, 276–301. [Google Scholar] [CrossRef]
- Høiby, N.; Bjarnsholt, T.; Givskov, M.; Molin, S.; Ciofu, O. Antibiotic resistance of bacterial biofilms. Int. J. Antimicrob. Agents 2010, 35, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Petrova, O.E.; Sauer, K. Escaping the biofilm in more than one way: Desorption, detachment or dispersion. Curr. Opin. Microbiol. 2016, 30, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Akers, K.S.; Cardile, A.; Wenke, J.C.; Murray, C.K. Biofilm formation by clinical isolates and its relevance to clinical infections. Adv. Exp. Med. Biol. 2015, 830, 1–28. [Google Scholar] [CrossRef] [PubMed]
- del Pozo, J.L. Biofilm-related disease. Expert Rev. Anti Infect. Ther. 2018, 16, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Trautner, B.W.; Darouiche, R.O. Role of biofilm in catheter-associated urinary tract infection. Am. J. Infect. Control 2004, 32, 177–183. [Google Scholar] [CrossRef]
- Steenackers, H.P.; Parijs, I.; Dubey, A.; Foster, K.R.; Vanderleyden, J. Experimental evolution in biofilm populations. FEMS Microbiol. Rev. 2016, 40, 373–397. [Google Scholar] [CrossRef] [PubMed]
- Taghizadeh, L.; Karimi, A.; Presterl, E.; Heitzinger, C. Bayesian inversion for a biofilm model including quorum sensing. Comput. Biol. Med. 2020, 117, 103582. [Google Scholar] [CrossRef]
- Li, Z.; Ding, Z.; Liu, Y.; Jin, X.; Xie, J.; Li, T.; Zeng, Z.; Wang, Z.; Liu, J. Phenotypic and genotypic characteristics of biofilm formation in clinical isolates of Acinetobacter baumannii. Infect. Drug Resist. 2021, 14, 2613–2624. [Google Scholar] [CrossRef]
- Park, H.R.; Hong, M.K.; Hwang, S.; Park, Y.; Kwon, K.H.; Yoon, J.W.; Shin, S.; Kim, J.H.; Park, Y.H. Characterisation of Pseudomonas aeruginosa related to bovine mastitis. Acta Vet. Hung. 2014, 62, 1–12. [Google Scholar] [CrossRef]
- Ciofu, O.; Tolker-Nielsen, T. Tolerance and resistance of Pseudomonas aeruginosa biofilms to antimicrobial agents-How P. aeruginosa can escape antibiotics. Front. Microbiol. 2019, 10, 913. [Google Scholar] [CrossRef]
- Anderl, J.N.; Franklin, M.J.; Stewart, P.S. Role of antibiotic penetration limitation in Klebsiella pneumoniae biofilm resistance to ampicillin and ciprofloxacin. Antimicrob. Agents Chemoter. 2000, 44, 1818–1824. [Google Scholar] [CrossRef] [PubMed]
- Suci, P.A.; Mittelman, M.W.; Yu, F.P.; Geesey, G.G. Investigation of ciprofloxacin penetration into Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 1994, 38, 2125–2133. [Google Scholar] [CrossRef] [PubMed]
- Billings, N.; Millan, M.; Caldara, M.; Rusconi, R.; Tarasova, Y.; Stocker, R.; Ribbeck, K. The extracellular matrix component Psl provides fast-acting antibiotic defense in Pseudomonas aeruginosa biofilms. PLoS Pathog. 2013, 9, e1003526. [Google Scholar] [CrossRef] [PubMed]
- Colvin, K.M.; Gordon, V.D.; Murakami, K.; Borlee, B.R.; Wozniak, D.J.; Wong, G.C.; Parsek, M.R. The Pel polysaccharide can serve a structural and protective role in the biofilm matrix of Pseudomonas aeruginosa. PLoS Pathog. 2011, 7, e1001264. [Google Scholar] [CrossRef] [PubMed]
- Becker, P.; Hufnagle, W.; Peters, G.; Herrmann, M. Detection of differential gene expression in biofilm-forming versus planktonic populations of Staphylococcus aureus using micro-representational-difference analysis. Appl. Environ. Microbiol. 2001, 67, 2958–2965. [Google Scholar] [CrossRef] [PubMed]
