
Targeting Progression in Pulmonary Fibrosis: An Overview of Underlying Mechanisms, Molecular Biomarkers, and Therapeutic Intervention

 and
and
Abstract
1. Introduction
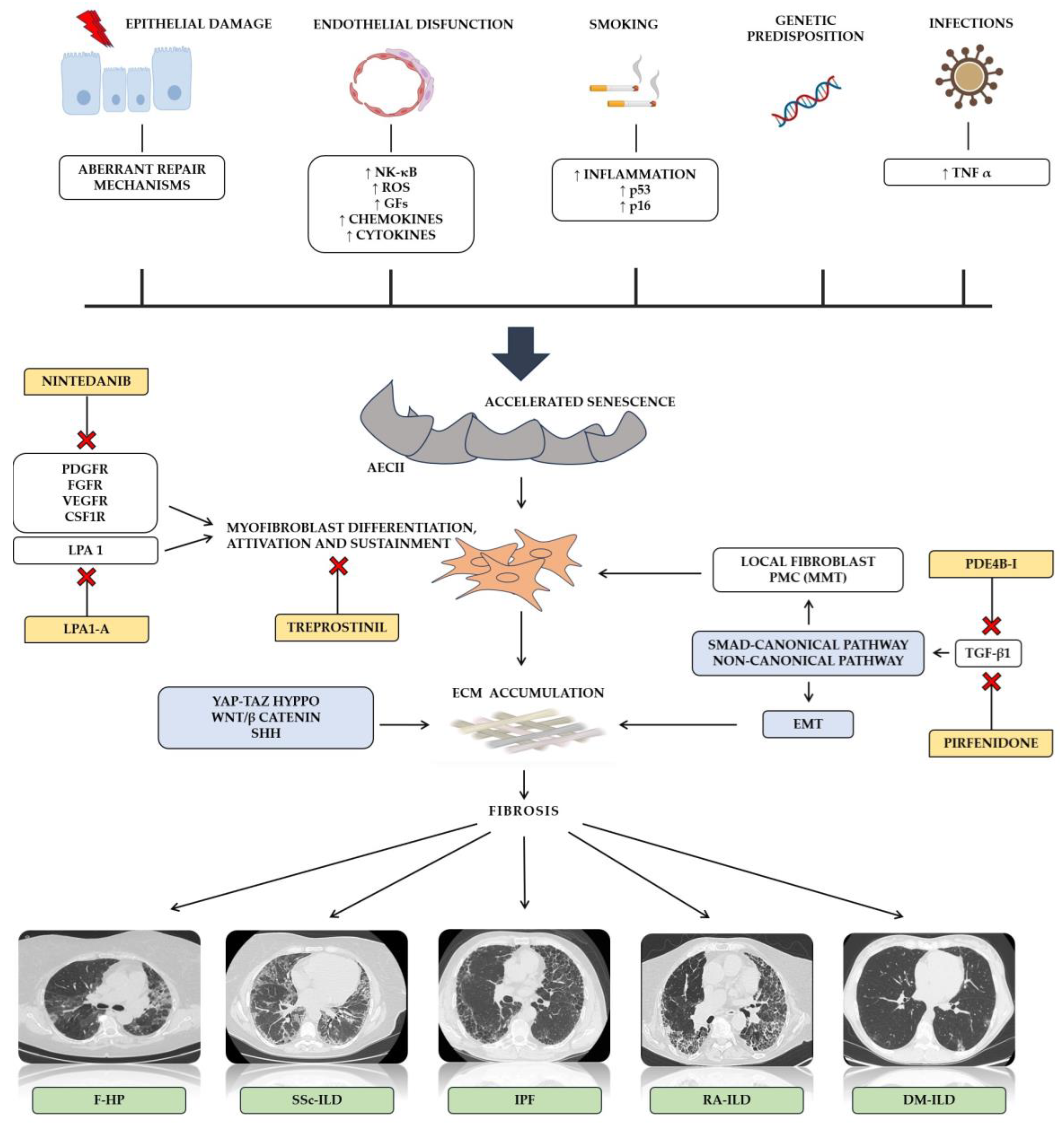
2. Molecular Mechanisms of Pulmonary Fibrosis
3. Serum Biomarkers for Diagnosing and Monitoring Pulmonary Fibrosis
3.1. Idiopathic Pulmonary Fibrosis (IPF)
3.2. CTD-ILDs
3.3. Other Fibrosing ILDs
4. Therapeutic Strategies in PPF
4.1. Idiopathic Pulmonary Fibrosis (IPF)
4.2. CTD-ILDs
4.3. Sarcoidosis
4.4. Fibrotic HP
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Travis, W.D.; Costabel, U.; Hansell, D.M.; King, T.E.J.; Lynch, D.A.; Nicholson, A.G.; Ryerson, C.J.; Ryu, J.H.; Selman, M.; Wells, A.U.; et al. An Official American Thoracic Society/European Respiratory Society Statement: Update of the International Multidisciplinary Classification of the Idiopathic Interstitial Pneumonias. Am. J. Respir. Crit. Care Med. 2013, 188, 733–748. [Google Scholar] [CrossRef]
- Valeyre, D.; Freynet, O.; Dion, G.; Bouvry, D.; Annesi-Maesano, I.; Nunes, H. [Epidemiology of interstitial lung diseases]. Presse Med. 2010, 39, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Piciucchi, S.; Tomassetti, S.; Ravaglia, C.; Gurioli, C.; Gurioli, C.; Dubini, A.; Carloni, A.; Chilosi, M.; Colby, T.V.; Poletti, V. From “Traction Bronchiectasis” to Honeycombing in Idiopathic Pulmonary Fibrosis: A Spectrum of Bronchiolar Remodeling Also in Radiology? BMC Pulm. Med. 2016, 16, 87. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Remy-Jardin, M.; Richeldi, L.; Thomson, C.C.; Inoue, Y.; Johkoh, T.; Kreuter, M.; Lynch, D.A.; Maher, T.M.; Martinez, F.J.; et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2022, 205, e18–e47. [Google Scholar] [CrossRef] [PubMed]
- Khine, N.; Mudawi, D.; Rivera-Ortega, P.; Leonard, C.; Chaudhuri, N.; Margaritopoulos, G.A. Rapidly Non-Ipf Progressive Fibrosing Interstitial Lung Disease: A Phenotype with an Ipf-like Behavior. Sarcoidosis Vasc. Diffus. Lung Dis. Off. J. WASOG 2020, 37, 231–233. [Google Scholar]
- Olson, A.L.; Gifford, A.H.; Inase, N.; Fernández Pérez, E.R.; Suda, T. The Epidemiology of Idiopathic Pulmonary Fibrosis and Interstitial Lung Diseases at Risk of a Progressive-Fibrosing Phenotype. Eur. Respir. Rev. Off. J. Eur. Respir. Soc. 2018, 27, 180077. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann-Vold, A.-M.; Weigt, S.S.; Saggar, R.; Palchevskiy, V.; Volkmann, E.R.; Liang, L.L.; Ross, D.; Ardehali, A.; Lynch, J.P., 3rd; Belperio, J.A. Endotype-Phenotyping May Predict a Treatment Response in Progressive Fibrosing Interstitial Lung Disease. EBioMedicine 2019, 50, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.W.; Ryerson, C.J.; Guler, S.A. Progression of Fibrosing Interstitial Lung Disease. Respir. Res. 2020, 21, 32. [Google Scholar] [CrossRef] [PubMed]
- Maher, T.M.; Wuyts, W. Management of Fibrosing Interstitial Lung Diseases. Adv. Ther. 2019, 36, 1518–1531. [Google Scholar] [CrossRef]
- Cottin, V.; Hirani, N.A.; Hotchkin, D.L.; Nambiar, A.M.; Ogura, T.; Otaola, M.; Skowasch, D.; Park, J.S.; Poonyagariyagorn, H.K.; Wuyts, W.; et al. Presentation, Diagnosis and Clinical Course of the Spectrum of Progressive-Fibrosing Interstitial Lung Diseases. Eur. Respir. Rev. Off. J. Eur. Respir. Soc. 2018, 27, 180076. [Google Scholar] [CrossRef]
- Nalysnyk, L.; Cid-Ruzafa, J.; Rotella, P.; Esser, D. Incidence and Prevalence of Idiopathic Pulmonary Fibrosis: Review of the Literature. Eur. Respir. Rev. Off. J. Eur. Respir. Soc. 2012, 21, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Ley, B.; Collard, H.R.; King, T.E.J. Clinical Course and Prediction of Survival in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2011, 183, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Hambly, N.; Farooqi, M.M.; Dvorkin-Gheva, A.; Donohoe, K.; Garlick, K.; Scallan, C.; Chong, S.G.; MacIsaac, S.; Assayag, D.; Johannson, K.A.; et al. Prevalence and Characteristics of Progressive Fibrosing Interstitial Lung Disease in a Prospective Registry. Eur. Respir. J. 2022, 60, 2102571. [Google Scholar] [CrossRef]
- Cottin, V.; Teague, R.; Nicholson, L.; Langham, S.; Baldwin, M. The Burden of Progressive-Fibrosing Interstitial Lung Diseases. Front. Med. 2022, 9, 799912. [Google Scholar] [CrossRef] [PubMed]
- Nasser, M.; Larrieu, S.; Si-Mohamed, S.; Ahmad, K.; Boussel, L.; Brevet, M.; Chalabreysse, L.; Fabre, C.; Marque, S.; Revel, D.; et al. Progressive Fibrosing Interstitial Lung Disease: A Clinical Cohort (the PROGRESS Study). Eur. Respir. J. 2021, 57, 2002718. [Google Scholar] [CrossRef]
- Faverio, P.; Piluso, M.; De Giacomi, F.; Della Zoppa, M.; Cassandro, R.; Harari, S.; Luppi, F.; Pesci, A. Progressive Fibrosing Interstitial Lung Diseases: Prevalence and Characterization in Two Italian Referral Centers. Respiration 2020, 99, 838–845. [Google Scholar] [CrossRef]
- Ryerson, C.J.; Vittinghoff, E.; Ley, B.; Lee, J.S.; Mooney, J.J.; Jones, K.D.; Elicker, B.M.; Wolters, P.J.; Koth, L.L.; King, T.E.J.; et al. Predicting Survival across Chronic Interstitial Lung Disease: The ILD-GAP Model. Chest 2014, 145, 723–728. [Google Scholar] [CrossRef]
- Samarelli, A.V.; Tonelli, R.; Marchioni, A.; Bruzzi, G.; Gozzi, F.; Andrisani, D.; Castaniere, I.; Manicardi, L.; Moretti, A.; Tabbì, L.; et al. Fibrotic Idiopathic Interstitial Lung Disease: The Molecular and Cellular Key Players. Int. J. Mol. Sci. 2021, 22, 8952. [Google Scholar] [CrossRef]
- Nyunoya, T.; Monick, M.M.; Klingelhutz, A.; Yarovinsky, T.O.; Cagley, J.R.; Hunninghake, G.W. Cigarette Smoke Induces Cellular Senescence. Am. J. Respir. Cell Mol. Biol. 2006, 35, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Bloom, S.I.; Islam, M.T.; Lesniewski, L.A.; Donato, A.J. Mechanisms and Consequences of Endothelial Cell Senescence. Nat. Rev. Cardiol. 2023, 20, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Schulz, L.; Hornung, F.; Häder, A.; Radosa, L.; Brakhage, A.A.; Löffler, B.; Deinhardt-Emmer, S. Influenza Virus-Induced Paracrine Cellular Senescence of the Lung Contributes to Enhanced Viral Load. Aging Dis. 2023, 14, 1331–1348. [Google Scholar] [CrossRef]
