
Review of Predicting Synergistic Drug Combinations


Abstract
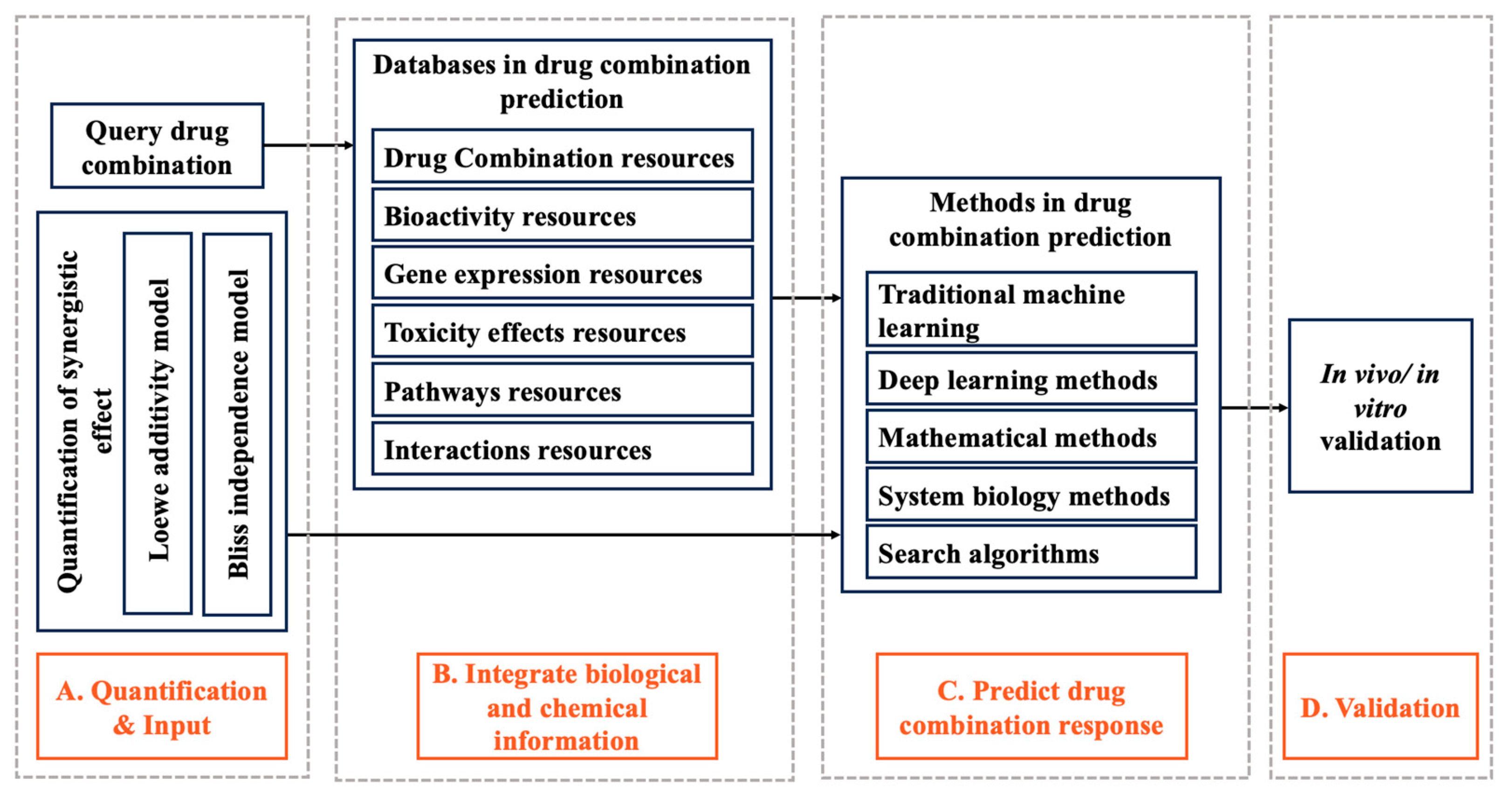
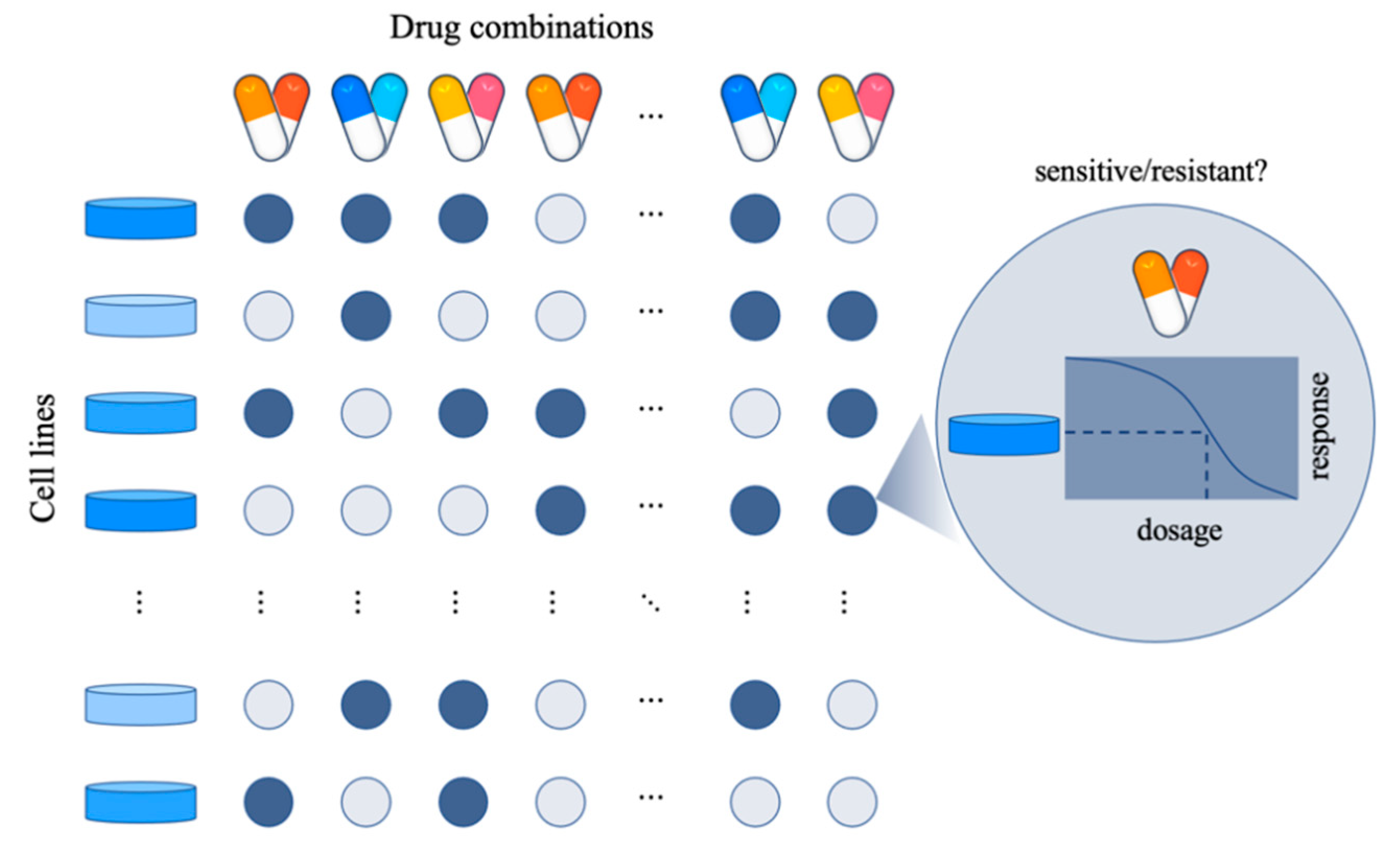
1. Introduction
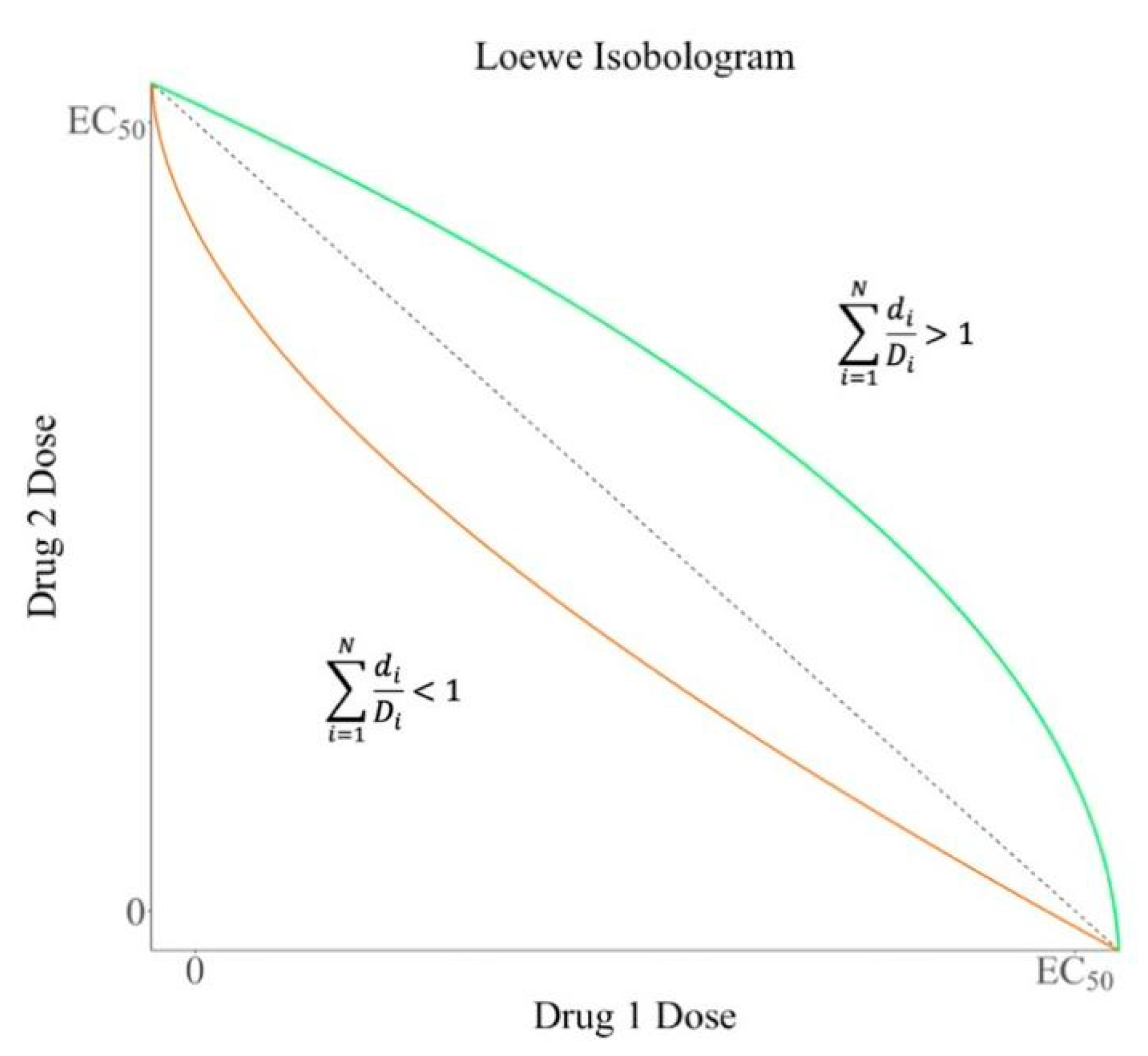
2. Quantification of Synergistic Effect
2.1. Loewe Additivity Model
2.2. Bliss Independence Model
3. Databases in Drug Combination Prediction
3.1. Drug Combination Resources
3.2. Bioactivity Resources
3.3. Gene Expression Resources
3.4. Toxicity Effects Resources
3.5. Pathways Resources
3.6. Interactions Resources
4. Methods in Drug Combination Prediction
4.1. Application of Traditional Machine Learning in Drug Combination Prediction
4.2. Application of Deep Learning Methods in Drug Combination Prediction
4.3. Application of Mathematical Methods in Drug Combination Prediction
4.4. Application of Search Algorithms in Drug Combination Prediction
4.5. Application of Systems Biology Methods in Drug Combination Prediction
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nair, N.U.; Greninger, P.; Zhang, X.H.; Friedman, A.A.; Amzallag, A.; Cortez, E.; Das Sahu, A.; Lee, J.S.; Dastur, A.; Egan, R.K.; et al. A landscape of response to drug combinations in non-small cell lung cancer. Nat. Commun. 2023, 14, 3830. [Google Scholar] [CrossRef] [PubMed]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Gorre, M.E.; Mohammed, M.; Ellwood, K.; Hsu, N.; Paquette, R.; Rao, P.N.; Sawyers, C.L. Clinical Resistance to STI-571 Cancer Therapy Caused by BCR-ABL Gene Mutation or Amplification. Science 2001, 293, 876–880. [Google Scholar] [CrossRef] [PubMed]
- Chang, G.; Roth, C.B. Structure of MsbA from E. coli: A Homolog of the Multidrug Resistance ATP Binding Cassette (ABC) Transporters. Science 2001, 293, 1793–1800. [Google Scholar] [CrossRef]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.-M.; Zhao, X.; Christensen, J.; et al. MET Amplification Leads to Gefitinib Resistance in Lung Cancer by Activating ERBB3 Signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef]
