
Integrating Artificial Intelligence for Drug Discovery in the Context of Revolutionizing Drug Delivery



Abstract
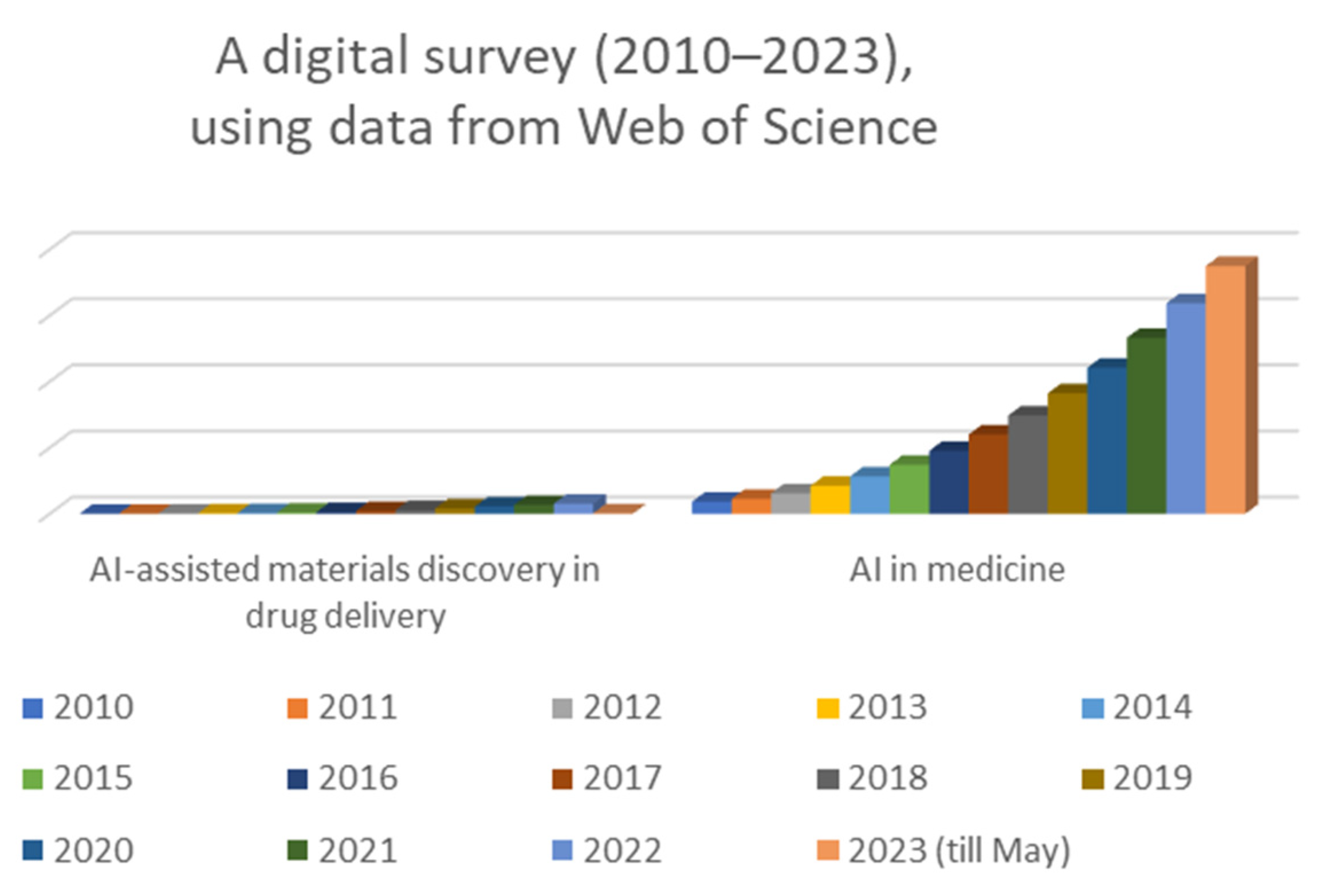
1. Introduction
1.1. Historical Background and the Concept of AI in Medicine
1.2. Statement of Significance
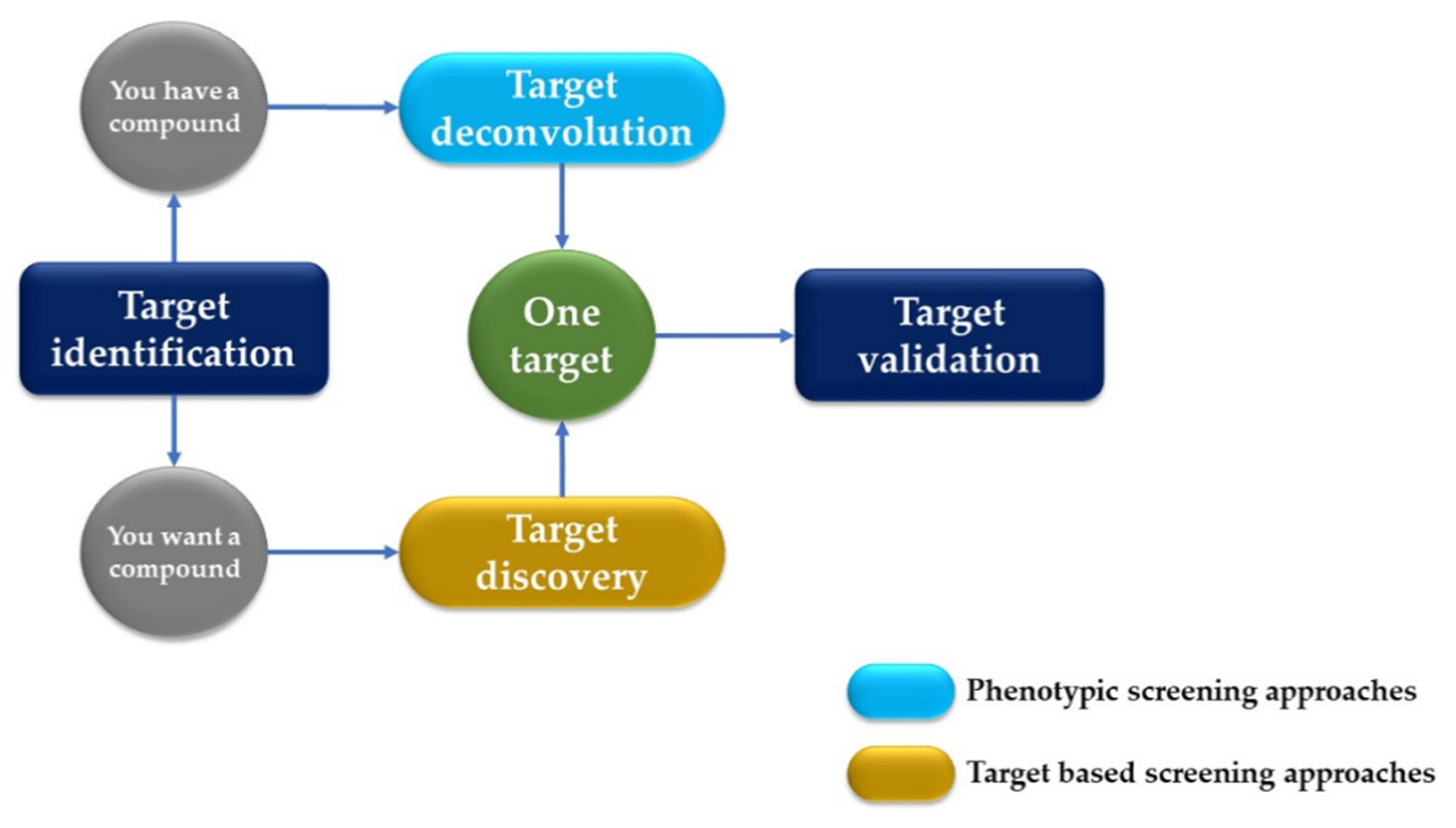
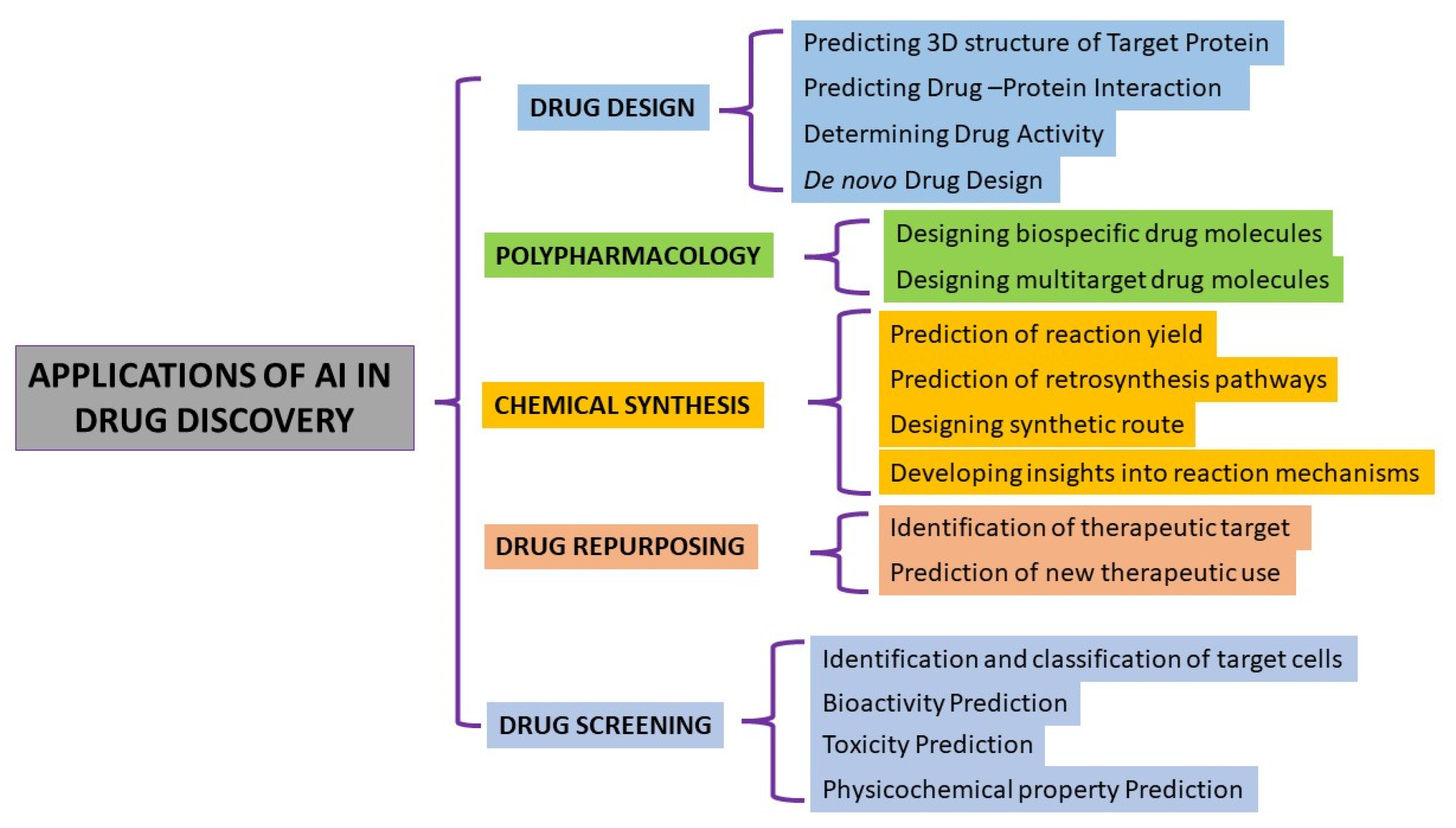
2. AI in Discovering New Drugs
- (i)
- Target Identification and Validation:
- (ii)
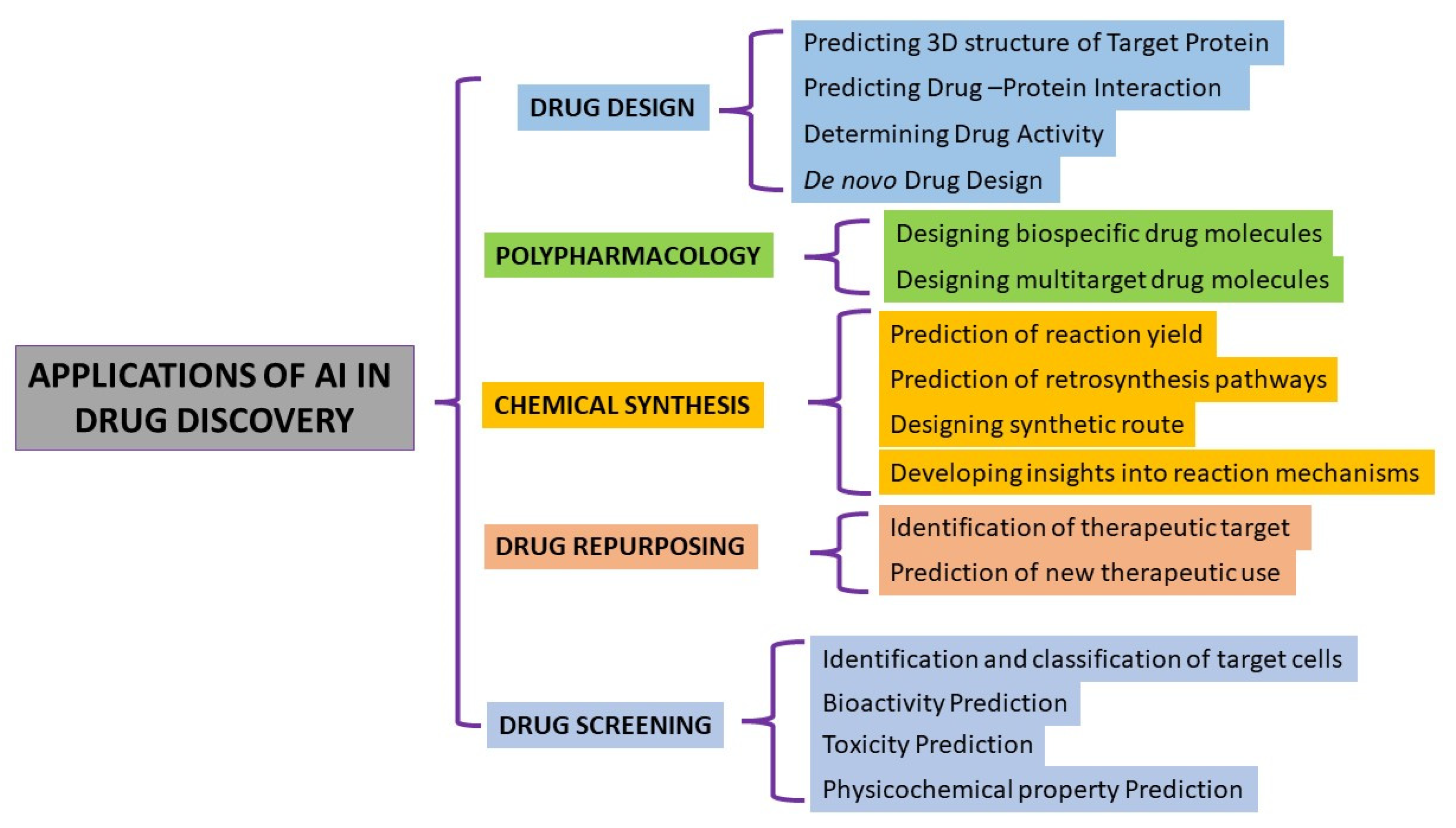
- Virtual Screening and Drug Design:
- (iii)
- Prediction of Drug Properties
- (iv)
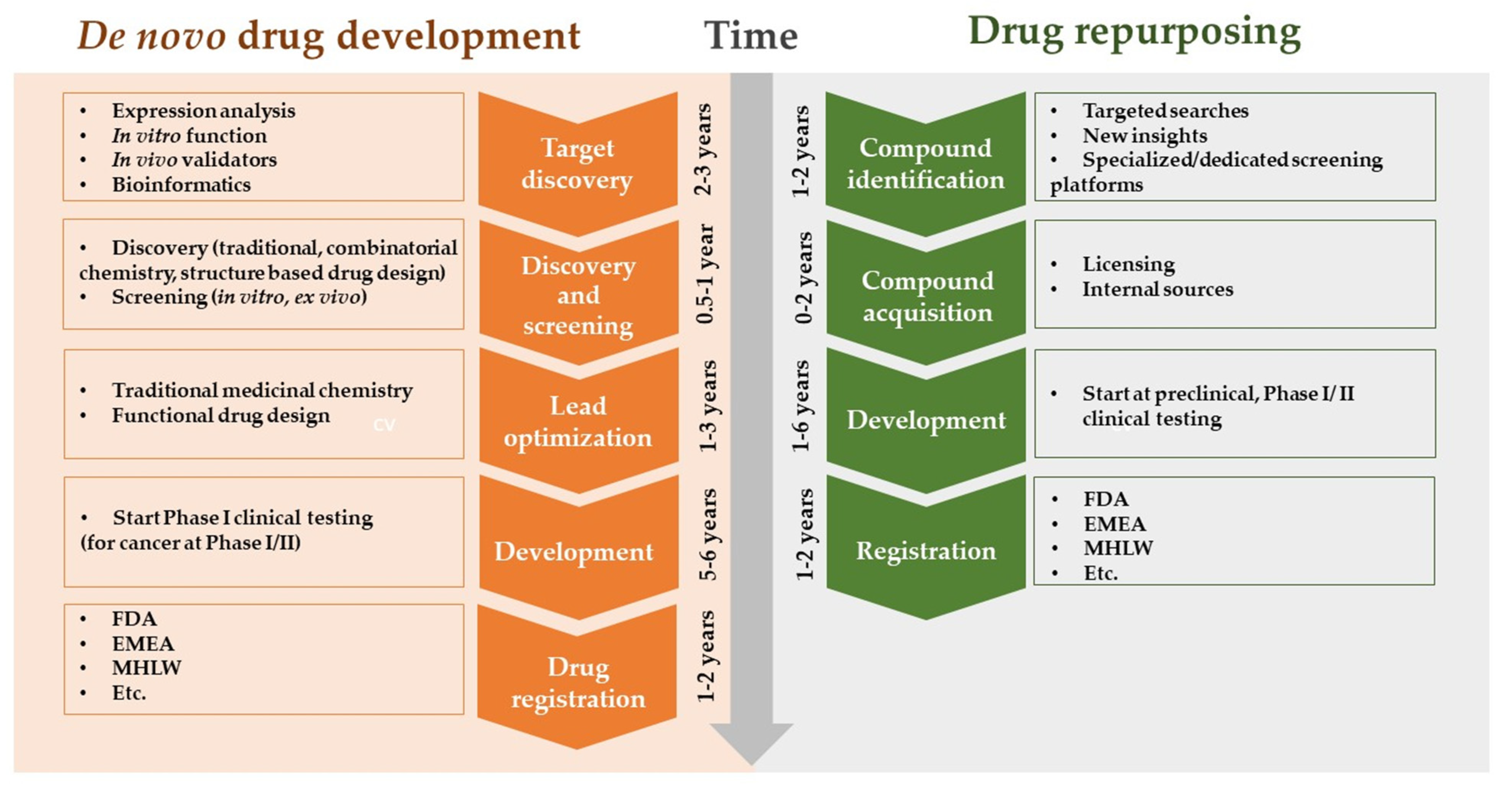
- Repositioning of Existing Drugs
2.1. AI Techniques Used in Material Discovery
2.1.1. Supervised Learning Methods
2.1.2. Unsupervised Learning Methods
2.1.3. AI Algorithms Used in Drug Discovery
Machine Learning and Deep Learning
- (i)
- Machine Learning
- (ii)
- Deep Learning
- (iii)
- High-throughput Density Functional Theory (DFT)
- (iv)
- Natural Language Processing (NLP)
- (v)
- Text mining
- (vi)
- Generative Adversarial Networks (GANs)
- (vii)
- Transfer Learning
- (viii)
- Active Learning
3. Studies of AI-Assisted Drug Discovery
3.1. AI Programs and Platforms Used for Drug Discovery
3.2. Example of AI in Drug Design
3.3. Examples Cases of AI Used in Polypharmacology
3.4. Example Cases of AI in Drug Chemical Synthesis
3.5. Case Examples of AI Used in Drug Screening
3.6. AI in Drug Discovery and Repurposing
4. The Market Prospects of AI-Based Drug Development
5. Challenges and Limitations in AI-Assisted Drug Discovery
6. Future Directions and Opportunities in AI-Assisted Drugs Discovery
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AI | Artificial intelligence |
| DL | Deep learning |
| ML | Machine learning |
| ANN | Artificial neural network |
| CT | Computed Tomography Scan |
| MRI | Magnetic resonance imaging |
| ML | Machine Learning |
| KronRLS | Kronecker-regularized least squares |
| DTBA | Drug target binding affinity |
| GPCRs | G protein-coupled receptors |
| MLR | Multiple linear regression |
| DT | Drug target |
| LR | Logistic regression |
| SVM | Support vector machine |
| CNN | Convolution neural network |
| RNN | Recurrent neural network |
| GAN | Generative adversarial network |
| PCA | Principal-component analysis |
| t-SNE | t-distributed stochastic neighbor embedding |
| SVR | Support vector regression |
| Lasso | Least absolute shrinkage and selection operator |
| PBAEs | poly(beta-amino ester)s |
| DFT | High-throughput density functional theory |
| ADMET | Absorption, distribution, metabolism, excretion, and toxicity |
| QSAR | Quantitative structure-activity relationship |
| NLP | Natural language processing |
References
- Kaul, V.; Enslin, S.; Gross, S.A. History of artificial intelligence in medicine. Gastrointest. Endosc. 2020, 92, 807–812. [Google Scholar] [CrossRef]
- Rudrapal, M.; Kirboga, K.K.; Abdalla, M.; Maji, S. Explainable artificial intelligence-assisted virtual screening and bioinformatics approaches for effective bioactivity prediction of phenolic cyclooxygenase-2 (COX-2) inhibitors using PubChem molecular fingerprints. Mol. Divers. 2024. [Google Scholar] [CrossRef] [PubMed]
- Blanco-González, A.; Cabezón, A.; Seco-González, A.; Conde-Torres, D.; Antelo-Riveiro, P.; Piñeiro, Á.; Garcia-Fandino, R. The Role of AI in Drug Discovery: Challenges, Opportunities, and Strategies. Pharmaceuticals 2023, 16, 891. [Google Scholar] [CrossRef]
- Malik, P.; Pathania, M.; Rathaur, V.K. Overview of artificial intelligence in medicine. J. Fam. Med. Prim. Care 2019, 8, 2328–2331. [Google Scholar] [CrossRef]
- Briganti, G.; Le Moine, O. Artificial Intelligence in Medicine: Today and Tomorrow. Front. Med. 2020, 7, 27. Available online: https://www.frontiersin.org/articles/10.3389/fmed.2020.00027 (accessed on 20 October 2023). [CrossRef]
- Yoon, H.J.; Jeong, Y.J.; Kang, H.; Jeong, J.E.; Kang, D.-Y. Medical Image Analysis Using Artificial Intelligence. Prog. Med. Phys. 2019, 30, 49–58. [Google Scholar] [CrossRef]
- Wu, J.; Chen, J.; Cai, J. Application of Artificial Intelligence in Gastrointestinal Endoscopy. J. Clin. Gastroenterol. 2021, 55, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Kaur, T.; Diwakar, A.; Mirpuri, P.; Tripathi, M.; Chandra, P.S.; Gandhi, T.K. Artificial Intelligence in Epilepsy. Neurol. India 2021, 69, 560. [Google Scholar] [CrossRef]
- Sohail, A. Genetic Algorithms in the Fields of Artificial Intelligence and Data Sciences. Ann. Data Sci. 2023, 10, 1007–1018. [Google Scholar] [CrossRef]
- Lee, S.; Kim, H.-S. Prospect of Artificial Intelligence Based on Electronic Medical Record. J. Lipid Atheroscler. 2021, 10, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Contreras, I.; Vehi, J. Artificial Intelligence for Diabetes Management and Decision Support: Literature Review. J. Med. Internet Res. 2018, 20, e10775. [Google Scholar] [CrossRef]
- Davis, C.R.; Murphy, K.J.; Curtis, R.G.; Maher, C.A. A Process Evaluation Examining the Performance, Adherence, and Acceptability of a Physical Activity and Diet Artificial Intelligence Virtual Health Assistant. Int. J. Environ. Res. Public Health 2020, 17, 9137. [Google Scholar] [CrossRef]
- Bhatt, T.K.; Nimesh, S. The Design and Development of Novel Drugs and Vaccines: Principles and Protocols; Academic Press: Cambridge, MA, USA, 2021; ISBN 978-0-12-821475-6. [Google Scholar]