- Albera, C.; Verri, G.; Sciarrone, F.; Sitia, E.; Mangiapia, M.; Solidoro, P. Progressive Fibrosing Interstitial Lung Diseases: A Current Perspective. Biomedicines 2021, 9, 1237. [Google Scholar] [CrossRef]
- Phan, S.H. Biology of Fibroblasts and Myofibroblasts. Proc. Am. Thorac. Soc. 2008, 5, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B. Mechanical Aspects of Lung Fibrosis: A Spotlight on the Myofibroblast. Proc. Am. Thorac. Soc. 2012, 9, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Karki, S.; Surolia, R.; Hock, T.D.; Guroji, P.; Zolak, J.S.; Duggal, R.; Ye, T.; Thannickal, V.J.; Antony, V.B. Wilms’ Tumor 1 (Wt1) Regulates Pleural Mesothelial Cell Plasticity and Transition into Myofibroblasts in Idiopathic Pulmonary Fibrosis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2014, 28, 1122–1131. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Du, Y.; Shen, Y.; He, Y.; Zhao, H.; Li, Z. TGF-Beta 1 Induced Fibroblast Proliferation Is Mediated by the FGF-2/ERK Pathway. Front. Biosci. Landmark Ed. 2012, 17, 2667–2674. [Google Scholar] [CrossRef] [PubMed]
- Kulasekaran, P.; Scavone, C.A.; Rogers, D.S.; Arenberg, D.A.; Thannickal, V.J.; Horowitz, J.C. Endothelin-1 and Transforming Growth Factor-Beta1 Independently Induce Fibroblast Resistance to Apoptosis via AKT Activation. Am. J. Respir. Cell Mol. Biol. 2009, 41, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Liu, Y.; Kahn, M.; Ann, D.K.; Han, A.; Wang, H.; Nguyen, C.; Flodby, P.; Zhong, Q.; Krishnaveni, M.S.; et al. Interactions between β-Catenin and Transforming Growth Factor-β Signaling Pathways Mediate Epithelial-Mesenchymal Transition and Are Dependent on the Transcriptional Co-Activator CAMP-Response Element-Binding Protein (CREB)-Binding Protein (CBP). J. Biol. Chem. 2012, 287, 7026–7038. [Google Scholar] [CrossRef]
- Königshoff, M.; Balsara, N.; Pfaff, E.-M.; Kramer, M.; Chrobak, I.; Seeger, W.; Eickelberg, O. Functional Wnt Signaling Is Increased in Idiopathic Pulmonary Fibrosis. PLoS ONE 2008, 3, e2142. [Google Scholar] [CrossRef] [PubMed]
- Meuten, T.; Hickey, A.; Franklin, K.; Grossi, B.; Tobias, J.; Newman, D.R.; Jennings, S.H.; Correa, M.; Sannes, P.L. WNT7B in Fibroblastic Foci of Idiopathic Pulmonary Fibrosis. Respir. Res. 2012, 13, 62. [Google Scholar] [CrossRef]
- Cao, H.; Chen, X.; Hou, J.; Wang, C.; Xiang, Z.; Shen, Y.; Han, X. The Shh/Gli Signaling Cascade Regulates Myofibroblastic Activation of Lung-Resident Mesenchymal Stem Cells via the Modulation of Wnt10a Expression during Pulmonary Fibrogenesis. Lab. Investig. 2020, 100, 363–377. [Google Scholar] [CrossRef]
- Lehmann, M.; Hu, Q.; Hu, Y.; Hafner, K.; Costa, R.; van den Berg, A.; Königshoff, M. Chronic WNT/β-Catenin Signaling Induces Cellular Senescence in Lung Epithelial Cells. Cell. Signal. 2020, 70, 109588. [Google Scholar] [CrossRef]
- Noguchi, S.; Saito, A.; Nagase, T. YAP/TAZ Signaling as a Molecular Link between Fibrosis and Cancer. Int. J. Mol. Sci. 2018, 19, 3674. [Google Scholar] [CrossRef]
- Samara, K.D.; Antoniou, K.M.; Karagiannis, K.; Margaritopoulos, G.; Lasithiotaki, I.; Koutala, E.; Siafakas, N.M. Expression Profiles of Toll-like Receptors in Non-Small Cell Lung Cancer and Idiopathic Pulmonary Fibrosis. Int. J. Oncol. 2012, 40, 1397–1404. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-Z.; Cui, B.; Liu, H.-Z.; Chen, Z.-R.; Yan, H.-M.; Hua, F.; Hu, Z.-W. Targeting TLR2 Attenuates Pulmonary Inflammation and Fibrosis by Reversion of Suppressive Immune Microenvironment. J. Immunol. 2009, 182, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Larson-Casey, J.L.; Deshane, J.S.; Ryan, A.J.; Thannickal, V.J.; Carter, A.B. Macrophage Akt1 Kinase-Mediated Mitophagy Modulates Apoptosis Resistance and Pulmonary Fibrosis. Immunity 2016, 44, 582–596. [Google Scholar] [CrossRef] [PubMed]
- Homma, S.; Nagaoka, I.; Abe, H.; Takahashi, K.; Seyama, K.; Nukiwa, T.; Kira, S. Localization of Platelet-Derived Growth Factor and Insulin-like Growth Factor I in the Fibrotic Lung. Am. J. Respir. Crit. Care Med. 1995, 152, 2084–2089. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Liu, L.; Mandal, J.; Molino, A.; Stolz, D.; Tamm, M.; Lu, S.; Roth, M. PDGF-BB Induces PRMT1 Expression through ERK1/2 Dependent STAT1 Activation and Regulates Remodeling in Primary Human Lung Fibroblasts. Cell. Signal. 2022, 89, 110114. [Google Scholar] [CrossRef] [PubMed]
- Grotendorst, G.R. Connective Tissue Growth Factor: A Mediator of TGF-Beta Action on Fibroblasts. Cytokine Growth Factor Rev. 1997, 8, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Selman, M.; Pardo, A. When Things Go Wrong: Exploring Possible Mechanisms Driving the Progressive Fibrosis Phenotype in Interstitial Lung Diseases. Eur. Respir. J. 2021, 58, 2004507. [Google Scholar] [CrossRef] [PubMed]
- Zanin-Silva, D.C.; Santana-Gonçalves, M.; Kawashima-Vasconcelos, M.Y.; Oliveira, M.C. Management of Endothelial Dysfunction in Systemic Sclerosis: Current and Developing Strategies. Front. Med. 2021, 8, 788250. [Google Scholar] [CrossRef]
- Garrett, S.M.; Hsu, E.; Thomas, J.M.; Pilewski, J.M.; Feghali-Bostwick, C. Insulin-like Growth Factor (IGF)-II-Mediated Fibrosis in Pathogenic Lung Conditions. PLoS ONE 2019, 14, e0225422. [Google Scholar] [CrossRef]
- Klinkhammer, B.M.; Floege, J.; Boor, P. PDGF in Organ Fibrosis. Mol. Aspects Med. 2018, 62, 44–62. [Google Scholar] [CrossRef]
- Ludwicka, A.; Ohba, T.; Trojanowska, M.; Yamakage, A.; Strange, C.; Smith, E.A.; Leroy, E.C.; Sutherland, S.; Silver, R.M. Elevated Levels of Platelet Derived Growth Factor and Transforming Growth Factor-Beta 1 in Bronchoalveolar Lavage Fluid from Patients with Scleroderma. J. Rheumatol. 1995, 22, 1876–1883. [Google Scholar]
- de Araújo, R.; Lôbo, M.; Trindade, K.; Silva, D.F.; Pereira, N. Fibroblast Growth Factors: A Controlling Mechanism of Skin Aging. Skin Pharmacol. Physiol. 2019, 32, 275–282. [Google Scholar] [CrossRef]
- Spagnolo, P.; Lee, J.S.; Sverzellati, N.; Rossi, G.; Cottin, V. The Lung in Rheumatoid Arthritis: Focus on Interstitial Lung Disease. Arthritis Rheumatol. 2018, 70, 1544–1554. [Google Scholar] [CrossRef]