- Mokhtari, R.B.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Liang, H.; Butte, A.J.; Eng, C.; Nussinov, R. Personal Mutanomes Meet Modern Oncology Drug Discovery and Precision Health. Pharmacol. Rev. 2019, 71, 1–19. [Google Scholar] [CrossRef]
- Argelaguet, R.; Velten, B.; Arnol, D.; Dietrich, S.; Zenz, T.; Marioni, J.C.; Buettner, F.; Huber, W.; Stegle, O. Multi-Omics Factor Analysis-a framework for unsupervised integration of multi-omics data sets. Mol. Syst. Biol. 2018, 14, e8124. [Google Scholar] [CrossRef] [PubMed]
- Cokol, M.; Chua, H.N.; Tasan, M.; Mutlu, B.; Weinstein, Z.B.; Suzuki, Y.; Nergiz, M.E.; Costanzo, M.; Baryshnikova, A.; Giaever, G.; et al. Systematic exploration of synergistic drug pairs. Mol. Syst. Biol. 2011, 7, 544. [Google Scholar] [CrossRef]
- Morris, M.; Clarke, D.; Osimiri, L.; Lauffenburger, D. Systematic Analysis of Quantitative Logic Model Ensembles Predicts Drug Combination Effects on Cell Signaling Networks. CPT Pharmacomet. Syst. Pharmacol. 2016, 5, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Kim, D.G.; Bae, T.J.; Rho, K.; Kim, J.-T.; Lee, J.-J.; Jang, Y.; Kim, B.C.; Park, K.M.; Kim, S. CDA: Combinatorial Drug Discovery Using Transcriptional Response Modules. PLoS ONE 2012, 7, e42573. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Wen, Y.; Leng, D.; Zhang, Q.; Dai, C.; Wang, Z.; Liu, Z.; Yan, B.; Zhang, Y.; Wang, J.; et al. Machine learning methods, databases and tools for drug combination prediction. Brief. Bioinform. 2022, 23, bbab355. [Google Scholar] [CrossRef] [PubMed]
- Loewe, S. The problem of synergism and antagonism of combined drugs. Arzneimittelforschung 1953, 3, 285–290. [Google Scholar]
- Bliss, C.I. The Toxicity of Poisons Applied Jointly. Ann. Appl. Biol. 1939, 26, 585–615. [Google Scholar] [CrossRef]
- Vakil, V.; Trappe, W. Drug Combinations: Mathematical Modeling and Networking Methods. Pharmaceutics 2019, 11, 208. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. What is synergy? Scientist 2007, 21, 15. [Google Scholar]