- Paul, D.; Sanap, G.; Shenoy, S.; Kalyane, D.; Kalia, K.; Tekade, R.K. Artificial intelligence in drug discovery and development. Drug Discov. Today 2021, 26, 80–93. [Google Scholar] [CrossRef]
- Mathai, N.; Chen, Y.; Kirchmair, J. Validation strategies for target prediction methods. Brief. Bioinform. 2020, 21, 791–802. [Google Scholar] [CrossRef]
- Gupta, R.; Srivastava, D.; Sahu, M.; Tiwari, S.; Ambasta, R.K.; Kumar, P. Artificial intelligence to deep learning: Machine intelligence approach for drug discovery. Mol. Divers. 2021, 25, 1315–1360. [Google Scholar] [CrossRef]
- Yaseen, B.T.; Kurnaz, S. Drug–target interaction prediction using artificial intelligence. Appl. Nanosci. 2023, 13, 3335–3345. [Google Scholar] [CrossRef]
- Kumar, R.; Sharma, A.; Siddiqui, M.H.; Tiwari, R.K. Prediction of Drug-Plasma Protein Binding Using Artificial Intelligence Based Algorithms. Comb. Chem. High Throughput Screen. 2018, 21, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Romeo-Guitart, D.; Forés, J.; Herrando-Grabulosa, M.; Valls, R.; Leiva-Rodríguez, T.; Galea, E.; González-Pérez, F.; Navarro, X.; Petegnief, V.; Bosch, A.; et al. Neuroprotective Drug for Nerve Trauma Revealed Using Artificial Intelligence. Sci. Rep. 2018, 8, 1879. [Google Scholar] [CrossRef] [PubMed]
- Rifaioglu, A.S.; Atas, H.; Martin, M.J.; Cetin-Atalay, R.; Atalay, V.; Doğan, T. Recent applications of deep learning and machine intelligence on in silico drug discovery: Methods, tools and databases. Brief. Bioinform. 2019, 20, 1878–1912. [Google Scholar] [CrossRef] [PubMed]
- Zhang, O.; Zhang, J.; Jin, J.; Zhang, X.; Hu, R.; Shen, C.; Cao, H.; Du, H.; Kang, Y.; Deng, Y.; et al. ResGen is a pocket-aware 3D molecular generation model based on parallel multiscale modelling. Nat. Mach. Intell. 2023, 5, 1020–1030. [Google Scholar] [CrossRef]
- Gentile, F.; Yaacoub, J.C.; Gleave, J.; Fernandez, M.; Ton, A.-T.; Ban, F.; Stern, A.; Cherkasov, A. Artificial intelligence–enabled virtual screening of ultra-large chemical libraries with deep docking. Nat. Protoc. 2022, 17, 672–697. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, K.A.; Huang, X. Machine Learning-based Virtual Screening and Its Applications to Alzheimer’s Drug Discovery: A Review. Curr. Pharm. Des. 2018, 24, 3347–3358. [Google Scholar] [CrossRef]
- Jiménez-Luna, J.; Grisoni, F.; Schneider, G. Drug discovery with explainable artificial intelligence. Nat. Mach. Intell. 2020, 2, 573–584. [Google Scholar] [CrossRef]
- Selvaraj, C.; Chandra, I.; Singh, S.K. Artificial intelligence and machine learning approaches for drug design: Challenges and opportunities for the pharmaceutical industries. Mol. Divers. 2022, 26, 1893–1913. [Google Scholar] [CrossRef] [PubMed]
- Sadybekov, A.V.; Katritch, V. Computational approaches streamlining drug discovery. Nature 2023, 616, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Boobier, S.; Hose, D.R.J.; Blacker, A.J.; Nguyen, B.N. Machine learning with physicochemical relationships: Solubility prediction in organic solvents and water. Nat. Commun. 2020, 11, 5753. [Google Scholar] [CrossRef]
- Fagerholm, U.; Hellberg, S.; Spjuth, O. Advances in Predictions of Oral Bioavailability of Candidate Drugs in Man with New Machine Learning Methodology. Molecules 2021, 26, 2572. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, H.; Ai, H.; Hu, H.; Li, S.; Zhao, J.; Liu, H. Applications of Machine Learning Methods in Drug Toxicity Prediction. Curr. Top. Med. Chem. 2018, 18, 987–997. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Rani, P.; Dutta, K.; Kumar, V. Artificial intelligence techniques for prediction of drug synergy in malignant diseases: Past, present, and future. Comput. Biol. Med. 2022, 144, 105334. [Google Scholar] [CrossRef]
- Gaweda, A.E.; Aronoff, G.R.; Brier, M.E. Use of Artificial Intelligence/Machine Learning for Individualization of Drug Dosing in Dialysis Patients. In Technological Advances in Care of Patients with Kidney Diseases; Saggi, S.J., Salifu, M.O., Eds.; Springer International Publishing: Cham, Switzerland, 2022; pp. 179–187. [Google Scholar] [CrossRef]
- Vadapalli, S.; Abdelhalim, H.; Zeeshan, S.; Ahmed, Z. Artificial intelligence and machine learning approaches using gene expression and variant data for personalized medicine. Brief. Bioinform. 2022, 23, bbac191. [Google Scholar] [CrossRef] [PubMed]
- Vo, T.H.; Nguyen, N.T.K.; Kha, Q.H.; Le, N.Q.K. On the road to explainable AI in drug-drug interactions prediction: A systematic review. Comput. Struct. Biotechnol. J. 2022, 20, 2112–2123. [Google Scholar] [CrossRef] [PubMed]
- Dara, S.; Dhamercherla, S.; Jadav, S.S.; Babu, C.M.; Ahsan, M.J. Machine Learning in Drug Discovery: A Review. Artif. Intell. Rev. 2022, 55, 1947–1999. [Google Scholar] [CrossRef]
- Zeng, X.; Xiang, H.; Yu, L.; Wang, J.; Li, K.; Nussinov, R.; Cheng, F. Accurate prediction of molecular properties and drug targets using a self-supervised image representation learning framework. Nat. Mach. Intell. 2022, 4, 1004–1016. [Google Scholar] [CrossRef]
- Brito-Pacheco, C.; Brito-Loeza, C.; Martin-Gonzalez, A. A regularized logistic regression based model for supervised learning. J. Algorithms Comput. Technol. 2020, 14, 1748302620971535. [Google Scholar] [CrossRef]
- Pisner, D.A.; Schnyer, D.M. Chapter 6—Support vector machine. In Machine Learning; Mechelli, A., Vieira, S., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 101–121. [Google Scholar] [CrossRef]