- Brito, Y.; Glassberg, M.K.; Ascherman, D.P. Rheumatoid Arthritis-Associated Interstitial Lung Disease: Current Concepts. Curr. Rheumatol. Rep. 2017, 19, 79. [Google Scholar] [CrossRef] [PubMed]
- Yanagihara, T.; Inoue, Y. Insights into Pathogenesis and Clinical Implications in Myositis-Associated Interstitial Lung Diseases. Curr. Opin. Pulm. Med. 2020, 26, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Casciola-Rosen, L.; Andrade, F.; Ulanet, D.; Wong, W.B.; Rosen, A. Cleavage by Granzyme B Is Strongly Predictive of Autoantigen Status: Implications for Initiation of Autoimmunity. J. Exp. Med. 1999, 190, 815–826. [Google Scholar] [CrossRef]
- Grundtman, C.; Lundberg, I.E. Pathogenesis of Idiopathic Inflammatory Myopathies. Curr. Rheumatol. Rep. 2006, 8, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, X.-X.; Nishimoto, T.; Takihara, T.; Mlakar, L.; Bradshaw, A.D.; Feghali-Bostwick, C. Lysyl Oxidase Directly Contributes to Extracellular Matrix Production and Fibrosis in Systemic Sclerosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 320, L29–L40. [Google Scholar] [CrossRef] [PubMed]
- Vadasz, Z.; Balbir Gurman, A.; Meroni, P.; Farge, D.; Levi, Y.; Ingegnoli, F.; Braun-Moscovici, Y.; Rosner, I.; Slobodin, G.; Rozenbaum, M.; et al. Lysyl Oxidase-a Possible Role in Systemic Sclerosis-Associated Pulmonary Hypertension: A Multicentre Study. Rheumatology 2019, 58, 1547–1555. [Google Scholar] [CrossRef]
- Enomoto, Y.; Suzuki, Y.; Hozumi, H.; Mori, K.; Kono, M.; Karayama, M.; Furuhashi, K.; Fujisawa, T.; Enomoto, N.; Nakamura, Y.; et al. Clinical Significance of Soluble CD163 in Polymyositis-Related or Dermatomyositis-Related Interstitial Lung Disease. Arthritis Res. Ther. 2017, 19, 9. [Google Scholar] [CrossRef]
- Horiike, Y.; Suzuki, Y.; Fujisawa, T.; Yasui, H.; Karayama, M.; Hozumi, H.; Furuhashi, K.; Enomoto, N.; Nakamura, Y.; Inui, N.; et al. Successful Classification of Macrophage-Mannose Receptor CD206 in Severity of Anti-MDA5 Antibody Positive Dermatomyositis Associated ILD. Rheumatology 2019, 58, 2143–2152. [Google Scholar] [CrossRef]
- Weigold, F.; Günther, J.; Pfeiffenberger, M.; Cabral-Marques, O.; Siegert, E.; Dragun, D.; Philippe, A.; Regensburger, A.-K.; Recke, A.; Yu, X.; et al. Antibodies against Chemokine Receptors CXCR3 and CXCR4 Predict Progressive Deterioration of Lung Function in Patients with Systemic Sclerosis. Arthritis Res. Ther. 2018, 20, 52. [Google Scholar] [CrossRef] [PubMed]
- Wangkaew, S.; Euathrongchit, J.; Wattanawittawas, P.; Kasitanon, N.; Louthrenoo, W. Incidence and Predictors of Interstitial Lung Disease (ILD) in Thai Patients with Early Systemic Sclerosis: Inception Cohort Study. Mod. Rheumatol. 2016, 26, 588–593. [Google Scholar] [CrossRef]
- Nagy, A.; Nagy, T.; Kolonics-Farkas, A.M.; Eszes, N.; Vincze, K.; Barczi, E.; Tarnoki, A.D.; Tarnoki, D.L.; Nagy, G.; Kiss, E.; et al. Autoimmune Progressive Fibrosing Interstitial Lung Disease: Predictors of Fast Decline. Front. Pharmacol. 2021, 12, 778649. [Google Scholar] [CrossRef]
- Watanabe, M.; Horimasu, Y.; Iwamoto, H.; Yamaguchi, K.; Sakamoto, S.; Masuda, T.; Nakashima, T.; Miyamoto, S.; Ohshimo, S.; Fujitaka, K.; et al. C-C Motif Chemokine Ligand 15 May Be a Useful Biomarker for Predicting the Prognosis of Patients with Chronic Hypersensitivity Pneumonitis. Respiration 2019, 98, 212–220. [Google Scholar] [CrossRef]
- Prasse, A.; Probst, C.; Bargagli, E.; Zissel, G.; Toews, G.B.; Flaherty, K.R.; Olschewski, M.; Rottoli, P.; Müller-Quernheim, J. Serum CC-Chemokine Ligand 18 Concentration Predicts Outcome in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2009, 179, 717–723. [Google Scholar] [CrossRef]
- Tiev, K.P.; Hua-Huy, T.; Kettaneh, A.; Gain, M.; Duong-Quy, S.; Tolédano, C.; Cabane, J.; Dinh-Xuan, A.T. Serum CC Chemokine Ligand-18 Predicts Lung Disease Worsening in Systemic Sclerosis. Eur. Respir. J. 2011, 38, 1355–1360. [Google Scholar] [CrossRef] [PubMed]
- Khanna, D.; Denton, C.P.; Jahreis, A.; van Laar, J.M.; Frech, T.M.; Anderson, M.E.; Baron, M.; Chung, L.; Fierlbeck, G.; Lakshminarayanan, S.; et al. Safety and Efficacy of Subcutaneous Tocilizumab in Adults with Systemic Sclerosis (FaSScinate): A Phase 2, Randomised, Controlled Trial. Lancet 2016, 387, 2630–2640. [Google Scholar] [CrossRef]
- De Lauretis, A.; Sestini, P.; Pantelidis, P.; Hoyles, R.; Hansell, D.M.; Goh, N.S.L.; Zappala, C.J.; Visca, D.; Maher, T.M.; Denton, C.P.; et al. Serum Interleukin 6 Is Predictive of Early Functional Decline and Mortality in Interstitial Lung Disease Associated with Systemic Sclerosis. J. Rheumatol. 2013, 40, 435–446. [Google Scholar] [CrossRef]
- Wakamatsu, K.; Nagata, N.; Kumazoe, H.; Oda, K.; Ishimoto, H.; Yoshimi, M.; Takata, S.; Hamada, M.; Koreeda, Y.; Takakura, K.; et al. Prognostic Value of Serial Serum KL-6 Measurements in Patients with Idiopathic Pulmonary Fibrosis. Respir. Investig. 2017, 55, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Kuwana, M.; Shirai, Y.; Takeuchi, T. Elevated Serum Krebs von Den Lungen-6 in Early Disease Predicts Subsequent Deterioration of Pulmonary Function in Patients with Systemic Sclerosis and Interstitial Lung Disease. J. Rheumatol. 2016, 43, 1825–1831. [Google Scholar] [CrossRef]
- Kim, H.C.; Choi, K.H.; Jacob, J.; Song, J.W. Prognostic Role of Blood KL-6 in Rheumatoid Arthritis-Associated Interstitial Lung Disease. PLoS ONE 2020, 15, e0229997. [Google Scholar] [CrossRef]
- Sánchez-Díez, S.; Munoz, X.; Ojanguren, I.; Romero-Mesones, C.; Espejo, D.; Villar, A.; Gómez-Olles, S.; Cruz, M.-J. YKL-40 and KL-6 Levels in Serum and Sputum of Patients Diagnosed with Hypersensitivity Pneumonitis. J. Allergy Clin. Immunol. Pract. 2022, 10, 2414–2423. [Google Scholar] [CrossRef]
- Ejima, M.; Okamoto, T.; Suzuki, T.; Miyazaki, Y. Role of Serum Surfactant Protein-D as a Prognostic Predictor in Fibrotic Hypersensitivity Pneumonitis. Respir. Investig. 2022, 60, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Tzouvelekis, A.; Herazo-Maya, J.D.; Slade, M.; Chu, J.-H.; Deiuliis, G.; Ryu, C.; Li, Q.; Sakamoto, K.; Ibarra, G.; Pan, H.; et al. Validation of the Prognostic Value of MMP-7 in Idiopathic Pulmonary Fibrosis. Respirology 2017, 22, 486–493. [Google Scholar] [CrossRef]