- Goldoni, M.; Johansson, C. A mathematical approach to study combined effects of toxicants in vitro: Evaluation of the Bliss independence criterion and the Loewe additivity model. Toxicol. Vitr. 2007, 21, 759–769. [Google Scholar] [CrossRef]
- Laskey, S.B.; Siliciano, R.F. A mechanistic theory to explain the efficacy of antiretroviral therapy. Nat. Rev. Microbiol. 2014, 12, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Chevereau, G.; Bollenbach, T. Systematic discovery of drug interaction mechanisms. Mol. Syst. Biol. 2015, 11, 807. [Google Scholar] [CrossRef] [PubMed]
- Tonekaboni, S.A.M.; Ghoraie, L.S.; Manem, V.S.K.; Haibe-Kains, B. Predictive approaches for drug combination discovery in cancer. Brief. Bioinform. 2018, 19, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Aldahdooh, J.; Shadbahr, T.; Wang, Y.; Aldahdooh, D.; Bao, J.; Wang, W.; Tang, J. DrugComb update: A more comprehensive drug sensitivity data repository and analysis portal. Nucleic Acids Res. 2021, 49, W174–W184. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, W.; Zou, B.; Wang, J.; Deng, Y.; Deng, L. DrugCombDB: A comprehensive database of drug combinations toward the discovery of combinatorial therapy. Nucleic Acids Res. 2020, 48, D871–D881. [Google Scholar]
- Holbeck, S.L.; Camalier, R.; Crowell, J.A.; Govindharajulu, J.P.; Hollingshead, M.; Anderson, L.W.; Polley, E.; Rubinstein, L.; Srivastava, A.; Wilsker, D.; et al. The National Cancer Institute ALMANAC: A Comprehensive Screening Resource for the Detection of Anticancer Drug Pairs with Enhanced Therapeutic Activity. Cancer Res. 2017, 77, 3564–3576. [Google Scholar] [CrossRef]
- Seo, H.; Tkachuk, D.; Ho, C.; Mammoliti, A.; Rezaie, A.; Tonekaboni, S.A.M.; Haibe-Kains, B. SYNERGxDB: An integrative pharmacogenomic portal to identify synergistic drug combinations for precision oncology. Nucleic Acids Res. 2020, 48, W494–W501. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2023 update. Nucleic Acids Res. 2023, 51, D1373–D1380. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.-P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using Gene-Expression Signatures to Connect Small Molecules, Genes, and Disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M.; Letunic, I.; Jensen, L.J.; Bork, P. The SIDER database of drugs and side effects. Nucleic Acids Res. 2016, 44, D1075–D1079. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Yan, B.; Han, J.; Li, R.; Xiao, J.; He, S.; Bo, X. TOXRIC: A comprehensive database of toxicological data and benchmarks. Nucleic Acids Res. 2023, 51, D1432–D1445. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, B.; Auerbach, S.S.; Houck, K.A.; Kleinstreuer, N.C. Tox21BodyMap: A webtool to map chemical effects on the human body. Nucleic Acids Res. 2020, 48, W472–W476. [Google Scholar]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Li, C.; Marcu, A.; Badran, H.; Pon, A.; Budinski, Z.; Patron, J.; Lipton, D.; Cao, X.; Oler, E.; et al. PathBank: A comprehensive pathway database for model organisms. Nucleic Acids Res. 2020, 48, D470–D478. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, Y.; Lian, X.; Li, F.; Wang, C.; Zhu, F.; Qiu, Y.; Chen, Y. Therapeutic target database update 2022: Facilitating drug discovery with enriched comparative data of targeted agents. Nucleic Acids Res. 2022, 50, D1398–D1407. [Google Scholar] [CrossRef] [PubMed]
- Gilson, M.K.; Liu, T.; Baitaluk, M.; Nicola, G.; Hwang, L.; Chong, J. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016, 44, D1045–D1053. [Google Scholar] [CrossRef]
- Prasad, T.S.K.; Goel, R.; Kandasamy, K.; Keerthikumar, S.; Kumar, S.; Mathivanan, S.; Telikicherla, D.; Raju, R.; Shafreen, B.; Venugopal, A.; et al. Human Protein Reference Database—2009 update. Nucleic Acids Res. 2009, 37, D767–D772. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Santos, A.; von Mering, C.; Jensen, L.J.; Bork, P.; Kuhn, M. STITCH 5: Augmenting protein-chemical interaction networks with tissue and affinity data. Nucleic Acids Res. 2016, 44, D380–D384. [Google Scholar] [CrossRef]