- Booth, S.C.; Workentine, M.L.; Wen, J.; Shaykhutdinov, R.; Vogel, H.J.; Ceri, H.; Turner, R.J.; Weljie, A.M. Differences in metabolism between the biofilm and planktonic response to metal stress. J. Proteome Res. 2011, 10, 3190–3199. [Google Scholar] [CrossRef]
- Crumplin, G.C.; Smith, J.T. Nalidixic acid: An antibacterial paradox. Antimicrob. Agents Chemother. 1975, 8, 251–261. [Google Scholar] [CrossRef]
- Uruén, C.; Chopo-Escuin, G.; Tommassen, J.; Mainar-Jaime, R.C.; Arenas, J. Biofilms as promoters of bacterial antibiotic resistance and tolerance. Antibiotics 2020, 10, 3. [Google Scholar] [CrossRef]
- Kolpen, M.; Lerche, C.J.; Kragh, K.N.; Sams, T.; Koren, K.; Jensen, A.S.; Line, L.; Bjarnsholt, T.; Ciofu, O.; Moser, C.; et al. Hyperbaric oxygen sensitizes anoxic Pseudomonas aeruginosa biofilm to ciprofloxacin. Antimicrob. Agents Chemother. 2017, 61, e01024-17. [Google Scholar] [CrossRef]
- Soares, A.; Roussel, V.; Pestel-Caron, M.; Barreau, M.; Caron, F.; Bouffartigues, E.; Chevalier, S.; Etienne, M. Understanding ciprofloxacin failure in Pseudomonas aeruginosa biofilm: Persister cells survive matrix disruption. Front. Microbiol. 2019, 10, 2603. [Google Scholar] [CrossRef]
- Clerch, B.; Rivera, E.; Llagostera, M. Identification of a pKM101 region which confers a slow growth rate and interferes with susceptibility to quinolone in Escherichia coli AB1157. J. Bacteriol. 1996, 178, 5568–5572. [Google Scholar] [CrossRef][Green Version]
- Cheng, G.; Hao, H.; Dai, M.; Liu, Z.; Yuan, Z. Antibacterial action of quinolones: From target to network. Eur. J. Med. Chem. 2013, 66, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Drlica, K.; Malik, M.; Kerns, R.J.; Zhao, X. Quinolone-mediated bacterial death. Antimicrob. Agents Chemother. 2008, 52, 385–392. [Google Scholar] [CrossRef]
- Pacios, O.; Blasco, L.; Bleriot, I.; Fernandez-Garcia, L.; Ambroa, A.; López, M.; Bou, G.; Cantón, R.; Garcia-Contreras, R.; Wood, T.K.; et al. (p)ppGpp and its role in bacterial persistence: New challenges. Antimicrob. Agents Chemother. 2020, 64, e01283-20. [Google Scholar] [CrossRef] [PubMed]
- Sonika, S.; Singh, S.; Mishra, S.; Verma, S. Toxin-antitoxin systems in bacterial pathogenesis. Heliyon 2023, 9, e14220. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; King, K.; Alqahtani, M.; Worden, M.; Muthuraman, P.; Cioffi, C.L.; Bakshi, C.S.; Malik, M. Stringent response governs the oxidative stress resistance and virulence of Francisella tularensis. PLoS ONE 2019, 14, e0224094. [Google Scholar] [CrossRef] [PubMed]
- Potrykus, K.; Cashel, M. (p)ppGpp: Still magical? Annu. Rev. Microbiol. 2008, 62, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Liu, W.; Du, Q.; Zhang, H.; Han, D. Recent advances in bacterial persistence mechanisms. Int. J. Mol. Sci. 2023, 24, 14311. [Google Scholar] [CrossRef]
- Das, B.; Bhadra, R.K. (p)ppGpp Metabolism and antimicrobial resistance in bacterial pathogens. Front. Microbiol. 2020, 11, 563944. [Google Scholar] [CrossRef]
- Viducic, D.; Ono, T.; Murakami, K.; Susilowati, H.; Kayama, S.; Hirota, K.; Miyake, Y. Functional analysis of spoT, relA and dksA genes on quinolone tolerance in Pseudomonas aeruginosa under nongrowing condition. Microbiol. Immunol. 2006, 50, 349–357. [Google Scholar] [CrossRef]