- Fraser, E.; Denney, L.; Antanaviciute, A.; Blirando, K.; Vuppusetty, C.; Zheng, Y.; Repapi, E.; Iotchkova, V.; Taylor, S.; Ashley, N.; et al. Multi-Modal Characterization of Monocytes in Idiopathic Pulmonary Fibrosis Reveals a Primed Type I Interferon Immune Phenotype. Front. Immunol. 2021, 12, 623430. [Google Scholar] [CrossRef] [PubMed]
- Kreuter, M.; Lee, J.S.; Tzouvelekis, A.; Oldham, J.M.; Molyneaux, P.L.; Weycker, D.; Atwood, M.; Kirchgaessler, K.-U.; Maher, T.M. Monocyte Count as a Prognostic Biomarker in Patients with Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2021, 204, 74–81. [Google Scholar] [CrossRef]
- Achaiah, A.; Rathnapala, A.; Pereira, A.; Bothwell, H.; Dwivedi, K.; Barker, R.; Iotchkova, V.; Benamore, R.; Hoyles, R.K.; Ho, L.-P. Neutrophil Lymphocyte Ratio as an Indicator for Disease Progression in Idiopathic Pulmonary Fibrosis. BMJ Open Respir. Res. 2022, 9, e001202. [Google Scholar] [CrossRef]
- Naik, P.K.; Bozyk, P.D.; Bentley, J.K.; Popova, A.P.; Birch, C.M.; Wilke, C.A.; Fry, C.D.; White, E.S.; Sisson, T.H.; Tayob, N.; et al. Periostin Promotes Fibrosis and Predicts Progression in Patients with Idiopathic Pulmonary Fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L1046–L1056. [Google Scholar] [CrossRef]
- Chen, J.; Chen, Y.; Liu, D.; Lin, Y.; Zhu, L.; Song, S.; Hu, Y.; Liang, T.; Liu, Y.; Liu, W.; et al. Predictors of Long-Term Prognosis in Rheumatoid Arthritis-Related Interstitial Lung Disease. Sci. Rep. 2022, 12, 9469. [Google Scholar] [CrossRef]
- Takahashi, H.; Fujishima, T.; Koba, H.; Murakami, S.; Kurokawa, K.; Shibuya, Y.; Shiratori, M.; Kuroki, Y.; Abe, S. Serum Surfactant Proteins A and D as Prognostic Factors in Idiopathic Pulmonary Fibrosis and Their Relationship to Disease Extent. Am. J. Respir. Crit. Care Med. 2000, 162, 1109–1114. [Google Scholar] [CrossRef]
- Hamai, K.; Iwamoto, H.; Ishikawa, N.; Horimasu, Y.; Masuda, T.; Miyamoto, S.; Nakashima, T.; Ohshimo, S.; Fujitaka, K.; Hamada, H.; et al. Comparative Study of Circulating MMP-7, CCL18, KL-6, SP-A, and SP-D as Disease Markers of Idiopathic Pulmonary Fibrosis. Dis. Markers 2016, 2016, 4759040. [Google Scholar] [CrossRef]
- Kohno, N.; Kyoizumi, S.; Awaya, Y.; Fukuhara, H.; Yamakido, M.; Akiyama, M. New Serum Indicator of Interstitial Pneumonitis Activity. Sialylated Carbohydrate Antigen KL-6. Chest 1989, 96, 68–73. [Google Scholar] [CrossRef]
- Ohshimo, S.; Yokoyama, A.; Hattori, N.; Ishikawa, N.; Hirasawa, Y.; Kohno, N. KL-6, a Human MUC1 Mucin, Promotes Proliferation and Survival of Lung Fibroblasts. Biochem. Biophys. Res. Commun. 2005, 338, 1845–1852. [Google Scholar] [CrossRef]
- Guo, L.; Yang, Y.; Liu, F.; Jiang, C.; Yang, Y.; Pu, H.; Li, W.; Zhong, Z. Clinical Research on Prognostic Evaluation of Subjects with IPF by Peripheral Blood Biomarkers, Quantitative Imaging Characteristics and Pulmonary Function Parameters. Arch. Bronconeumol. 2020, 56, 365–372. [Google Scholar] [CrossRef]
- Zheng, P.; Zheng, X.; Takehiro, H.; Cheng, Z.J.; Wang, J.; Xue, M.; Lin, Q.; Huang, Z.; Huang, H.; Liao, C.; et al. The Prognostic Value of Krebs von Den Lungen-6 and Surfactant Protein-A Levels in the Patients with Interstitial Lung Disease. J. Transl. Intern. Med. 2021, 9, 212–222. [Google Scholar] [CrossRef]
- Ikeda, K.; Chiba, H.; Nishikiori, H.; Azuma, A.; Kondoh, Y.; Ogura, T.; Taguchi, Y.; Ebina, M.; Sakaguchi, H.; Miyazawa, S.; et al. Serum Surfactant Protein D as a Predictive Biomarker for the Efficacy of Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis: A Post-Hoc Analysis of the Phase 3 Trial in Japan. Respir. Res. 2020, 21, 316. [Google Scholar] [CrossRef]
- Bauer, Y.; White, E.S.; de Bernard, S.; Cornelisse, P.; Leconte, I.; Morganti, A.; Roux, S.; Nayler, O. MMP-7 Is a Predictive Biomarker of Disease Progression in Patients with Idiopathic Pulmonary Fibrosis. ERJ Open Res. 2017, 3, 00074–2016. [Google Scholar] [CrossRef]
- Ando, M.; Miyazaki, E.; Ito, T.; Hiroshige, S.; Nureki, S.; Ueno, T.; Takenaka, R.; Fukami, T.; Kumamoto, T. Significance of Serum Vascular Endothelial Growth Factor Level in Patients with Idiopathic Pulmonary Fibrosis. Lung 2010, 188, 247–252. [Google Scholar] [CrossRef]
- Bonhomme, O.; André, B.; Gester, F.; de Seny, D.; Moermans, C.; Struman, I.; Louis, R.; Malaise, M.; Guiot, J. Biomarkers in Systemic Sclerosis-Associated Interstitial Lung Disease: Review of the Literature. Rheumatology 2019, 58, 1534–1546. [Google Scholar] [CrossRef]
- Tyker, A.; Ventura, I.B.; Lee, C.T.; Strykowski, R.; Garcia, N.; Guzy, R.; Jablonski, R.; Vij, R.; Strek, M.E.; Chung, J.H.; et al. High-Titer Rheumatoid Factor Seropositivity Predicts Mediastinal Lymphadenopathy and Mortality in Rheumatoid Arthritis-Related Interstitial Lung Disease. Sci. Rep. 2021, 11, 22821. [Google Scholar] [CrossRef]
- Mouthon, L.; Bérezné, A.; Guillevin, L.; Valeyre, D. Therapeutic Options for Systemic Sclerosis Related Interstitial Lung Diseases. Respir. Med. 2010, 104 (Suppl. S1), S59–S69. [Google Scholar] [CrossRef]
- Distler, O.; Assassi, S.; Cottin, V.; Cutolo, M.; Danoff, S.K.; Denton, C.P.; Distler, J.H.W.; Hoffmann-Vold, A.-M.; Johnson, S.R.; Müller Ladner, U.; et al. Predictors of Progression in Systemic Sclerosis Patients with Interstitial Lung Disease. Eur. Respir. J. 2020, 55, 1902026. [Google Scholar] [CrossRef]
- Ashmore, P.; Tikly, M.; Wong, M.; Ickinger, C. Interstitial Lung Disease in South Africans with Systemic Sclerosis. Rheumatol. Int. 2018, 38, 657–662. [Google Scholar] [CrossRef]
- Ho, K.T.; Reveille, J.D. The Clinical Relevance of Autoantibodies in Scleroderma. Arthritis Res. Ther. 2003, 5, 80–93. [Google Scholar] [CrossRef]
- Schupp, J.; Becker, M.; Günther, J.; Müller-Quernheim, J.; Riemekasten, G.; Prasse, A. Serum CCL18 Is Predictive for Lung Disease Progression and Mortality in Systemic Sclerosis. Eur. Respir. J. 2014, 43, 1530–1532. [Google Scholar] [CrossRef]
- Hoffmann-Vold, A.-M.; Tennøe, A.H.; Garen, T.; Midtvedt, Ø.; Abraityte, A.; Aaløkken, T.M.; Lund, M.B.; Brunborg, C.; Aukrust, P.; Ueland, T.; et al. High Level of Chemokine CCL18 Is Associated with Pulmonary Function Deterioration, Lung Fibrosis Progression, and Reduced Survival in Systemic Sclerosis. Chest 2016, 150, 299–306. [Google Scholar] [CrossRef]