- Kuru, H.I.; Tastan, O.; Cicek, A.E. MatchMaker: A Deep Learning Framework for Drug Synergy Prediction. IEEE/ACM Trans. Comput. Biol. Bioinform. 2022, 19, 2334–2344. [Google Scholar] [CrossRef] [PubMed]
- Sidorov, P.; Naulaerts, S.; Ariey-Bonnet, J.; Pasquier, E.; Ballester, P.J. Predicting Synergism of Cancer Drug Combinations Using NCI-ALMANAC Data. Front. Chem. 2019, 7, 509. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Chen, F.; Zeng, J.; Gao, J.; Zhang, M.Q. ScaffComb: A Phenotype-Based Framework for Drug Combination Virtual Screening in Large-Scale Chemical Datasets. Adv. Sci. 2021, 8, e2102092. [Google Scholar] [CrossRef]
- Ke, J.; Li, M.T.; Huo, Y.J.; Cheng, Y.Q.; Guo, S.F.; Wu, Y.; Zhang, L.; Ma, J.; Liu, A.J.; Han, Y. The Synergistic Effect of Ginkgo biloba Extract 50 and Aspirin Against Platelet Aggregation. Drug Des. Dev. Ther. 2021, 15, 3543–3560. [Google Scholar] [CrossRef]
- Lv, Y.; Wu, L.; Jian, H.; Zhang, C.; Lou, Y.; Kang, Y.; Hou, M.; Li, Z.; Li, X.; Sun, B.; et al. Identification and characterization of aging/senescence-induced genes in osteosarcoma and predicting clinical prognosis. Front. Immunol. 2022, 13, 997765. [Google Scholar] [CrossRef]
- Aissa, A.F.; Islam, A.; Ariss, M.M.; Go, C.C.; Rader, A.E.; Conrardy, R.D.; Gajda, A.M.; Rubio-Perez, C.; Valyi-Nagy, K.; Pasquinelli, M.; et al. Single-cell transcriptional changes associated with drug tolerance and response to combination therapies in cancer. Nat. Commun. 2021, 12, 1628. [Google Scholar] [CrossRef]
- Jin, L.; Tu, J.; Jia, J.; An, W.; Tan, H.; Cui, Q.; Li, Z. Drug-repurposing identified the combination of Trolox C and Cytisine for the treatment of type 2 diabetes. J. Transl. Med. 2014, 12, 153. [Google Scholar] [CrossRef]