- Dörr, T.; Vulić, M.; Lewis, K. Ciprofloxacin causes persister formation by inducing the TisB toxin in Escherichia coli. PLoS Biol. 2010, 8, e1000317. [Google Scholar] [CrossRef]
- Muthuramalingam, M.; White, J.C.; Murphy, T.; Ames, J.R.; Bourne, C.R. The toxin from a ParDE toxin-antitoxin system found in Pseudomonas aeruginosa offers protection to cells challenged with anti-gyrase antibiotics. Mol. Microbiol. 2019, 111, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Kamruzzaman, M.; Iredell, J. A ParDE-family toxin antitoxin system in major resistance plasmids of Enterobacteriaceae confers antibiotic and heat tolerance. Sci. Rep. 2019, 9, 9872. [Google Scholar] [CrossRef]
- Kang, S.M.; Kim, D.H.; Jin, C.; Lee, B.J. A systematic overview of type II and III toxin-antitoxin systems with a focus on druggability. Toxins 2018, 10, 515. [Google Scholar] [CrossRef]
- Rittershaus, E.S.; Baek, S.H.; Sassetti, C.M. The normalcy of dormancy: Common themes in microbial quiescence. Cell Host Microbe 2013, 13, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Bush, N.G.; Evans-Roberts, K.; Maxwell, A. DNA Topoisomerases. EcoSal Plus 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Ocampo, P.S.; Lázár, V.; Papp, B.; Arnoldini, M.; Abel zur Wiesch, P.; Busa-Fekete, R.; Fekete, G.; Pál, C.; Ackermann, M.; Bonhoeffer, S. Antagonism between bacteriostatic and bactericidal antibiotics is prevalent. Antimicrob. Agents Chemother. 2014, 58, 4573–4582. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Thawng, C.N.; Choi, J.H.; Lee, K.; Cha, C.J. Polymorphism of antibiotic-inactivating enzyme driven by ecology expands the environmental resistome. ISME J. 2018, 12, 267–276. [Google Scholar] [CrossRef]
- Chávez-Jacobo, V.M.; Hernández-Ramírez, K.C.; Romo-Rodríguez, P.; Pérez-Gallardo, R.V.; Campos-García, J.; Gutiérrez-Corona, J.F.; García-Merinos, J.P.; Meza-Carmen, V.; Silva-Sánchez, J.; Ramírez-Díaz, M.I. CrpP is a novel ciprofloxacin-modifying enzyme encoded by the Pseudomonas aeruginosa pUM505 plasmid. Antimicrob. Agents Chemother. 2018, 62, e02629-17. [Google Scholar] [CrossRef]
- Martens, R.; Wetzstein, H.G.; Zadrazil, F.; Capelari, M.; Hoffmann, P.; Schmeer, N. Degradation of the fluoroquinolone enrofloxacin by wood-rotting fungi. Appl. Environ. Microbiol. 1996, 62, 4206–4209. [Google Scholar] [CrossRef]
- Ben Ayed, A.; Akrout, I.; Albert, Q.; Greff, S.; Simmler, C.; Armengaud, J.; Kielbasa, M.; Turbé-Doan, A.; Chaduli, D.; Navarro, D.; et al. Biotransformation of the fluoroquinolone, levofloxacin, by the white-rot fungus Coriolopsis gallica. J. Fungi 2022, 8, 965. [Google Scholar] [CrossRef]
- Čvančarová, M.; Moeder, M.; Filipová, A.; Cajthaml, T. Biotransformation of fluoroquinolone antibiotics by ligninolytic fungi—Metabolites, enzymes and residual antibacterial activity. Chemosphere 2015, 136, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Rusch, M.; Kauschat, A.; Spielmeyer, A.; Römpp, A.; Hausmann, H.; Zorn, H.; Hamscher, G. Biotransformation of the antibiotic danofloxacin by Xylaria longipes leads to an efficient reduction of its antibacterial activity. J. Agric. Food Chem. 2015, 63, 6897–6904. [Google Scholar] [CrossRef] [PubMed]