- Liu, X.; Mayes, M.D.; Pedroza, C.; Draeger, H.T.; Gonzalez, E.B.; Harper, B.E.; Reveille, J.D.; Assassi, S. Does C-Reactive Protein Predict the Long-Term Progression of Interstitial Lung Disease and Survival in Patients with Early Systemic Sclerosis? Arthritis Care Res. 2013, 65, 1375–1380. [Google Scholar] [CrossRef]
- Ross, L.; Stevens, W.; Rabusa, C.; Wilson, M.; Ferdowsi, N.; Walker, J.; Sahhar, J.; Ngian, G.-S.; Zochling, J.; Roddy, J.; et al. The Role of Inflammatory Markers in Assessment of Disease Activity in Systemic Sclerosis. Clin. Exp. Rheumatol. 2018, 36 (Suppl. S1), 126–134. [Google Scholar]
- Volkmann, E.R.; Tashkin, D.P.; Roth, M.D.; Clements, P.J.; Khanna, D.; Furst, D.E.; Mayes, M.; Charles, J.; Tseng, C.-H.; Elashoff, R.M.; et al. Changes in Plasma CXCL4 Levels Are Associated with Improvements in Lung Function in Patients Receiving Immunosuppressive Therapy for Systemic Sclerosis-Related Interstitial Lung Disease. Arthritis Res. Ther. 2016, 18, 305. [Google Scholar] [CrossRef]
- Doishita, S.; Inokuma, S.; Asashima, H.; Nakachi, S.; Matsuo, Y.; Rokutanda, R.; Kobayashi, S.; Hagiwara, K.; Satoh, T.; Akiyama, O. Serum KL-6 Level as an Indicator of Active or Inactive Interstitial Pneumonitis Associated with Connective Tissue Diseases. Intern. Med. 2011, 50, 2889–2892. [Google Scholar] [CrossRef]
- Hu, Y.; Wang, L.-S.; Jin, Y.-P.; Du, S.-S.; Du, Y.-K.; He, X.; Weng, D.; Zhou, Y.; Li, Q.-H.; Shen, L.; et al. Serum Krebs von Den Lungen-6 Level as a Diagnostic Biomarker for Interstitial Lung Disease in Chinese Patients. Clin. Respir. J. 2017, 11, 337–345. [Google Scholar] [CrossRef]
- Sumida, H.; Asano, Y.; Tamaki, Z.; Aozasa, N.; Taniguchi, T.; Toyama, T.; Takahashi, T.; Ichimura, Y.; Noda, S.; Akamata, K.; et al. Prediction of Therapeutic Response before and during i.v. Cyclophosphamide Pulse Therapy for Interstitial Lung Disease in Systemic Sclerosis: A Longitudinal Observational Study. J. Dermatol. 2018, 45, 1425–1433. [Google Scholar] [CrossRef]
- Elhai, M.; Hoffmann-Vold, A.M.; Avouac, J.; Pezet, S.; Cauvet, A.; Leblond, A.; Fretheim, H.; Garen, T.; Kuwana, M.; Molberg, Ø.; et al. Performance of Candidate Serum Biomarkers for Systemic Sclerosis-Associated Interstitial Lung Disease. Arthritis Rheumatol. 2019, 71, 972–982. [Google Scholar] [CrossRef]
- Satoh, H.; Kurishima, K.; Ishikawa, H.; Ohtsuka, M. Increased Levels of KL-6 and Subsequent Mortality in Patients with Interstitial Lung Diseases. J. Intern. Med. 2006, 260, 429–434. [Google Scholar] [CrossRef]
- Yanaba, K.; Hasegawa, M.; Hamaguchi, Y.; Fujimoto, M.; Takehara, K.; Sato, S. Longitudinal Analysis of Serum KL-6 Levels in Patients with Systemic Sclerosis: Association with the Activity of Pulmonary Fibrosis. Clin. Exp. Rheumatol. 2003, 21, 429–436. [Google Scholar]
- Fukaya, S.; Oshima, H.; Kato, K.; Komatsu, Y.; Matsumura, H.; Ishii, K.; Miyama, H.; Nagai, T.; Tanaka, I.; Mizutani, A.; et al. KL-6 as a Novel Marker for Activities of Interstitial Pneumonia in Connective Tissue Diseases. Rheumatol. Int. 2000, 19, 223–225. [Google Scholar] [CrossRef]
- Nakanishi, Y.; Horimasu, Y.; Yamaguchi, K.; Sakamoto, S.; Masuda, T.; Nakashima, T.; Miyamoto, S.; Iwamoto, H.; Ohshimo, S.; Fujitaka, K.; et al. IL-18 Binding Protein Can Be a Prognostic Biomarker for Idiopathic Pulmonary Fibrosis. PLoS ONE 2021, 16, e0252594. [Google Scholar] [CrossRef]
- Wu, C.-Y.; Yang, H.-Y.; Luo, S.-F.; Lai, J.-H. From Rheumatoid Factor to Anti-Citrullinated Protein Antibodies and Anti-Carbamylated Protein Antibodies for Diagnosis and Prognosis Prediction in Patients with Rheumatoid Arthritis. Int. J. Mol. Sci. 2021, 22, 686. [Google Scholar] [CrossRef]
- Sato, S.; Nagaoka, T.; Hasegawa, M.; Nishijima, C.; Takehara, K. Elevated Serum KL-6 Levels in Patients with Systemic Sclerosis: Association with the Severity of Pulmonary Fibrosis. Dermatology 2000, 200, 196–201. [Google Scholar] [CrossRef]
- Nell, V.P.K.; Machold, K.P.; Stamm, T.A.; Eberl, G.; Heinzl, H.; Uffmann, M.; Smolen, J.S.; Steiner, G. Autoantibody Profiling as Early Diagnostic and Prognostic Tool for Rheumatoid Arthritis. Ann. Rheum. Dis. 2005, 64, 1731–1736. [Google Scholar] [CrossRef]
- Mena-Vázquez, N.; Godoy-Navarrete, F.J.; Lisbona-Montañez, J.M.; Redondo-Rodriguez, R.; Manrique-Arija, S.; Rioja, J.; Mucientes, A.; Ruiz-Limón, P.; Garcia-Studer, A.; Ortiz-Márquez, F.; et al. Inflammatory Biomarkers in the Diagnosis and Prognosis of Rheumatoid Arthritis-Associated Interstitial Lung Disease. Int. J. Mol. Sci. 2023, 24, 6800. [Google Scholar] [CrossRef]
- Avouac, J.; Cauvet, A.; Steelandt, A.; Shirai, Y.; Elhai, M.; Kuwana, M.; Distler, O.; Allanore, Y. Improving Risk-Stratification of Rheumatoid Arthritis Patients for Interstitial Lung Disease. PLoS ONE 2020, 15, e0232978. [Google Scholar] [CrossRef]
- Nakashita, T.; Ando, K.; Kaneko, N.; Takahashi, K.; Motojima, S. Potential Risk of TNF Inhibitors on the Progression of Interstitial Lung Disease in Patients with Rheumatoid Arthritis. BMJ Open 2014, 4, e005615. [Google Scholar] [CrossRef]
- Lee, Y.S.; Kim, H.C.; Lee, B.Y.; Lee, C.K.; Kim, M.-Y.; Jang, S.J.; Lee, H.S.; Moon, J.; Colby, T.V.; Kim, D.S. The Value of Biomarkers as Predictors of Outcome in Patients with Rheumatoid Arthritis-Associated Usual Interstitial Pneumonia. Sarcoidosis, Vasc. Diffus. Lung Dis. Off. J. WASOG 2016, 33, 216–223. [Google Scholar]
- He, J.; Zhang, J.; Ren, X. Krebs von Den Lungen-6 as a Clinical Marker for Hypersensitivity Pneumonitis: A Meta-Analysis and Bioinformatics Analysis. Front. Immunol. 2022, 13, 1041098. [Google Scholar] [CrossRef]
- Long, X.; He, X.; Ohshimo, S.; Griese, M.; Sarria, R.; Guzman, J.; Costabel, U.; Bonella, F. Serum YKL-40 as Predictor of Outcome in Hypersensitivity Pneumonitis. Eur. Respir. J. 2017, 49, 1501924. [Google Scholar] [CrossRef]
- Johansen, J.S.; Milman, N.; Hansen, M.; Garbarsch, C.; Price, P.A.; Graudal, N. Increased Serum YKL-40 in Patients with Pulmonary Sarcoidosis--a Potential Marker of Disease Activity? Respir. Med. 2005, 99, 396–402. [Google Scholar] [CrossRef]