- Smaldone, A.M.; Kyro, G.W.; Batista, V.S. Quantum Convolutional Neural Networks for Multi-Channel Supervised Learning. arXiv 2023, arXiv:2305.18961. [Google Scholar] [CrossRef]
- Kaur, M.; Mohta, A. A Review of Deep Learning with Recurrent Neural Network. In Proceedings of the 2019 International Conference on Smart Systems and Inventive Technology (ICSSIT), Tirunelveli, India, 27–29 November 2019; IEEE: Tirunelveli, India, 2019; pp. 460–465. [Google Scholar] [CrossRef]
- Lim, H.; Chon, K.-W.; Kim, M.-S. Active learning using Generative Adversarial Networks for improving generalization and avoiding distractor points. Expert Syst. Appl. 2023, 227, 120193. [Google Scholar] [CrossRef]
- Sinaga, K.P.; Yang, M.-S. Unsupervised K-Means Clustering Algorithm. IEEE Access 2020, 8, 80716–80727. [Google Scholar] [CrossRef]
- Lakshmi, B.S.S.S.; Ravi Kiran Varma, P. Machine Learning for Drug Discovery Using Agglomerative Hierarchical Clustering. In Soft Computing and Signal Processing; Reddy, V.S., Prasad, V.K., Wang, J., Reddy, K.T.V., Eds.; Smart Innovation, Systems and Technologies Book Series; Springer Nature: Singapore, 2023; pp. 127–137. [Google Scholar] [CrossRef]
- Lever, J.; Krzywinski, M.; Altman, N. Points of Significance: Principal component analysis. Nat. Methods 2017, 14, 641–643. [Google Scholar] [CrossRef]
- Balamurali, M. t-Distributed Stochastic Neighbor Embedding. In Encyclopedia of Mathematical Geosciences; Daya Sagar, B.S., Cheng, Q., McKinley, J., Agterberg, F., Eds.; Encyclopedia of Earth Sciences Series; Springer International Publishing: Cham, Switzerland, 2020; pp. 1–9. [Google Scholar] [CrossRef]
- Rickert, C.A.; Lieleg, O. Machine learning approaches for biomolecular, biophysical, and biomaterials research. Biophys. Rev. 2022, 3, 021306. [Google Scholar] [CrossRef]
- Stern, M.; Arinze, C.; Perez, L.; Palmer, S.E.; Murugan, A. Supervised learning through physical changes in a mechanical system. Proc. Natl. Acad. Sci. USA 2020, 117, 14843–14850. [Google Scholar] [CrossRef] [PubMed]
- Usama, M.; Qadir, J.; Raza, A.; Arif, H.; Yau, K.A.; Elkhatib, Y.; Hussain, A.; Al-Fuqaha, A. Unsupervised Machine Learning for Networking: Techniques, Applications and Research Challenges. IEEE Access 2019, 7, 65579–65615. [Google Scholar] [CrossRef]
- Talevi, A.; Morales, J.F.; Hather, G.; Podichetty, J.T.; Kim, S.; Bloomingdale, P.C.; Kim, S.; Burton, J.; Brown, J.D.; Winterstein, A.G.; et al. Machine Learning in Drug Discovery and Development Part 1: A Primer. CPT Pharmacomet. Syst. Pharmacol. 2020, 9, 129–142. [Google Scholar] [CrossRef]
- Zhou, Z.; Li, X.; Zare, R.N. Optimizing Chemical Reactions with Deep Reinforcement Learning. ACS Cent. Sci. 2017, 3, 1337–1344. [Google Scholar] [CrossRef]
- Kramer, O. Scikit-Learn. In Machine Learning for Evolution Strategies; Kramer, O., Ed.; Studies in Big Data; Springer International Publishing: Cham, Switzerland, 2016; pp. 45–53. [Google Scholar] [CrossRef]
- Imambi, S.; Prakash, K.B.; Kanagachidambaresan, G.R. PyTorch. In Programming with TensorFlow: Solution for Edge Computing Applications; Prakash, K.B., Kanagachidambaresan, G.R., Eds.; EAI/Springer Innovations in Communication and Computing; Springer International Publishing: Cham, Switzerland, 2021; pp. 87–104. [Google Scholar] [CrossRef]
- Gulli, A.; Pal, S. Deep Learning with Keras; Packt Publishing Ltd.: Birmingham, UK, 2017. [Google Scholar]
- Priya, S.; Tripathi, G.; Singh, D.B.; Jain, P.; Kumar, A. Machine learning approaches and their applications in drug discovery and design. Chem. Biol. Drug Des. 2022, 100, 136–153. [Google Scholar] [CrossRef]
- Pandis, N. Linear regression. Am. J. Orthod. Dentofac. Orthop. 2016, 149, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Taskinen, J.; Yliruusi, J. Prediction of physicochemical properties based on neural network modelling. Adv. Drug Deliv. Rev. 2003, 55, 1163–1183. [Google Scholar] [CrossRef]
- Suprapto, S.; Nikmah, Y.L. Ridge and Lasso Regression for Feature Selection of Overlapping Ibuprofen and Paracetamol UV Spectra. Moroc. J. Chem. 2023, 11, 11–229. [Google Scholar] [CrossRef]
- Roozbeh, M.; Arashi, M.; Hamzah, N.A. Generalized Cross-Validation for Simultaneous Optimization of Tuning Parameters in Ridge Regression. Iran. J. Sci. Technol. Trans. Sci. 2020, 44, 473–485. [Google Scholar] [CrossRef]
- Kim, Y.; Hao, J.; Mallavarapu, T.; Park, J.; Kang, M. Hi-LASSO: High-Dimensional LASSO. IEEE Access 2019, 7, 44562–44573. [Google Scholar] [CrossRef]
- Abdulhafedh, A. Comparison between Common Statistical Modeling Techniques Used in Research, Including: Discriminant Analysis vs Logistic Regression, Ridge Regression vs LASSO, and Decision Tree vs Random Forest. Open Access Libr. J. 2022, 9, 1–19. [Google Scholar] [CrossRef]
- Patel, L.; Shukla, T.; Huang, X.; Ussery, D.W.; Wang, S. Machine Learning Methods in Drug Discovery. Molecules 2020, 25, 5277. [Google Scholar] [CrossRef] [PubMed]
- Peña-Guerrero, J.; Nguewa, P.A.; García-Sosa, A.T. Machine learning, artificial intelligence, and data science breaking into drug design and neglected diseases. WIREs Comput. Mol. Sci. 2021, 11, e1513. [Google Scholar] [CrossRef]
- Ozdemir, S.; Susarla, D. Feature Engineering Made Easy: Identify Unique Features from Your Dataset in Order to Build Powerful Machine Learning Systems; Packt Publishing Ltd.: Birmingham, UK, 2018. [Google Scholar]