- Wetzstein, H.G.; Stadler, M.; Tichy, H.V.; Dalhoff, A.; Karl, W. Degradation of ciprofloxacin by basidiomycetes and identification of metabolites generated by the brown rot fungus Gloeophyllum striatum. Appl. Environ. Microbiol. 1999, 65, 1556–1563. [Google Scholar] [CrossRef] [PubMed]
- Gao, N.; Liu, C.X.; Xu, Q.M.; Cheng, J.S.; Yuan, Y.J. Simultaneous removal of ciprofloxacin, norfloxacin, sulfamethoxazole by co-producing oxidative enzymes system of Phanerochaete chrysosporium and Pycnoporus sanguineus. Chemosphere 2018, 195, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Suryadi, H.; Judono, J.J.; Putri, M.R.; Eclessia, A.D.; Ulhaq, J.M.; Agustina, D.N.; Sumiati, T. Biodelignification of lignocellulose using ligninolytic enzymes from white-rot fungi. Heliyon 2022, 8, e08865. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Li, X.; Hu, M.; Li, S.; Zhai, Q.; Jiang, Y. Efficient enzymatic degradation used as pre-stage treatment for norfloxacin removal by activated sludge. Bioprocess. Biosyst. Eng. 2017, 40, 1261–1270. [Google Scholar] [CrossRef]
- Chen, Z.; Li, N.; Lan, Q.; Zhang, X.; Wu, L.; Liu, J.; Yang, R. Laccase inducer Mn2+ inhibited the intracellular degradation of norfloxacin by Phanerochaete chrysosporium. Int. Biodeterior. Biodegrad. 2021, 164, 105300. [Google Scholar] [CrossRef]
- Adjei, M.D.; Heinze, T.M.; Deck, J.; Freeman, J.P.; Williams, A.J.; Sutherland, J.B. Transformation of the antibacterial agent norfloxacin by environmental mycobacteria. Appl. Environ. Microbiol. 2006, 72, 5790–5793. [Google Scholar] [CrossRef]
- Amorim, C.L.; Moreira, I.S.; Maia, A.S.; Tiritan, M.E.; Castro, P.M. Biodegradation of ofloxacin, norfloxacin, and ciprofloxacin as single and mixed substrates by Labrys portucalensis F11. Appl. Microbiol. Biotechnol. 2014, 98, 3181–3190. [Google Scholar] [CrossRef]
- Kim, D.W.; Heinze, T.M.; Kim, B.S.; Schnackenberg, L.K.; Woodling, K.A.; Sutherland, J.B. Modification of norfloxacin by a Microbacterium sp. strain isolated from a wastewater treatment plant. Appl. Environ. Microbiol. 2011, 77, 6100–6108. [Google Scholar] [CrossRef]
- Pan, L.J.; Li, J.; Li, C.X.; Tang, X.D.; Yu, G.W.; Wang, Y. Study of ciprofloxacin biodegradation by a Thermus sp. isolated from pharmaceutical sludge. J. Hazard. Mater. 2018, 343, 59–67. [Google Scholar] [CrossRef]
- Chen, Y.; Rosazza, J.P.; Reese, C.P.; Chang, H.Y.; Nowakowski, M.A.; Kiplinger, J.P. Microbial models of soil metabolism: Biotransformations of danofloxacin. J. Ind. Microbiol. Biotechnol. 1997, 19, 378–384. [Google Scholar] [CrossRef]
- Kim, D.W.; Feng, J.; Chen, H.; Kweon, O.; Gao, Y.; Yu, L.R.; Burrowes, V.J.; Sutherland, J.B. Identification of the enzyme responsible for N-acetylation of norfloxacin by Microbacterium sp. strain 4N2-2. Appl. Environ. Microbiol. 2013, 79, 314–321. [Google Scholar] [CrossRef]
- Millanao, A.R.; Mora, A.Y.; Saavedra, C.; Villagra, N.A.; Mora, G.C.; Hidalgo, A.A. Inactivation of glutamine synthetase-coding gene glnA increases susceptibility to quinolones through increasing outer membrane protein F in Salmonella enterica Serovar Typhi. Front. Microbiol. 2020, 11, 428. [Google Scholar] [CrossRef]