- Vorselaars, A.D.M.; van Moorsel, C.H.M.; Zanen, P.; Ruven, H.J.T.; Claessen, A.M.E.; van Velzen-Blad, H.; Grutters, J.C. ACE and SIL-2R Correlate with Lung Function Improvement in Sarcoidosis during Methotrexate Therapy. Respir. Med. 2015, 109, 279–285. [Google Scholar] [CrossRef]
- Cai, M.; Bonella, F.; He, X.; Sixt, S.U.; Sarria, R.; Guzman, J.; Costabel, U. CCL18 in Serum, BAL Fluid and Alveolar Macrophage Culture Supernatant in Interstitial Lung Diseases. Respir. Med. 2013, 107, 1444–1452. [Google Scholar] [CrossRef]
- Bargagli, E.; Magi, B.; Olivieri, C.; Bianchi, N.; Landi, C.; Rottoli, P. Analysis of Serum Amyloid A in Sarcoidosis Patients. Respir. Med. 2011, 105, 775–780. [Google Scholar] [CrossRef]
- Di Francesco, A.M.; Verrecchia, E.; Sicignano, L.L.; Massaro, M.G.; Antuzzi, D.; Covino, M.; Pasciuto, G.; Richeldi, L.; Manna, R. The Use of Chitotriosidase as a Marker of Active Sarcoidosis and in the Diagnosis of Fever of Unknown Origin (FUO). J. Clin. Med. 2021, 10, 5283. [Google Scholar] [CrossRef]
- Kraaijvanger, R.; Janssen Bonás, M.; Vorselaars, A.D.M.; Veltkamp, M. Biomarkers in the Diagnosis and Prognosis of Sarcoidosis: Current Use and Future Prospects. Front. Immunol. 2020, 11, 1443. [Google Scholar] [CrossRef]
- Nombel, A.; Fabien, N.; Coutant, F. Dermatomyositis with Anti-MDA5 Antibodies: Bioclinical Features, Pathogenesis and Emerging Therapies. Front. Immunol. 2021, 12, 773352. [Google Scholar] [CrossRef]
- Li, X.; Liu, Y.; Cheng, L.; Huang, Y.; Yan, S.; Li, H.; Zhan, H.; Li, Y. Roles of Biomarkers in Anti-MDA5-Positive Dermatomyositis, Associated Interstitial Lung Disease, and Rapidly Progressive Interstitial Lung Disease. J. Clin. Lab. Anal. 2022, 36, e24726. [Google Scholar] [CrossRef]
- Porse, S.; Hoyer, N.; Shaker, S.B. Impact of Reduction in Antifibrotic Treatment on Mortality in Idiopathic Pulmonary Fibrosis. Respir. Med. 2022, 204, 107015. [Google Scholar] [CrossRef]
- Richeldi, L.; Kolb, M.; Jouneau, S.; Wuyts, W.A.; Schinzel, B.; Stowasser, S.; Quaresma, M.; Raghu, G. Efficacy and Safety of Nintedanib in Patients with Advanced Idiopathic Pulmonary Fibrosis. BMC Pulm. Med. 2020, 20, 3. [Google Scholar] [CrossRef]
- Vianello, A.; Molena, B.; Turato, C.; Braccioni, F.; Arcaro, G.; Paladini, L.; Andretta, M.; Saetta, M. Pirfenidone Improves the Survival of Patients with Idiopathic Pulmonary Fibrosis Hospitalized for Acute Exacerbation. Curr. Med. Res. Opin. 2019, 35, 1187–1190. [Google Scholar] [CrossRef]
- Nathan, S.D.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glaspole, I.; Glassberg, M.K.; Kardatzke, D.R.; Daigl, M.; Kirchgaessler, K.-U.; Lancaster, L.H.; et al. Effect of Pirfenidone on Mortality: Pooled Analyses and Meta-Analyses of Clinical Trials in Idiopathic Pulmonary Fibrosis. Lancet Respir. Med. 2017, 5, 33–41. [Google Scholar] [CrossRef]
- Fernández Pérez, E.R.; Crooks, J.L.; Lynch, D.A.; Humphries, S.M.; Koelsch, T.L.; Swigris, J.J.; Solomon, J.J.; Mohning, M.P.; Groshong, S.D.; Fier, K. Pirfenidone in Fibrotic Hypersensitivity Pneumonitis: A Double-Blind, Randomised Clinical Trial of Efficacy and Safety. Thorax 2023, 78, 1097–1104. [Google Scholar] [CrossRef]
- Herrmann, F.E.; Hesslinger, C.; Wollin, L.; Nickolaus, P. BI 1015550 Is a PDE4B Inhibitor and a Clinical Drug Candidate for the Oral Treatment of Idiopathic Pulmonary Fibrosis. Front. Pharmacol. 2022, 13, 838449. [Google Scholar] [CrossRef]
- Richeldi, L.; Azuma, A.; Cottin, V.; Hesslinger, C.; Stowasser, S.; Valenzuela, C.; Wijsenbeek, M.S.; Zoz, D.F.; Voss, F.; Maher, T.M. Trial of a Preferential Phosphodiesterase 4B Inhibitor for Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2022, 386, 2178–2187. [Google Scholar] [CrossRef]
- Richeldi, L.; Azuma, A.; Cottin, V.; Kreuter, M.; Maher, T.M.; Martinez, F.J.; Oldham, J.M.; Valenzuela, C.; Gordat, M.; Liu, Y.; et al. Design of a Phase III, Double-Blind, Randomised, Placebo-Controlled Trial of BI 1015550 in Patients with Idiopathic Pulmonary Fibrosis (FIBRONEER-IPF). BMJ Open Respir. Res. 2023, 10, e001563. [Google Scholar] [CrossRef]
- Clapp, L.H.; Gurung, R. The Mechanistic Basis of Prostacyclin and Its Stable Analogues in Pulmonary Arterial Hypertension: Role of Membrane versus Nuclear Receptors. Prostaglandins Other Lipid Mediat. 2015, 120, 56–71. [Google Scholar] [CrossRef]
- Nathan, S.D.; Waxman, A.; Rajagopal, S.; Case, A.; Johri, S.; DuBrock, H.; De La Zerda, D.J.; Sahay, S.; King, C.; Melendres-Groves, L.; et al. Inhaled Treprostinil and Forced Vital Capacity in Patients with Interstitial Lung Disease and Associated Pulmonary Hypertension: A Post-Hoc Analysis of the INCREASE Study. Lancet Respir. Med. 2021, 9, 1266–1274. [Google Scholar] [CrossRef]
- Nathan, S.D.; Behr, J.; Cottin, V.; Lancaster, L.; Smith, P.; Deng, C.Q.; Pearce, N.; Bell, H.; Peterson, L.; Flaherty, K.R. Study Design and Rationale for the TETON Phase 3, Randomised, Controlled Clinical Trials of Inhaled Treprostinil in the Treatment of Idiopathic Pulmonary Fibrosis. BMJ Open Respir. Res. 2022, 9, e001310. [Google Scholar] [CrossRef]
- Corte, T.J.; Lancaster, L.; Swigris, J.J.; Maher, T.M.; Goldin, J.G.; Palmer, S.M.; Suda, T.; Ogura, T.; Minnich, A.; Zhan, X.; et al. Phase 2 Trial Design of BMS-986278, a Lysophosphatidic Acid Receptor 1 (LPA(1)) Antagonist, in Patients with Idiopathic Pulmonary Fibrosis (IPF) or Progressive Fibrotic Interstitial Lung Disease (PF-ILD). BMJ Open Respir. Res. 2021, 8, e001026. [Google Scholar] [CrossRef] [PubMed]
- Maher, T.M.; Ford, P.; Brown, K.K.; Costabel, U.; Cottin, V.; Danoff, S.K.; Groenveld, I.; Helmer, E.; Jenkins, R.G.; Milner, J.; et al. Ziritaxestat, a Novel Autotaxin Inhibitor, and Lung Function in Idiopathic Pulmonary Fibrosis: The ISABELA 1 and 2 Randomized Clinical Trials. JAMA 2023, 329, 1567–1578. [Google Scholar] [CrossRef] [PubMed]
- Zephyrus II: A Phase 3, Randomized, Double-Blind, Placebo-Controlled Efficacy and Safety Study of Pamrevlumab in Subjects with Idiopathic Pulmonary Fibrosis (IPF). Available online: https://www.clinicaltrials.gov/study/NCT04419558 (accessed on 6 January 2024).