- Prinz, J.; Koohi-Moghadam, M.; Sun, H.; Kocher, J.A.; Wang, J. Novel Neural Network Approach to Predict Drug-Target Interactions Based on Drug Side Effects and Genome-Wide Association Studies. Hum. Hered. 2018, 83, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Chamberlin, S.R.; Blucher, A.; Wu, G.; Shinto, L.; Choonoo, G.; Kulesz-Martin, M.; McWeeney, S. Natural Product Target Network Reveals Potential for Cancer Combination Therapies. Front. Pharmacol. 2019, 10, 557. [Google Scholar] [CrossRef]
- Li, P.; Chen, J.; Wang, J.; Zhou, W.; Wang, X.; Li, B.; Tao, W.; Wang, W.; Wang, Y.; Yang, L. Systems pharmacology strategies for drug discovery and combination with applications to cardiovascular diseases. J. Ethnopharmacol. 2014, 151, 93–107. [Google Scholar] [CrossRef]
- Wang, T.; Chen, L.; Zhao, X. Prediction of Drug Combinations with a Network Embedding Method. Comb. Chem. High Throughput Screen. 2018, 21, 789–797. [Google Scholar] [CrossRef]
- Gertrudes, J.C.; Maltarollo, V.G.; Silva, R.A.; Oliveira, P.R.; Honorio, K.M.; da Silva, A.B. Machine Learning Techniques and Drug Design. Curr. Med. Chem. 2012, 19, 4289–4297. [Google Scholar] [CrossRef] [PubMed]
- Bajorath, J.; Kearnes, S.; Walters, W.P.; Meanwell, N.A.; Georg, G.I.; Wang, S. Artificial Intelligence in Drug Discovery: Into the Great Wide Open. J. Med. Chem. 2020, 63, 8651–8652. [Google Scholar] [CrossRef]
- Talevi, A.; Morales, J.F.; Hather, G.; Podichetty, J.T.; Kim, S.; Bloomingdale, P.C.; Kim, S.; Burton, J.; Brown, J.D.; Winterstein, A.G.; et al. Machine Learning in Drug Discovery and Development Part 1: A Primer. CPT Pharmacomet. Syst. Pharmacol. 2020, 9, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Tsigelny, I.F. Artificial intelligence in drug combination therapy. Brief. Bioinform. 2019, 20, 1434–1448. [Google Scholar] [CrossRef]
- Sheng, Z.; Sun, Y.; Yin, Z.; Tang, K.; Cao, Z. Advances in computational approaches in identifying synergistic drug combinations. Brief. Bioinform. 2018, 19, 1172–1182. [Google Scholar] [CrossRef] [PubMed]
- Ryall, K.A.; Tan, A.C. Systems biology approaches for advancing the discovery of effective drug combinations. J. Cheminform. 2015, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Noble, W.S. What is a support vector machine? Nat. Biotechnol. 2006, 24, 1565–1567. [Google Scholar] [CrossRef] [PubMed]