- Chauhan, N.K.; Singh, K. A Review on Conventional Machine Learning vs Deep Learning. In Proceedings of the 2018 International Conference on Computing, Power and Communication Technologies (GUCON), Greater Noida, India, 28–29 September 2018; IEEE: Greater Noida, India, 2018; pp. 347–352. [Google Scholar] [CrossRef]
- Janiesch, C.; Zschech, P.; Heinrich, K. Machine learning and deep learning. Electron. Mark. 2021, 31, 685–695. [Google Scholar] [CrossRef]
- Gong, D.; Ben-Akiva, E.; Singh, A.; Yamagata, H.; Est-Witte, S.; Shade, J.K.; Trayanova, N.A.; Green, J.J. Machine learning guided structure function predictions enable in silico nanoparticle screening for polymeric gene delivery. Acta Biomater. 2022, 154, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Taye, M.M. Understanding of Machine Learning with Deep Learning: Architectures, Workflow, Applications and Future Directions. Computers 2023, 12, 91. [Google Scholar] [CrossRef]
- Choudhary, K.; Kalish, I.; Beams, R.; Tavazza, F. High-throughput Identification and Characterization of Two-dimensional Materials using Density functional theory. Sci. Rep. 2017, 7, 5179. [Google Scholar] [CrossRef]
- Friedman, R. Computational studies of protein–drug binding affinity changes upon mutations in the drug target. WIREs Comput. Mol. Sci. 2022, 12, e1563. [Google Scholar] [CrossRef]
- Varadharajan, V.; Arumugam, G.S.; Shanmugam, S. Isatin-based virtual high throughput screening, molecular docking, DFT, QM/MM, MD and MM-PBSA study of novel inhibitors of SARS-CoV-2 main protease. J. Biomol. Struct. Dyn. 2022, 40, 7852–7867. [Google Scholar] [CrossRef]
- Joseph Sahayarayan, J.; Soundar Rajan, K.; Nachiappan, M.; Prabhu, D.; Guru Raj Rao, R.; Jeyakanthan, J.; Hossam Mahmoud, A.; Mohammed, O.B.; Morgan, A.M.A. Identification of potential drug target in malarial disease using molecular docking analysis. Saudi J. Biol. Sci. 2020, 27, 3327–3333. [Google Scholar] [CrossRef]
- Faris, A.; Ibrahim, I.M.; Hadni, H.; Elhallaoui, M. High-throughput virtual screening of phenylpyrimidine derivatives as selective JAK3 antagonists using computational methods. J. Biomol. Struct. Dyn. 2023, 1–26. [Google Scholar] [CrossRef]
- Sohlenius-Sternbeck, A.-K.; Terelius, Y. Evaluation of ADMET Predictor in Early Discovery Drug Metabolism and Pharmacokinetics Project Work. Drug Metab. Dispos. 2022, 50, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Öeren, M.; Walton, P.J.; Hunt, P.A.; Ponting, D.J.; Segall, M.D. Predicting reactivity to drug metabolism: Beyond P450s—Modelling FMOs and UGTs. J. Comput. Aided Mol. Des. 2021, 35, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Rydberg, P.; Jørgensen, F.S.; Olsen, L. Use of density functional theory in drug metabolism studies. Expert Opin. Drug Metab. Toxicol. 2014, 10, 215–227. [Google Scholar] [CrossRef]
- Smirnova, A.; Yablonskiy, M.; Petrov, V.; Mitrofanov, A. DFT Prediction of Radiolytic Stability of Conformationally Flexible Ligands. Energies 2023, 16, 257. [Google Scholar] [CrossRef]
- Alzain, A.A.; Elbadwi, F.A.; Alsamani, F.O. Discovery of novel TMPRSS2 inhibitors for COVID-19 using in silico fragment-based drug design, molecular docking, molecular dynamics, and quantum mechanics studies. Inform. Med. Unlocked 2022, 29, 100870. [Google Scholar] [CrossRef]
- Bhatnagar, R.; Sardar, S.; Beheshti, M.; Podichetty, J.T. How can natural language processing help model informed drug development?: A review. JAMIA Open 2022, 5, ooac043. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Dharssi, S.; Wu, M.; Li, J.; Lu, Z. Text Mining for Drug Discovery. In Bioinformatics and Drug Discovery; Larson, R.S., Oprea, T.I., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2019; pp. 231–252. [Google Scholar] [CrossRef]
- Dumitriu, A.; Molony, C.; Daluwatte, C. Graph-Based Natural Language Processing for the Pharmaceutical Industry. In Provenance in Data Science: From Data Models to Context-Aware Knowledge Graphs; Sikos, L.F., Seneviratne, O.W., McGuinness, D.L., Eds.; Advanced Information and Knowledge Processing; Springer International Publishing: Cham, Switzerland, 2021; pp. 75–110. [Google Scholar] [CrossRef]
- Study of the Drug-Related Adverse Events with the Help of Electronic Health Records and Natural Language Processing—ProQuest. Available online: https://www.proquest.com/openview/e0e053ffe5b850bd656912f47db18b77/1?pq-origsite=gscholar&cbl=5444811 (accessed on 23 October 2023).
- Corcoran, C.M.; Mittal, V.A.; Bearden, C.E.; Gur, R.E.; Hitczenko, K.; Bilgrami, Z.; Savic, A.; Cecchi, G.A.; Wolff, P. Language as a biomarker for psychosis: A natural language processing approach. Schizophr. Res. 2020, 226, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Jang, G.; Lee, T.; Hwang, S.; Park, C.; Ahn, J.; Seo, S.; Hwang, Y.; Yoon, Y. PISTON: Predicting drug indications and side effects using topic modeling and natural language processing. J. Biomed. Inform. 2018, 87, 96–107. [Google Scholar] [CrossRef]
- Piñero, J.; Bravo, À.; Queralt-Rosinach, N.; Gutiérrez-Sacristán, A.; Deu-Pons, J.; Centeno, E.; García-García, J.; Sanz, F.; Furlong, L.I. DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2017, 45, D833–D839. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Santos, A.; von Mering, C.; Jensen, L.J.; Bork, P.; Kuhn, M. STITCH 5: Augmenting protein–chemical interaction networks with tissue and affinity data. Nucleic Acids Res. 2016, 44, D380–D384. [Google Scholar] [CrossRef]