- Raghu, G.; Hamblin, M.J.; Brown, A.W.; Golden, J.A.; Ho, L.A.; Wijsenbeek, M.S.; Vasakova, M.; Pesci, A.; Antin-Ozerkis, D.E.; Meyer, K.C.; et al. Long-Term Evaluation of the Safety and Efficacy of Recombinant Human Pentraxin-2 (RhPTX-2) in Patients with Idiopathic Pulmonary Fibrosis (IPF): An Open-Label Extension Study. Respir. Res. 2022, 23, 129. [Google Scholar] [CrossRef]
- Richeldi, L.; Anstrom, K.J.; Behr, J.; Corte, T.J.; Cottin, V.; Jenkins, G.; Kamath, N.; Inoue, Y.; Islam, L.; Nathan, S.D.; et al. Recombinant Human Pentraxin-2 for Idiopathic Pulmonary Fibrosis: Design of STARSCAPE-OLE, a Phase III Open Label Extension Study. Eur. Respir. J. 2021, 58, PA461. [Google Scholar] [CrossRef]
- Flaherty, K.R.; Wells, A.U.; Cottin, V.; Devaraj, A.; Walsh, S.L.F.; Inoue, Y.; Richeldi, L.; Kolb, M.; Tetzlaff, K.; Stowasser, S.; et al. Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. N. Engl. J. Med. 2019, 381, 1718–1727. [Google Scholar] [CrossRef]
- Distler, O.; Highland, K.B.; Gahlemann, M.; Azuma, A.; Fischer, A.; Mayes, M.D.; Raghu, G.; Sauter, W.; Girard, M.; Alves, M.; et al. Nintedanib for Systemic Sclerosis-Associated Interstitial Lung Disease. N. Engl. J. Med. 2019, 380, 2518–2528. [Google Scholar] [CrossRef]
- Solomon, J.J.; Danoff, S.K.; Woodhead, F.A.; Hurwitz, S.; Maurer, R.; Glaspole, I.; Dellaripa, P.F.; Gooptu, B.; Vassallo, R.; Cox, P.G.; et al. Safety, Tolerability, and Efficacy of Pirfenidone in Patients with Rheumatoid Arthritis-Associated Interstitial Lung Disease: A Randomised, Double-Blind, Placebo-Controlled, Phase 2 Study. Lancet Respir. Med. 2023, 11, 87–96. [Google Scholar] [CrossRef]
- Cottin, V.; Brown, K.K. Interstitial lung disease associated with systemic sclerosis (SSc-ILD). Respir. Res. 2019, 20, 13. [Google Scholar] [CrossRef]
- Udwadia, Z.F.; Mullerpattan, J.B.; Balakrishnan, C.; Richeldi, L. Improved Pulmonary Function Following Pirfenidone Treatment in a Patient with Progressive Interstitial Lung Disease Associated with Systemic Sclerosis. Lung India 2015, 32, 50–52. [Google Scholar] [CrossRef]
- Khanna, D.; Albera, C.; Fischer, A.; Khalidi, N.; Raghu, G.; Chung, L.; Chen, D.; Schiopu, E.; Tagliaferri, M.; Seibold, J.R.; et al. An Open-Label, Phase II Study of the Safety and Tolerability of Pirfenidone in Patients with Scleroderma-Associated Interstitial Lung Disease: The LOTUSS Trial. J. Rheumatol. 2016, 43, 1672–1679. [Google Scholar] [CrossRef]
- Miura, Y.; Saito, T.; Fujita, K.; Tsunoda, Y.; Tanaka, T.; Takoi, H.; Yatagai, Y.; Rin, S.; Sekine, A.; Hayashihara, K.; et al. Clinical Experience with Pirfenidone in Five Patients with Scleroderma-Related Interstitial Lung Disease. Sarcoidosis Vasc. Diffus. Lung Dis. Off. J. WASOG 2014, 31, 235–238. [Google Scholar]
- Acharya, N.; Sharma, S.K.; Mishra, D.; Dhooria, S.; Dhir, V.; Jain, S. Efficacy and Safety of Pirfenidone in Systemic Sclerosis-Related Interstitial Lung Disease-a Randomised Controlled Trial. Rheumatol. Int. 2020, 40, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Khanna, D.; Spino, C.; Bernstein, E.; Goldin, J.; Tashkin, D.; Roth, M. Combination Therapy of Mycophenolate Mofetil and Pirfenidone vs. Mycophenolate Alone: Results from the Scleroderma Lung Study III. In Arthritis & Rheumatology; Wiley: Hoboken, NJ, USA, 2022; Volume 0520. [Google Scholar]
- Kur-Zalewska, J.; Kisiel, B.; Kania-Pudło, M.; Tłustochowicz, M.; Chciałowski, A.; Tłustochowicz, W. A Dose-Dependent Beneficial Effect of Methotrexate on the Risk of Interstitial Lung Disease in Rheumatoid Arthritis Patients. PLoS ONE 2021, 16, e0250339. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Woo, A.; Park, Y.; Yong, S.H.; Lee, S.H.; Lee, S.H.; Leem, A.Y.; Kim, S.Y.; Chung, K.S.; Kim, E.Y.; et al. Protective Effect of Methotrexate on Lung Function and Mortality in Rheumatoid Arthritis-Related Interstitial Lung Disease: A Retrospective Cohort Study. Ther. Adv. Respir. Dis. 2022, 16, 17534666221135314. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Xiao, L.; Zhu, J.; Qin, Q.; Fang, Y.; Zhang, J.-A. Methotrexate Use Reduces Mortality Risk in Rheumatoid Arthritis: A Systematic Review and Meta-Analysis of Cohort Studies. Semin. Arthritis Rheum. 2022, 55, 152031. [Google Scholar] [CrossRef]
- Baker, M.C.; Liu, Y.; Lu, R.; Lin, J.; Melehani, J.; Robinson, W.H. Incidence of Interstitial Lung Disease in Patients with Rheumatoid Arthritis Treated with Biologic and Targeted Synthetic Disease-Modifying Antirheumatic Drugs. JAMA Netw. Open 2023, 6, e233640. [Google Scholar] [CrossRef]
- Tashkin, D.P.; Elashoff, R.; Clements, P.J.; Goldin, J.; Roth, M.D.; Furst, D.E.; Arriola, E.; Silver, R.; Strange, C.; Bolster, M.; et al. Cyclophosphamide versus Placebo in Scleroderma Lung Disease. N. Engl. J. Med. 2006, 354, 2655–2666. [Google Scholar] [CrossRef]
- Tashkin, D.P.; Roth, M.D.; Clements, P.J.; Furst, D.E.; Khanna, D.; Kleerup, E.C.; Goldin, J.; Arriola, E.; Volkmann, E.R.; Kafaja, S.; et al. Mycophenolate Mofetil versus Oral Cyclophosphamide in Scleroderma-Related Interstitial Lung Disease (SLS II): A Randomised Controlled, Double-Blind, Parallel Group Trial. Lancet Respir. Med. 2016, 4, 708–719. [Google Scholar] [CrossRef]
- Sharma, S.; Mathew, J.; Kopp, C.; Dhir, V.; Dhooria, S.; Sinha, A.; Jain, S. A Randomized Controlled Trial to Compare the Efficacy and Safety of Tacrolimus with Mycophenolate Mofetil in Patients with Systemic Sclerosis—Interstitial Lung Disease (INSIST TRIAL) [Abstract]. Arthritis Rheumatol. 2023, 75 (Suppl. S9). Available online: https://acrabstracts.org/abstract/a-randomized-controlled-trial-to-compare-the-efficacy-and-safety-of-tacrolimus-with-mycophenolate-mofetil-in-patients-with-systemic-sclerosis-interstitial-lung-disease-insist-trial/ (accessed on 6 January 2024).
- Khanna, D.; Lin, C.J.F.; Furst, D.E.; Goldin, J.; Kim, G.; Kuwana, M.; Allanore, Y.; Matucci-Cerinic, M.; Distler, O.; Shima, Y.; et al. Tocilizumab in Systemic Sclerosis: A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Respir. Med. 2020, 8, 963–974. [Google Scholar] [CrossRef]
- Available online: https://rheumatology.org/interstitial-lung-disease-guideline (accessed on 6 January 2024).
- Fujisawa, T. Management of Myositis-Associated Interstitial Lung Disease. Medicina 2021, 57, 347. [Google Scholar] [CrossRef]
- Baughman, R.P.; Valeyre, D.; Korsten, P.; Mathioudakis, A.G.; Wuyts, W.A.; Wells, A.; Rottoli, P.; Nunes, H.; Lower, E.E.; Judson, M.A.; et al. ERS Clinical Practice Guidelines on Treatment of Sarcoidosis. Eur. Respir. J. 2021, 58, 2004079. [Google Scholar] [CrossRef] [PubMed]
- Baughman, R.P.; Gupta, R.; Judson, M.A.; Lower, E.E.; Birring, S.S.; Stewart, J.; Reeves, R.; Wells, A.U. Value of Pulmonary Function Testing Identifying Progressive Pulmonary Disease in Fibrotic Sarcoidosis: Results of a Prospective Feasibility Study. Sarcoidosis Vasc. Diffus. Lung Dis. Off. J. WASOG 2022, 39, e2022011. [Google Scholar] [CrossRef]
- Study Details|Efficacy and Safety of Intravenous Efzofitimod in Patients with Pulmonary Sarcoidosis. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT05415137 (accessed on 6 January 2024).
- Study of Efficacy, Safety and Tolerability of CMK389 in Patients with Chronic Pulmonary Sarcoidosis. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT04064242 (accessed on 6 January 2024).
- ClinicalTrials.gov. A Study to Assess the Efficacy and Safety of Risankizumab in Participants with Ulcerative Colitis. Available online: https://clinicaltrials.gov/study/NCT03398135 (accessed on 6 January 2024).