- Safavian, S.R.; Landgrebe, D. A survey of decision tree classifier methodology. IEEE Trans. Syst. Man Cybern. 1991, 21, 660–674. [Google Scholar] [CrossRef]
- Friedman, J.H. Stochastic gradient boosting. Comput. Stat. Data Anal. 2002, 38, 367–378. [Google Scholar] [CrossRef]
- Schmidhuber, J. Deep Learning in Neural Networks: An Overview. Neural Netw. 2015, 61, 85–117. [Google Scholar] [CrossRef]
- Kramer, M.A. Nonlinear principal component analysis using autoassociative neural networks. AIChE J. 1991, 37, 233–243. [Google Scholar] [CrossRef]
- Jiang, P.; Huang, S.; Fu, Z.; Sun, Z.; Lakowski, T.M.; Hu, P. Deep graph embedding for prioritizing synergistic anticancer drug combinations. Comput. Struct. Biotechnol. J. 2020, 18, 427–438. [Google Scholar] [CrossRef]
- Movahedi, F.; Coyle, J.L.; Sejdic, E. Deep Belief Networks for Electroencephalography: A Review of Recent Contributions and Future Outlooks. IEEE J. Biomed. Health Inform. 2018, 22, 642–652. [Google Scholar] [CrossRef] [PubMed]
- Barabasi, A.-L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 2011, 12, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.S.; Gao, Q.T.; Guo, R.W.; Zhang, X.; Zhou, Z.H.; Yu, Z.; Zhu, X.J.; Gao, Y.T.; Sun, X.H.; Gao, Y.Q.; et al. Immunomodulatory Effects of Combination Therapy with Bushen Formula plus Entecavir for Chronic Hepatitis B Patients. J. Immunol. Res. 2019, 2019, 8983903. [Google Scholar] [CrossRef]
- Kim, J.; Yoo, M.; Kang, J.; Tan, A.C. K-Map: Connecting kinases with therapeutics for drug repurposing and development. Hum. Genom. 2013, 7, 20. [Google Scholar] [CrossRef][Green Version]
- Huang, S.; Cai, N.; Pacheco, P.P.; Narandes, S.; Wang, Y.; Xu, W. Applications of Support Vector Machine (SVM) Learning in Cancer Genomics. Cancer Genom. Proteom. 2018, 15, 41–51. [Google Scholar]