- Salehi, P.; Chalechale, A.; Taghizadeh, M. Generative Adversarial Networks (GANs): An Overview of Theoretical Model, Evaluation Metrics, and Recent Developments. arXiv 2020, arXiv:2005.13178. [Google Scholar] [CrossRef]
- Blanchard, A.E.; Stanley, C.; Bhowmik, D. Using GANs with adaptive training data to search for new molecules. J. Cheminformatics 2021, 13, 14. [Google Scholar] [CrossRef]
- Abbasi, M.; Santos, B.P.; Pereira, T.C.; Sofia, R.; Monteiro, N.R.C.; Simões, C.J.V.; Brito, R.M.M.; Ribeiro, B.; Oliveira, J.L.; Arrais, J.P. Designing optimized drug candidates with Generative Adversarial Network. J. Cheminformatics 2022, 14, 40. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, F.; Guan, J.; Kong, Z.; Shi, L.; Zhou, S. GANs for Molecule Generation in Drug Design and Discovery. In Generative Adversarial Learning: Architectures and Applications; Razavi-Far, R., Ruiz-Garcia, A., Palade, V., Schmidhuber, J., Eds.; Intelligent Systems Reference Library; Springer International Publishing: Cham, Switzerland, 2022; pp. 233–273. [Google Scholar] [CrossRef]
- Barcelos, M.P.; Gomes, S.Q.; Federico, L.B.; Francischini, I.A.G.; Hage-Melim, L.I.d.S.; Silva, G.M.; de Paula da Silva, C.H.T. Lead Optimization in Drug Discovery. In Research Topics in Bioactivity, Environment and Energy: Experimental and Theoretical Tools; Taft, C.A., de Lazaro, S.R., Eds.; Engineering Materials; Springer International Publishing: Cham, Switzerland, 2022; pp. 481–500. [Google Scholar] [CrossRef]
- Lin, E.; Lin, C.-H.; Lane, H.-Y. Relevant Applications of Generative Adversarial Networks in Drug Design and Discovery: Molecular De Novo Design, Dimensionality Reduction, and De Novo Peptide and Protein Design. Molecules 2020, 25, 3250. [Google Scholar] [CrossRef]
- Wu, B.; Li, L.; Cui, Y.; Zheng, K. Cross-Adversarial Learning for Molecular Generation in Drug Design. Front. Pharmacol. 2022, 12, 827606. Available online: https://www.frontiersin.org/articles/10.3389/fphar.2021.827606 (accessed on 20 October 2023). [CrossRef] [PubMed]
- Zhao, L.; Wang, J.; Pang, L.; Liu, Y.; Zhang, J. GANsDTA: Predicting Drug-Target Binding Affinity Using GANs. Front. Genet. 2020, 10, 1243. Available online: https://www.frontiersin.org/articles/10.3389/fgene.2019.01243 (accessed on 20 October 2023). [CrossRef] [PubMed]
- Gan, J.; Liu, J.; Liu, Y.; Chen, S.; Dai, W.; Xiao, Z.-X.; Cao, Y. DrugRep: An automatic virtual screening server for drug repurposing. Acta Pharmacol. Sin. 2023, 44, 888–896. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.; Augustin, A.I.; Dunlop, A.; Sukumaran, R.; Dheer, S.; Zavalny, A.; Haslam, O.; Austin, T.; Donchez, J.; Tripathi, P.K.; et al. Recent advances and application of generative adversarial networks in drug discovery, development, and targeting. Artif. Intell. Life Sci. 2022, 2, 100045. [Google Scholar] [CrossRef]
- Sureyya Rifaioglu, A.; Nalbat, E.; Atalay, V.; Jesus Martin, M.; Cetin-Atalay, R.; Doğan, T. DEEPScreen: High performance drug–target interaction prediction with convolutional neural networks using 2-D structural compound representations. Chem. Sci. 2020, 11, 2531–2557. [Google Scholar] [CrossRef]
- Cai, C.; Wang, S.; Xu, Y.; Zhang, W.; Tang, K.; Ouyang, Q.; Lai, L.; Pei, J. Transfer Learning for Drug Discovery. J. Med. Chem. 2020, 63, 8683–8694. [Google Scholar] [CrossRef]
- Zhuang, D.; Ibrahim, A.K. Deep Learning for Drug Discovery: A Study of Identifying High Efficacy Drug Compounds Using a Cascade Transfer Learning Approach. Appl. Sci. 2021, 11, 7772. [Google Scholar] [CrossRef]
- Miller, E.; Hansch, C. Structure-Activity Analysis of Tetrahydrofolate Analogs Using Substituent Constants and Regression Analysis. J. Pharm. Sci. 1967, 56, 92–97. [Google Scholar] [CrossRef]
- Schneider, N.; Jäckels, C.; Andres, C.; Hutter, M.C. Gradual in Silico Filtering for Druglike Substances. J. Chem. Inf. Model. 2008, 48, 613–628. [Google Scholar] [CrossRef] [PubMed]
- Jorissen, R.N.; Gilson, M.K. Virtual Screening of Molecular Databases Using a Support Vector Machine. J. Chem. Inf. Model. 2005, 45, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Wang, J.; Li, Y. ADME Evaluation in Drug Discovery. 8. The Prediction of Human Intestinal Absorption by a Support Vector Machine. J. Chem. Inf. Model. 2007, 47, 2408–2415. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Ramsundar, B.; Feinberg, E.N.; Gomes, J.; Geniesse, C.; Pappu, A.S.; Leswing, K.; Pande, V. MoleculeNet: A benchmark for molecular machine learning. Chem. Sci. 2018, 9, 513–530. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Sheridan, R.P.; Liaw, A.; Dahl, G.E.; Svetnik, V. Deep Neural Nets as a Method for Quantitative Structure–Activity Relationships. J. Chem. Inf. Model. 2015, 55, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Swanson, K.; Jin, W.; Coley, C.; Eiden, P.; Gao, H.; Guzman-Perez, A.; Hopper, T.; Kelley, B.; Mathea, M.; et al. Analyzing Learned Molecular Representations for Property Prediction. J. Chem. Inf. Model. 2019, 59, 3370–3388. [Google Scholar] [CrossRef] [PubMed]