- Blánquez, A.; Guillén, F.; Rodríguez, J.; Arias, M.E.; Hernández, M. The degradation of two fluoroquinolone based antimicrobials by SilA, an alkaline laccase from Streptomyces ipomoeae. World J. Microbiol. Biotechnol. 2016, 32, 52. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, S.; Choudhary, P.; Chatterjee, S.; Akhter, Y. Comparative analysis of SilA-laccase mediated degradation of ciprofloxacin, norfloxacin and ofloxacin and interpretation of the possible catalytic mechanism. J. Biomol. Struct. Dyn. 2024, 42, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, G.A.; Griffin, C.M.; Hooper, D.C. Citrobacter spp. as a source of qnrB alleles. Antimicrob. Agents Chemother. 2011, 55, 4979–4984. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Xu, X.; Guo, Q.; Zhao, X.; Ye, X.; Guo, Y.; Wang, M. Prevalence of the oqxAB gene complex in Klebsiella pneumoniae and Escherichia coli clinical isolates. J. Antimicrob. Chemother. 2012, 67, 1655–1659. [Google Scholar] [CrossRef]
- Tavío, M.M.; Vila, J.; Ruiz, J.; Amicosante, G.; Franceschini, N.; Martín-Sánchez, A.M.; Jiménez de Anta, M.T. In vitro selected fluoroquinolone-resistant mutants of Citrobacter freundii: Analysis of the quinolone resistance acquisition. J. Antimicrob. Chemother. 2000, 45, 521–524. [Google Scholar] [CrossRef]
- Turkmani, A.; Psaroulaki, A.; Christidou, A.; Chochlakis, D.; Tabaa, D.; Tselentis, Y. In vitro-selected resistance to fluoroquinolones in two Brucella strains associated with mutational changes in gyrA. Int. J. Antimicrob. Agents 2008, 32, 227–232. [Google Scholar] [CrossRef]
- Tavío, M.M.; Vila, J.; Ruiz, J.; Martín Sánchez, A.M.; Jiménez de Anta, M.T. Decreased permeability and enhanced proton-dependent active efflux in the development of resistance to quinolones in Morganella morganii. Int. J. Antimicrob. Agents 2000, 14, 157–160. [Google Scholar] [CrossRef]
- Vinué, L.; Corcoran, M.A.; Hooper, D.C.; Jacoby, G.A. Mutations that enhance the ciprofloxacin resistance of Escherichia coli with qnrA1. Antimicrob. Agents Chemother. 2016, 60, 537–545. [Google Scholar] [CrossRef]
- Helling, R.B.; Janes, B.K.; Kimball, H.; Tran, T.; Bundesmann, M.; Check, P.; Phelan, D.; Miller, C. Toxic waste disposal in Escherichia coli. J. Bacteriol. 2002, 184, 3699–3703. [Google Scholar] [CrossRef]
- Webber, M.A.; Whitehead, R.N.; Mount, M.; Loman, N.J.; Pallen, M.J.; Piddock, L.J. Parallel evolutionary pathways to antibiotic resistance selected by biocide exposure. J. Antimicrob. Chemother. 2015, 70, 2241–2248. [Google Scholar] [CrossRef]
- Pietsch, F.; Bergman, J.M.; Brandis, G.; Marcusson, L.L.; Zorzet, A.; Huseby, D.L.; Hughes, D. Ciprofloxacin selects for RNA polymerase mutations with pleiotropic antibiotic resistance effects. J. Antimicrob. Chemother. 2017, 72, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Garoff, L.; Huseby, D.L.; Praski Alzrigat, L.; Hughes, D. Effect of aminoacyl-tRNA synthetase mutations on susceptibility to ciprofloxacin in Escherichia coli. J. Antimicrob. Chemother. 2018, 73, 3285–3292. [Google Scholar] [CrossRef] [PubMed]