- Raghu, G.; Remy-Jardin, M.; Ryerson, C.J.; Myers, J.L.; Kreuter, M.; Vasakova, M.; Bargagli, E.; Chung, J.H.; Collins, B.F.; Bendstrup, E.; et al. Diagnosis of Hypersensitivity Pneumonitis in Adults. An Official ATS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2020, 202, e36–e69. [Google Scholar] [CrossRef] [PubMed]
- Morisset, J.; Johannson, K.A.; Vittinghoff, E.; Aravena, C.; Elicker, B.M.; Jones, K.D.; Fell, C.D.; Manganas, H.; Dubé, B.-P.; Wolters, P.J.; et al. Use of Mycophenolate Mofetil or Azathioprine for the Management of Chronic Hypersensitivity Pneumonitis. Chest 2017, 151, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Fiddler, C.A.; Simler, N.; Thillai, M.; Parfrey, H. Use of Mycophenolate Mofetil and Azathioprine for the Treatment of Chronic Hypersensitivity Pneumonitis-A Single-Centre Experience. Clin. Respir. J. 2019, 13, 791–794. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, A.T.; Martins, N.; Raimundo, S.; Melo, N.; Catetano Mota, P.; Bastos, H.N.E.; Pereira, J.M.; Cunha, R.; Guimarães, S.; Souto Moura, C.; et al. Impact of Azathioprine Use in Chronic Hypersensitivity Pneumonitis Patients. Pulm. Pharmacol. Ther. 2020, 60, 101878. [Google Scholar] [CrossRef] [PubMed]
- Adegunsoye, A.; Oldham, J.M.; Fernández Pérez, E.R.; Hamblin, M.; Patel, N.; Tener, M.; Bhanot, D.; Robinson, L.; Bullick, S.; Chen, L.; et al. Outcomes of Immunosuppressive Therapy in Chronic Hypersensitivity Pneumonitis. ERJ Open Res. 2017, 3, 00016–2017. [Google Scholar] [CrossRef]
- Ferreira, M.; Borie, R.; Crestani, B.; Rigaud, P.; Wemeau, L.; Israel-Biet, D.; Leroy, S.; Quétant, S.; Plantier, L.; Dalphin, J.-C.; et al. Efficacy and Safety of Rituximab in Patients with Chronic Hypersensitivity Pneumonitis (CHP): A Retrospective, Multicentric, Observational Study. Respir. Med. 2020, 172, 106146. [Google Scholar] [CrossRef]
- Behr, J.; Prasse, A.; Kreuter, M.; Johow, J.; Rabe, K.F.; Bonella, F.; Bonnet, R.; Grohe, C.; Held, M.; Wilkens, H.; et al. Pirfenidone in Patients with Progressive Fibrotic Interstitial Lung Diseases Other than Idiopathic Pulmonary Fibrosis (RELIEF): A Double-Blind, Randomised, Placebo-Controlled, Phase 2b Trial. Lancet Respir. Med. 2021, 9, 476–486. [Google Scholar] [CrossRef]


{kind=link}
| Biomarker | Population | Findings | References |
|---|---|---|---|
| Anti-CXCR3/CXCR4 | SSc (n = 327) |
| Weigold et al. [55] |
| Anti-Scl 70 | SSc (n = 117) |
| Wangkaew et al. [56] |
| Anti-SS-A/Ro52+ | CTD/IPAF (n = 107) |
| Nagy et al. [57] |
| CCL15 | HP (n = 51) |
| Watanabe [58] |
| CCL18 | IPF (n = 72) |
| Prasse et al. [59] |
| SSc (n = 83) | Tiev et al. [60] | ||
| SSc (n = 87) |
| Khanna et al. [61] | |
| |||
| |||
| IL-6 | SSc (n = 286) |
| De Lauretis [62] |
| KL-6 | IPF (n = 89) |
| Wakamatsu et al. [63] |
| Early-SSc (n = 50) |
| Kuwana et al. [64] | |
| RA-ILD (n = 84) |
| Kim et al. [65] | |
| HP (n = 49) |
| Sánchez-Díez [66] | |
| f-HP (n = 185) |
| Ejima et al. [67] | |
| MMP7 | IPF (n = 97) |
| Tzouvelekis et al. [68] |
| Monocytes | IPF (n = 37) |
| Fraser et al. [69] |
| IPF (n = 2067) |
| Kreuter et al. [70] | |
| IPF (n = 128) |
| Achaiah et al. [71] | |
| Periostin | IPF (n = 54) |
| Naik et al. [72] |
| RF | RA (n = 60) |
| Chen et al. [73] |
| SP-D | IPF (n = 52) |
| Takahasi et al. [74] |
| |||
|
| Molecule | Route of Administration | Mechanism of Action | Key Trials | Status |
|---|---|---|---|---|
| BI 1015550 | Oral | ↓ PDE4b ↓ TNF-α ↓ IL-2 ↓ FGF ↓ myofibroblast transformation | FIBRONEER-IPF NCT05321069 NCT04419506 (Phase 2) | Under evaluation |
| BMS 986278 | Oral | LPAR1 antagonist ↓ myofibroblast activation | NCT04308681—Phase 2 | Under evaluation |
| Pirfenidone | Oral | ↓ TGF-β ↓ PDGF ↓ TNF-α ↓ IL-13 ↓ IL-4 ↑ MMPs | ASCEND—NCT01366209 Capacity 1—NCT00287729 Capacity 2—NCT00287716 SP3—Japanese | Approved |
| Nintedanib | Oral | FGFR inhibitor PDGFR inhibitor VGFR inhibitor | INPULSIS-1—NCT01335464 INPULSIS-2—NCT01335477 | Approved |
| Pamrevlumab | Intravenous | ↓ CTGF | ZEPHYRUS 2—NCT04419558 | Not approved |
| PRM-151 (rhPTX-2) | Intravenous | ↓ Inflammation ↓ Fibrosis | WA42404—NCT02550873 STARSCAPE—NCT04552899 | Not approved |
| Treprostinil | Inhaled | ↑ EP2 ↑ DP1 ↑ PPAR ↓ Cell proliferation ↓ Collagen synthesis ↓ Inflammation ↓ Fibroblast proliferation | INCREASE—NCT02630316 TETON—NCT04708782 | Under evaluation |
| Ziritaxestat | Oral | ↓ Autotaxin ↓ LPA | ISABELLA 1—NCT03711162 ISABELLA 2—NCT03733444 | Not approved |
| RA-ILD | |||
|---|---|---|---|
| Molecule | Key Trials | Recommendation | |
| BI 1015550 (PDE4Bi) | NCT05321082 | Under evaluation as per PPF | |
| Nintedanib | INBUILD—NCT02999178 | Recommended for ILD progression despite first-line treatment | |
| Pirfenidone | TRAIL 1—NCT02808871 | Not recommended | |
| SSc-ILD | |||
| Molecule | Key Trials | Recommendation | |
| BI 1015550 (PDE4Bi) | NCT05321082 | Under evaluation as per PPF | |
| Nintedanib | INBUILD—NCT02999178 SENSCIS—NCT02597933 | Recommended as additional first-line ILD option | |
| Pirfenidone | NCT03856853 SLSIII—NCT03221257 | Not recommended | |
| Tacrolimus | INSIST—CTRI/2021/11/037864 | Under evaluation | |
| Tocilizumab | focuSSced—NCT02453256 | Recommended as first-line preferred ILD option | |
| Other CTD-ILDs (Myositis, MCTD, Sjogren) | |||
| Molecule | Key Trial | Recommendation | |
| BI 1015550 (PDE4Bi) | NCT05321082 | Under evaluation as per PPF | |
| Nintedanib | INBUILD—NCT02999178 | Recommended for ILD progression despite first-line treatment | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Agnano, V.; Mariniello, D.F.; Ruotolo, M.; Quarcio, G.; Moriello, A.; Conte, S.; Sorrentino, A.; Sanduzzi Zamparelli, S.; Bianco, A.; Perrotta, F. Targeting Progression in Pulmonary Fibrosis: An Overview of Underlying Mechanisms, Molecular Biomarkers, and Therapeutic Intervention. Life 2024, 14, 229. https://doi.org/10.3390/life14020229
D’Agnano V, Mariniello DF, Ruotolo M, Quarcio G, Moriello A, Conte S, Sorrentino A, Sanduzzi Zamparelli S, Bianco A, Perrotta F. Targeting Progression in Pulmonary Fibrosis: An Overview of Underlying Mechanisms, Molecular Biomarkers, and Therapeutic Intervention. Life. 2024; 14(2):229. https://doi.org/10.3390/life14020229
Chicago/Turabian StyleD’Agnano, Vito, Domenica Francesca Mariniello, Michela Ruotolo, Gianluca Quarcio, Alessandro Moriello, Stefano Conte, Antonio Sorrentino, Stefano Sanduzzi Zamparelli, Andrea Bianco, and Fabio Perrotta. 2024. "Targeting Progression in Pulmonary Fibrosis: An Overview of Underlying Mechanisms, Molecular Biomarkers, and Therapeutic Intervention" Life 14, no. 2: 229. https://doi.org/10.3390/life14020229
APA StyleD’Agnano, V., Mariniello, D. F., Ruotolo, M., Quarcio, G., Moriello, A., Conte, S., Sorrentino, A., Sanduzzi Zamparelli, S., Bianco, A., & Perrotta, F. (2024). Targeting Progression in Pulmonary Fibrosis: An Overview of Underlying Mechanisms, Molecular Biomarkers, and Therapeutic Intervention. Life, 14(2), 229. https://doi.org/10.3390/life14020229