- Ragoza, M.; Hochuli, J.; Idrobo, E.; Sunseri, J.; Koes, D.R. Protein–Ligand Scoring with Convolutional Neural Networks. J. Chem. Inf. Model. 2017, 57, 942–957. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, Y. Improving scoring-docking-screening powers of protein–ligand scoring functions using random forest. J. Comput. Chem. 2017, 38, 169–177. [Google Scholar] [CrossRef]
- Segler, M.H.S.; Preuss, M.; Waller, M.P. Learning to Plan Chemical Syntheses. Nature 2018, 555, 604–610. [Google Scholar] [CrossRef]
- Coley, C.W.; Thomas, D.A.; Lummiss, J.A.M.; Jaworski, J.N.; Breen, C.P.; Schultz, V.; Hart, T.; Fishman, J.S.; Rogers, L.; Gao, H.; et al. A robotic platform for flow synthesis of organic compounds informed by AI planning. Science 2019, 365, eaax1566. [Google Scholar] [CrossRef]
- Xu, Y.; Lin, K.; Wang, S.; Wang, L.; Cai, C.; Song, C.; Lai, L.; Pei, J. Deep learning for molecular generation. Future Med. Chem. 2019, 11, 567–597. [Google Scholar] [CrossRef] [PubMed]
- Elton, D.C.; Boukouvalas, Z.; Fuge, M.D.; Chung, P.W. Deep learning for molecular design—A review of the state of the art. Mol. Syst. Des. Eng. 2019, 4, 828–849. [Google Scholar] [CrossRef]
- Dana, D.; Gadhiya, S.V.; St. Surin, L.G.; Li, D.; Naaz, F.; Ali, Q.; Paka, L.; Yamin, M.A.; Narayan, M.; Goldberg, I.D.; et al. Deep Learning in Drug Discovery and Medicine; Scratching the Surface. Molecules 2018, 23, 2384. [Google Scholar] [CrossRef] [PubMed]
- Kitchin, R.; McArdle, G. What makes Big Data, Big Data? Exploring the ontological characteristics of 26 datasets. Big Data Soc. 2016, 3, 2053951716631130. [Google Scholar] [CrossRef]
- Reker, D.; Schneider, G. Active-learning strategies in computer-assisted drug discovery. Drug Discov. Today 2015, 20, 458–465. [Google Scholar] [CrossRef] [PubMed]
- DeepChem. Available online: https://github.com/deepchem/deepchem (accessed on 20 October 2023).
- Merck/DeepNeuralNet-QSAR. Merck Sharp & Dohme Corp. a Subsidiary of Merck & Co., Inc. Available online: https://github.com/Merck/DeepNeuralNet-QSAR (accessed on 20 October 2023).
- Keenan, G. Croningp/ChemputerSoftware: Chemputer First Release; Version 0.1.1; Zenodo: Genève, Switzerland, 2018. [Google Scholar] [CrossRef]
- Mayr, A.; Klambauer, G.; Unterthiner, T.; Hochreiter, S. DeepTox: Toxicity Prediction using Deep Learning. Front. Environ. Sci. 2016, 3, 80. [Google Scholar] [CrossRef]
- AlphaFold. Available online: https://www.deepmind.com/research/highlighted-research/alphafold (accessed on 20 October 2023).
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- ORGANIC. The Matter Lab, Aspuru-Guzik Group Repo, 80 St. George Street Toronto, ON, M5S 3H6. Available online: https://github.com/aspuru-guzik-group/ORGANIC (accessed on 20 October 2023).
- Kwak, H.S.; An, Y.; Giesen, D.J.; Hughes, T.F.; Brown, C.T.; Leswing, K.; Abroshan, H.; Halls, M.D. Design of Organic Electronic Materials with a Goal-Directed Generative Model Powered by Deep Neural Networks and High-Throughput Molecular Simulations. Front. Chem. 2022, 9, 800370. Available online: https://www.frontiersin.org/articles/10.3389/fchem.2021.800370 (accessed on 20 October 2023). [CrossRef] [PubMed]
- Mostaghimi, M.; Rêgo, C.R.C.; Haldar, R.; Wöll, C.; Wenzel, W.; Kozlowska, M. Automated Virtual Design of Organic Semiconductors Based on Metal-Organic Frameworks. Front. Mater. 2022, 9, 840644. Available online: https://www.frontiersin.org/articles/10.3389/fmats.2022.840644 (accessed on 20 October 2023). [CrossRef]
- Chen, P.; Wang, Y.; Yan, H.; Gao, S.; Xu, Z.; Li, Y.; Mo, Q.; Huang, J.; Tao, J.; Pan, G.; et al. 3DStructGen: An interactive web-based 3D structure generation for non-periodic molecule and crystal. J. Cheminform. 2020, 12, 7. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, E.N.; Sur, D.; Wu, Z.; Husic, B.E.; Mai, H.; Li, Y.; Sun, S.; Yang, J.; Ramsundar, B.; Pande, V.S. PotentialNet for Molecular Property Prediction. ACS Cent. Sci. 2018, 4, 1520–1530. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.; Choi, J.; Park, S.; Ahn, J. Binding affinity prediction for protein–ligand complex using deep attention mechanism based on intermolecular interactions. BMC Bioinform. 2021, 22, 542. [Google Scholar] [CrossRef] [PubMed]
- Kandel, J.; Tayara, H.; Chong, K.T. PUResNet: Prediction of protein-ligand binding sites using deep residual neural network. J. Cheminform. 2021, 13, 65. [Google Scholar] [CrossRef]
- Wang, C. DeltaVina. Available online: https://github.com/chengwang88/deltavina (accessed on 20 October 2023).
- Kumar, S.; Kim, M. SMPLIP-Score: Predicting ligand binding affinity from simple and interpretable on-the-fly interaction fingerprint pattern descriptors. J. Cheminform. 2021, 13, 28. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, Y. Delta Machine Learning to Improve Scoring-Ranking-Screening Performances of Protein–Ligand Scoring Functions. J. Chem. Inf. Model. 2022, 62, 2696–2712. [Google Scholar] [CrossRef]
- Neural Graph Fingerprints. Formerly: Harvard Intelligent Probabilistic Systems Group—Now at Princeton. Available online: https://github.com/HIPS/neural-fingerprint (accessed on 20 October 2023).
- Wen, N.; Liu, G.; Zhang, J.; Zhang, R.; Fu, Y.; Han, X. A fingerprints based molecular property prediction method using the BERT model. J. Cheminform. 2022, 14, 71. [Google Scholar] [CrossRef]
- GastroPlus® PBPK & PBBM Modeling and Simulation. Simulations Plus. Available online: https://www.simulations-plus.com/software/gastroplus/ (accessed on 20 October 2023).