- Cesaro, A.; Bettoni, R.R.; Lascols, C.; Mérens, A.; Soussy, C.J.; Cambau, E. Low selection of topoisomerase mutants from strains of Escherichia coli harbouring plasmid-borne qnr genes. J. Antimicrob. Chemother. 2008, 61, 1007–1015. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Kawamura, K.; Arakawa, Y. Contribution of QnrA, a plasmid mediated quinolone resistance peptide, to survival of Escherichia coli exposed to a lethal ciprofloxacin concentration. Jpn. J. Infect. Dis. 2015, 68, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Helling, R.B.; Adams, B.S. Nalidixic acid-resistant auxotrophs of Escherichia coli. J. Bacteriol. 1970, 104, 1027–1029. [Google Scholar] [CrossRef]
- Kaspersen, H.; Sekse, C.; Zeyl Fiskebeck, E.; Slettemeås, J.S.; Simm, R.; Norström, M.; Urdahl, A.M.; Lagesen, K. Dissemination of quinolone-resistant Escherichia coli in the Norwegian broiler and pig production chains and possible persistence in the broiler production environment. Appl. Environ. Microbiol. 2020, 86, e02769-19. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.J.; Ma, C.J.; Kim, J.C.; Ahn, J. Proteomics-based discrimination of differentially expressed proteins in antibiotic-sensitive and antibiotic-resistant Salmonella Typhimurium, Klebsiella pneumoniae, and Staphylococcus aureus. Arch. Microbiol. 2019, 201, 1259–1275. [Google Scholar] [CrossRef]
- Moore, S.D.; Sauer, R.T. Revisiting the mechanism of macrolide-antibiotic resistance mediated by ribosomal protein L22. Proc. Natl. Acad. Sci. USA 2008, 105, 18261–18266. [Google Scholar] [CrossRef]
- Power, E.G.; Phillips, I. Correlation between umuC induction and Salmonella mutagenicity assay for quinolone antimicrobial agents. FEMS Microbiol. Lett. 1993, 112, 251–254. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ysern, P.; Clerch, B.; Castaño, M.; Gibert, I.; Barbé, J.; Llagostera, M. Induction of SOS genes in Escherichia coli and mutagenesis in Salmonella typhimurium by fluoroquinolones. Mutagenesis 1990, 5, 63–66. [Google Scholar] [CrossRef]
- Breidenstein, E.B.; Khaira, B.K.; Wiegand, I.; Overhage, J.; Hancock, R.E. Complex ciprofloxacin resistome revealed by screening a Pseudomonas aeruginosa mutant library for altered susceptibility. Antimicrob. Agents Chemother. 2008, 52, 4486–4491. [Google Scholar] [CrossRef]
- Agnello, M.; Finkel, S.E.; Wong-Beringer, A. Fitness cost of fluoroquinolone resistance in clinical isolates of Pseudomonas aeruginosa differs by type III secretion genotype. Front. Microbiol. 2016, 7, 1591. [Google Scholar] [CrossRef]
- Agnello, M.; Wong-Beringer, A. Differentiation in quinolone resistance by virulence genotype in Pseudomonas aeruginosa. PLoS ONE 2012, 7, e42973. [Google Scholar] [CrossRef]
- Horna, G.; Amaro, C.; Palacios, A.; Guerra, H.; Ruiz, J. High frequency of the exoU+/exoS+ genotype associated with multidrug-resistant “high-risk clones” of Pseudomonas aeruginosa clinical isolates from Peruvian hospitals. Sci. Rep. 2019, 9, 10874. [Google Scholar] [CrossRef]
- Lim, W.S.; Phang, K.K.; Tan, A.H.; Li, S.F.; Ow, D.S. Small colony variants and single nucleotide variations in Pf1 region of PB1 phage-resistant Pseudomonas aeruginosa. Front. Microbiol. 2016, 7, 282. [Google Scholar] [CrossRef]
- Ramiro, R.S.; Costa, H.; Gordo, I. Macrophage adaptation leads to parallel evolution of genetically diverse Escherichia coli small-colony variants with increased fitness in vivo and antibiotic collateral sensitivity. Evol. Appl. 2016, 9, 994–1004. [Google Scholar] [CrossRef] [PubMed]