- Hussain, A.; Afzal, O.; Yasmin, S.; Haider, N.; Altamimi, A.S.A.; Martinez, F.; Acree, W.E., Jr.; Ramzan, M. Preferential Solvation Study of Rosuvastatin in the {PEG400 (1) + Water (2)} Cosolvent Mixture and GastroPlus Software-Based In Vivo Predictions. ACS Omega 2023, 8, 12761–12772. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; Walters, W.P.; Plowright, A.T.; Sieroka, N.; Listgarten, J.; Goodnow, R.A.; Fisher, J.; Jansen, J.M.; Duca, J.S.; Rush, T.S.; et al. Rethinking drug design in the artificial intelligence era. Nat. Rev. Drug Discov. 2020, 19, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P.; Schuffenhauer, A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminform. 2009, 1, 8. [Google Scholar] [CrossRef]
- Chaudhari, R.; Fong, L.W.; Tan, Z.; Huang, B.; Zhang, S. An up-to-date overview of computational polypharmacology in modern drug discovery. Expert Opin. Drug Discov. 2020, 15, 1025–1044. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.S.; Zhang, S. Polypharmacology: Drug discovery for the future. Expert Rev. Clin. Pharmacol. 2013, 6, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Sirois, C.; Khoury, R.; Durand, A.; Deziel, P.-L.; Bukhtiyarova, O.; Chiu, Y.; Talbot, D.; Bureau, A.; Després, P.; Gagné, C.; et al. Exploring polypharmacy with artificial intelligence: Data analysis protocol. BMC Med. Inform. Decis. Mak. 2021, 21, 219. [Google Scholar] [CrossRef] [PubMed]
- Duch, W.; Swaminathan, K.; Meller, J. Artificial Intelligence Approaches for Rational Drug Design and Discovery. Curr. Pharm. Des. 2007, 13, 1497–1508. [Google Scholar] [CrossRef] [PubMed]
- Blasiak, A.; Khong, J.; Kee, T. CURATE.AI: Optimizing Personalized Medicine with Artificial Intelligence. SLAS Technol. Transl. Life Sci. Innov. 2020, 25, 95–105. [Google Scholar] [CrossRef]
- Baronzio, G.; Parmar, G.; Baronzio, M. Overview of Methods for Overcoming Hindrance to Drug Delivery to Tumors, with Special Attention to Tumor Interstitial Fluid. Front. Oncol. 2015, 5, 165. Available online: https://www.frontiersin.org/articles/10.3389/fonc.2015.00165 (accessed on 20 October 2023). [CrossRef]
- Nexocode. Available online: https://nexocode.com/ (accessed on 20 October 2023).
- Universität Basel. Available online: https://www.unibas.ch/de.html (accessed on 20 October 2023).
- Popular Mechanics—Product Reviews, How-To, Space, Military, Math, Science, and New Technology. Popular Mechanics. Available online: https://www.popularmechanics.com/ (accessed on 20 October 2023).
- IBM—United Kingdom. Available online: https://www.ibm.com/uk-en (accessed on 20 October 2023).
- Main Page. Wikipedia, the Free Encyclopedia. Available online: https://en.wikipedia.org/w/index.php?title=Main_Page&oldid=1114291180 (accessed on 20 October 2023).
- Zong, N.; Wen, A.; Moon, S.; Fu, S.; Wang, L.; Zhao, Y.; Yu, Y.; Huang, M.; Wang, Y.; Zheng, G.; et al. Computational drug repurposing based on electronic health records: A scoping review. Npj Digit. Med. 2022, 5, 77. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, F.; Tang, J.; Nussinov, R.; Cheng, F. Artificial intelligence in COVID-19 drug repurposing. Lancet Digit. Health 2020, 2, e667–e676. [Google Scholar] [CrossRef]
- Wang, Y.; Aldahdooh, J.; Hu, Y.; Yang, H.; Vähä-Koskela, M.; Tang, J.; Tanoli, Z. DrugRepo: A novel approach to repurposing drugs based on chemical and genomic features. Sci. Rep. 2022, 12, 21116. [Google Scholar] [CrossRef] [PubMed]
- Lejal, N.; Tarus, B.; Bouguyon, E.; Chenavas, S.; Bertho, N.; Delmas, B.; Ruigrok, R.W.H.; Di Primo, C.; Slama-Schwok, A. Structure-Based Discovery of the Novel Antiviral Properties of Naproxen against the Nucleoprotein of Influenza A Virus. Antimicrob. Agents Chemother. 2013, 57, 2231–2242. [Google Scholar] [CrossRef] [PubMed]
- Terrier, O.; Dilly, S.; Pizzorno, A.; Chalupska, D.; Humpolickova, J.; Bouřa, E.; Berenbaum, F.; Quideau, S.; Lina, B.; Fève, B.; et al. Antiviral Properties of the NSAID Drug Naproxen Targeting the Nucleoprotein of SARS-CoV-2 Coronavirus. Molecules 2021, 26, 2593. [Google Scholar] [CrossRef]
- Mostafa, A.; Kandeil, A.; Elshaier, Y.A.M.M.; Kutkat, O.; Moatasim, Y.; Rashad, A.A.; Shehata, M.; Gomaa, M.R.; Mahrous, N.; Mahmoud, S.H.; et al. FDA-Approved Drugs with Potent In Vitro Antiviral Activity against Severe Acute Respiratory Syndrome Coronavirus 2. Pharmaceuticals 2020, 13, 443. [Google Scholar] [CrossRef]
- Stebbing, J.; Krishnan, V.; de Bono, S.; Ottaviani, S.; Casalini, G.; Richardson, P.J.; Monteil, V.; Lauschke, V.M.; Mirazimi, A.; Youhanna, S.; et al. Mechanism of baricitinib supports artificial intelligence-predicted testing in COVID-19 patients. EMBO Mol. Med. 2020, 12, e12697. [Google Scholar] [CrossRef]
- Farghali, H.; Kutinová Canová, N.; Arora, M. The Potential Applications of Artificial Intelligence in Drug Discovery and Development. Physiol. Res. 2021, 70 (Suppl. 4), S715–S722. [Google Scholar] [CrossRef]
- Evaluation of Safety, Tolerability & PK of DSP-2230 in Healthy Subjects. Health Research Authority. Available online: https://www.hra.nhs.uk/planning-and-improving-research/application-summaries/research-summaries/evaluation-of-safetytolerability-pk-of-dsp-2230-in-healthy-subjects/ (accessed on 20 October 2023).
- Terranova, N.; Jansen, M.; Falk, M.; Hendriks, B.S. Population pharmacokinetics of ATR inhibitor berzosertib in phase I studies for different cancer types. Cancer Chemother. Pharmacol. 2021, 87, 185–196. [Google Scholar] [CrossRef]
- Plummer, R.; Dean, E.; Arkenau, H.-T.; Redfern, C.; Spira, A.I.; Melear, J.M.; Chung, K.Y.; Ferrer-Playan, J.; Goddemeier, T.; Locatelli, G.; et al. A phase 1b study evaluating the safety and preliminary efficacy of berzosertib in combination with gemcitabine in patients with advanced non-small cell lung cancer. Lung Cancer 2022, 163, 19–26. [Google Scholar] [CrossRef]
- Almallah, Z.; El-Lababidi, R.; Shamout, F.; Doyle, D.J. Artificial Intelligence: The New Alexander Fleming. Healthc. Inform. Res. 2021, 27, 168–171. [Google Scholar] [CrossRef] [PubMed]
- Valavanidis, A. Artificial Intelligence Application with Machine-Learning Algorithm Identified a Powerful Broad-Spectrum Antibiotic. Available online: http://chem-tox-ecotox.org/wp-content/uploads/2020/03/ANTIBIOTICS-HALICIN-ARTIFICIAL-INTELLIGENCE-2020.pdf (accessed on 2 January 2024).
- Markham, A. Evinacumab: First Approval. Drugs 2021, 81, 1101–1105. [Google Scholar] [CrossRef]
- Miller, M.; Tokgozoglu, L.; Parhofer, K.G.; Handelsman, Y.; Leiter, L.A.; Landmesser, U.; Brinton, E.A.; Catapano, A.L. Icosapent ethyl for reduction of persistent cardiovascular risk: A critical review of major medical society guidelines and statements. Expert Rev. Cardiovasc. Ther. 2022, 20, 609–625. [Google Scholar] [CrossRef]
- Ballantyne, C.M.; Manku, M.S.; Bays, H.E.; Philip, S.; Granowitz, C.; Doyle, R.T.; Juliano, R.A. Icosapent Ethyl Effects on Fatty Acid Profiles in Statin-Treated Patients with High Triglycerides: The Randomized, Placebo-controlled ANCHOR Study. Cardiol. Ther. 2019, 8, 79–90. [Google Scholar] [CrossRef] [PubMed]
- kgi-admin. Delpazolid by LegoChem Biosciences for Tuberculosis: Likelihood of Approval. Pharmaceutical Technology. Available online: https://www.pharmaceutical-technology.com/data-insights/delpazolid-legochem-biosciences-tuberculosis-likelihood-of-approval/ (accessed on 20 October 2023).
- Healthcare, G. First Drug Created by AI Enters Clinical Trials. Clinical Trials Arena. Available online: https://www.clinicaltrialsarena.com/comment/first-drug-created-ai-enters-trials/ (accessed on 20 October 2023).
- Evaxion Cleared by FDA to Begin Phase IIb Trial of Cancer Vaccine, Keytruda in Melanoma. Precision Medicine Online. Available online: https://www.precisionmedicineonline.com/cancer/evaxion-cleared-fda-begin-phase-iib-trial-cancer-vaccine-keytruda-melanoma (accessed on 23 October 2023).
- Arnold, C. Inside the nascent industry of AI-designed drugs. Nat. Med. 2023, 29, 1292–1295. [Google Scholar] [CrossRef]
- The Discovery of Amgen’s Novel Investigational KRAS(G12C) Inhibitor AMG 510 Published in Nature. Available online: https://www.amgen.com/newsroom/press-releases/2019/10/the-discovery-of-amgens-novel-investigational-krasg12c-inhibitor-amg-510-published-in-nature (accessed on 23 October 2023).
- Ghislaine PELLAT. Constantin Anghelache. Governance in the EU Member States in the Era of Big Data. In Proceedings of the 25th PGV Network Conference—International Scientific Conference, Bucharest, Romania, 12–13 September 2019. Available online: https://www.researchgate.net/profile/Grzegorz-Maciejewski/publication/335929459_Use_of_Big_Data_On_The_Food_Market_-_Areas_Applications_Examples/links/5d84bb29a6fdcc8fd6fda856/Use-of-Big-Data-On-The-Food-Market-Areas-Applications-Examples.pdf (accessed on 2 January 2024).
- Yang, X.; Wang, Y.; Byrne, R.; Schneider, G.; Yang, S. Concepts of Artificial Intelligence for Computer-Assisted Drug Discovery. Chem. Rev. 2019, 119, 10520–10594. [Google Scholar] [CrossRef] [PubMed]
- How to Navigate the Patenting Challenges of AI-Assisted Drug Discovery. Available online: https://www.pharmaceuticalonline.com/doc/how-to-navigate-the-patenting-challenges-of-ai-assisted-drug-discovery-0001 (accessed on 23 October 2023).
- Freedman, D.H. Hunting for New Drugs with AI. Nature 2019, 576, S49–S53. [Google Scholar] [CrossRef] [PubMed]
- How AI Is Aiming at the Bad Math of Drug Development. Bloomberg.com, 29 November 2021. Available online: https://www.bloomberg.com/news/articles/2021-11-29/how-ai-is-aiming-at-the-bad-math-of-drug-development-quicktake (accessed on 23 October 2023).
- DiNuzzo, M. How artificial intelligence enables modeling and simulation of biological networks to accelerate drug discovery. Front. Drug Discov. 2022, 2, 1019706. Available online: https://www.frontiersin.org/articles/10.3389/fddsv.2022.1019706 (accessed on 23 October 2023). [CrossRef]
- Fleming, N. How artificial intelligence is changing drug discovery. Nature 2018, 557, S55–S57. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Algorithms | Reference | |
|---|---|---|---|
| Supervised learning | Regression analysis | MLR | [35] |
| DT | [36] | ||
| LR | [37] | ||
| Classification | SVM | [38] | |
| CNN | [39] | ||
| RNN | [40] | ||
| GAN | [41] | ||
| Unsupervised learning | Clustering | k-means | [42] |
| Hierarchical | [43] | ||
| Dimensionality reduction | PCA | [44] | |
| t-SNE | [45] |
| Pharmaceutical Company | AI Provider | Project Details |
|---|---|---|
| AstraZeneca | BenevolentAI | Selected the first AI-generated drug target for chronic kidney disease (CKD) |
| Pfizer | AI Technology | Used AI for COVID-19 vaccine trials and streamlined distribution. Also used AI and predictive analytics to modernize, streamline, and simplify the development of medicines. |
| Pfizer | XtalPi | Developed a hybrid physics and AI-powered software platform for the accurate molecular modeling of drug-like small molecules. |
| Pfizer | Insilico Medicine | Mined data for drug targets. |
| Pfizer | ConcertAI | Expanded partnership to improve study design and diversify clinical trials with the aid of AI. |
| Pfizer | Janssen Research and Development (Johnson & Johnson) | Collaborated to apply AI in the identification and selection of new targets and disease subsets to aid therapeutic programs. |
| Eli Lilly, Bayer, Bridge Biotherapeutics | Atomwise | Assisted in structure-based small-molecule drug discovery. |
| Boehringer Ingelheim | Google Quantum AI | Working together to leverage quantum computing to accelerate and optimize the discovery of future new medicines. |
| Boehringer Ingelheim | Insilico Medicine | Partnered to use AI technology in identifying potential therapeutic targets. |
| BMS | Exscientia | Contract led to the selection of an AI-designed immune-modulating drug candidate. |
| GSK | Exscientia | Developed the first-ever AI-powered treatment for COPD. |
| Roche, Sanofi, Bayer | Exscientia | Working with these big pharma players. |
| AstraZeneca | Eko, BERG, Renalytix AI, Mila-Quebec AI Institute | Using AI algorithms and supercomputers for drug discovery. |
| Optellum | J&J Lung Cancer Initiative | Applied AI-powered clinical decision support platform to transform early lung cancer treatment. |
| Program/Platform | Description | Primary Use | Accession |
|---|---|---|---|
| DeepChem | Python-based AI system using MLP model | Candidate selection in drug discovery | https://github.com/deepchem/deepchem (accessed on 1 February 2024) |
| DeepNeuralNetQSAR | Python-based AI system | Can aid the detection of the molecular activity of compounds | https://github.com/Merck/DeepNeuralNet-QSAR (accessed on 1 February 2024) |
| Chemputer | Combination of Monte Carlo tree search and symbolic AI, including DNNs | Synthesize organic molecules | https://zenodo.org/record/1481731 (accessed on 1 February 2024) |
| DeepTox | AI system using DL | Chemical toxicity prediction | www.bioinf.jku.at/research/DeepTox (accessed on 1 February 2024) |
| AlphaFold | AI system using DL | Predicts the 3D structures of proteins | https://alphafold.ebi.ac.uk/ (accessed on 1 February 2024) |
| ORGANIC | Generative ML approaches and DNNs | Novel molecular materials | https://github.com/aspuru-guzik-group/ORGANIC (accessed on 1 February 2024) |
| PotentialNet | Neural networks, deep attention mechanisms and descriptor embeddings | The binding affinity of ligands in protein–ligand complexes. | https://www.genesistherapeutics.ai/platform.html (accessed on 1 February 2024) |
| Hit Dexter | ML technique, CNNs and ANNs | For predicting molecules that might respond to biochemical assays | http://hitdexter2.zbh.uni-hamburg.de (accessed on 1 February 2024) |
| DeltaVina | ML algorithms, including XGBoost and random forest | Scoring protein–ligand binding affinity | https://github.com/chengwang88/deltavina (accessed on 1 February 2024) |
| Neural graph fingerprint | CNNs | Predict properties of novel molecules | https://github.com/HIPS/neural-fingerprint (accessed on 1 February 2024) |
| GastroPlus | AI and predictive modeling | For pharmaceutical products (dosage form) in many animal models | https://www.simulations-plus.com/software/gastroplus/# (accessed on 1 February 2024) |
| Company | AI Use | Collaboration with the Pharmaceutical Industry | Application/Agents for Clinical Trials |
|---|---|---|---|
| IBM Watson Health Cambridge | AI for evaluating clinical and health-related data | Novartis | Real-time patient monitoring to improve breast cancer patient intervention outcomes |
| Pfizer | Accelerating immuno-oncology medication discovery efforts | ||
| Benevolent AI | AI-enabled Judgement Augmented Cognition System (JACS) for developing new drugs effective against neurodegenerative diseases | Janssen | Such partnership will lead to the advancement of new medicinal molecules. |
| Using AI, new clinical lead agents for chronic renal diseases are being developed. | AstraZeneca | During Phase 2b clinical trials, a drug candidate was assessed as a primary agent for treating chronic renal diseases. | |
| Microsoft | AI for image processing and therapeutic interventions using cells and genes | Novartis | Creating an AI innovation lab to improve medication research and commercialization processes |
| Numerate | AI-enabled drug design for oncology and gastrointestinal specialties | Takeda | Phase 1 clinical trial of drug S48168 for Ryanodine Receptor 2 |
| Servier | Drug development for conditions of the central nervous system, the digestive system, and cancer | ||
| Owkin | Clinical testing by means of ML | Roche | Created and improved the Owkin’s Studio platform using artificial intelligence |
| XtalPi | A target identification and validation package integrating quantum mechanics and ML techniques | Pfizer | Preparation and improvement of crystalline drug candidate entities for use in early drug screening |
| Exscientia | AI-enabled drug discovery and lead refinement | Sanofi | Agent DSP-1181 is currently undergoing Phase I clinical testing. Advancement of the Centaur ChemistTM drug discovery AI system Drug discovery in obsessive-convulsive disorder |
| Merck and BenevolentAI | New clinical development drug candidates in key therapeutic areas of oncology, neurology, and immunology | ||
| Atomwise | AI-enabled structural modeling | Lilly | Agent BBT-401 in Phase 2 of clinical testing |
| Bridge Biotherapeutics | Augmentation of Pellino Inhibitor Pipeline Agent BBT-401 evaluated in Phase-2a of clinical testing | ||
| Sensyne Health | Clinical AI schemes | Bayer | Created and improved the specialized clinical AI technology suite for Sensyne Health. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Visan, A.I.; Negut, I. Integrating Artificial Intelligence for Drug Discovery in the Context of Revolutionizing Drug Delivery. Life 2024, 14, 233. https://doi.org/10.3390/life14020233
Visan AI, Negut I. Integrating Artificial Intelligence for Drug Discovery in the Context of Revolutionizing Drug Delivery. Life. 2024; 14(2):233. https://doi.org/10.3390/life14020233
Chicago/Turabian StyleVisan, Anita Ioana, and Irina Negut. 2024. "Integrating Artificial Intelligence for Drug Discovery in the Context of Revolutionizing Drug Delivery" Life 14, no. 2: 233. https://doi.org/10.3390/life14020233
APA StyleVisan, A. I., & Negut, I. (2024). Integrating Artificial Intelligence for Drug Discovery in the Context of Revolutionizing Drug Delivery. Life, 14(2), 233. https://doi.org/10.3390/life14020233