- Curtis, T.D.; Gram, L.; Knudsen, G.M. The small colony variant of Listeria monocytogenes is more tolerant to antibiotics and has altered survival in RAW 264.7 murine macrophages. Front. Microbiol. 2016, 7, 1056. [Google Scholar] [CrossRef] [PubMed]
- Mitsuyama, J.; Yamada, H.; Maehana, J.; Fukuda, Y.; Kurose, S.; Minami, S.; Todo, Y.; Watanabe, Y.; Narita, H. Characteristics of quinolone-induced small colony variants in Staphylococcus aureus. J. Antimicrob. Chemother. 1997, 39, 697–705. [Google Scholar] [CrossRef]
- Al-Maleki, A.R.; Vellasamy, K.M.; Mariappan, V.; Venkatraman, G.; Tay, S.T.; Vadivelu, J. Transcriptome analysis of Burkholderia pseudomallei SCV reveals an association with virulence, stress resistance and intracellular persistence. Genomics 2020, 112, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Sandoz, K.M.; Popham, D.L.; Beare, P.A.; Sturdevant, D.E.; Hansen, B.; Nair, V.; Heinzen, R.A. Transcriptional profiling of Coxiella burnetii reveals extensive cell wall remodeling in the small cell variant developmental form. PLoS ONE 2016, 11, e0149957. [Google Scholar] [CrossRef] [PubMed]
- Brouillette, E.; Martinez, A.; Boyll, B.J.; Allen, N.E.; Malouin, F. Persistence of a Staphylococcus aureus small-colony variant under antibiotic pressure in vivo. FEMS Immunol. Med. Microbiol. 2004, 41, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.A.; Olsen, R.J.; Long, S.W.; Rosato, A.E.; Musser, J.M. Identification of point mutations in clinical Staphylococcus aureus strains that produce small-colony variants auxotrophic for menadione. Infect. Immun. 2014, 82, 1600–1605. [Google Scholar] [CrossRef]
- de Souza, D.C.; Cogo, L.L.; Palmeiro, J.K.; Dalla-Costa, L.M.; de Oliveira Tomaz, A.P.; Riedi, C.A.; Rosario Filho, N.A. Thymidine-auxotrophic Staphylococcus aureus small-colony variant bacteremia in a patient with cystic fibrosis. Pediatr. Pulmonol. 2020, 55, 1388–1393. [Google Scholar] [CrossRef]
- Hubbard, A.T.M.; Bulgasim, I.; Roberts, A.P. A novel hemA mutation is responsible for a small-colony-variant phenotype in Escherichia coli. Microbiology 2021, 167, 000962. [Google Scholar] [CrossRef]
- Irvine, S.; Bunk, B.; Bayes, H.K.; Spröer, C.; Connolly, J.P.R.; Six, A.; Evans, T.J.; Roe, A.J.; Overmann, J.; Walker, D. Genomic and transcriptomic characterization of Pseudomonas aeruginosa small colony variants derived from a chronic infection model. Microb. Genom. 2019, 5, e000262. [Google Scholar] [CrossRef]
- Precit, M.R.; Wolter, D.J.; Griffith, A.; Emerson, J.; Burns, J.L.; Hoffman, L.R. Optimized in vitro antibiotic susceptibility testing method for small-colony variant Staphylococcus aureus. Antimicrob. Agents Chemother. 2016, 60, 1725–1735. [Google Scholar] [CrossRef] [PubMed]
- Eggertsson, G.; Söll, D. Transfer ribonucleic acid-mediated suppression of termination codons in Escherichia coli. Microbiol. Rev. 1988, 52, 354–374. [Google Scholar] [CrossRef] [PubMed]
- Minarini, L.A.R.; Darini, A.L.C. Mutations in the quinolone resistance-determining regions of gyrA and parC in Enterobacteriaceae isolates from Brazil. Braz. J. Microbiol. 2012, 43, 1309–1314. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).