- Wu, L.; Gao, J.; Zhang, Y.; Sui, B.; Wen, Y.; Wu, Q.; Liu, K.; He, S.; Bo, X. A hybrid deep forest-based method for predicting synergistic drug combinations. Cell Rep. Methods 2023, 3, 100411. [Google Scholar] [CrossRef]
- Galal, A.; Talal, M.; Moustafa, A. Applications of machine learning in metabolomics: Disease modeling and classification. Front. Genet. 2022, 13, 1017340. [Google Scholar] [CrossRef]
- Xu, Q.; Xiong, Y.; Dai, H.; Kumari, K.M.; Xu, Q.; Ou, H.Y.; Wei, D.-Q. PDC-SGB: Prediction of effective drug combinations using a stochastic gradient boosting algorithm. J. Theor. Biol. 2017, 417, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Yao, S.; Zhang, Z.; Cao, B.; Wilson, C.M.; Kalos, D.; Kuan, P.F.; Zhu, R.; Wang, X. Efficient gradient boosting for prognostic biomarker discovery. Bioinformatics 2022, 38, 1631–1638. [Google Scholar] [CrossRef]
- Tsai, P.-L.; Chang, H.H.; Chen, P.S. Predicting the Treatment Outcomes of Antidepressants Using a Deep Neural Network of Deep Learning in Drug-Naïve Major Depressive Patients. J. Pers. Med. 2022, 12, 693. [Google Scholar] [CrossRef] [PubMed]
- Kriegeskorte, N.; Golan, T. Neural network models and deep learning. Curr. Biol. 2019, 29, R231–R236. [Google Scholar] [CrossRef]
- Wang, D.; Gu, J. VASC: Dimension Reduction and Visualization of Single-cell RNA-seq Data by Deep Variational Autoencoder. Genom. Proteom. Bioinform. 2018, 16, 320–331. [Google Scholar] [CrossRef]
- Liu, Q.; Xie, L. TranSynergy: Mechanism-driven interpretable deep neural network for the synergistic prediction and pathway deconvolution of drug combinations. PLOS Comput. Biol. 2021, 17, e1008653. [Google Scholar] [CrossRef]
- Xu, J.; Xu, J.; Meng, Y.; Lu, C.; Cai, L.; Zeng, X.; Nussinov, R.; Cheng, F. Graph embedding and Gaussian mixture variational autoencoder network for end-to-end analysis of single-cell RNA sequencing data. Cell Rep. Methods 2023, 3, 100382. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, L.; Zhong, F.; Wang, D.; Jiang, J.; Zhang, S.; Jiang, H.; Zheng, M.; Li, X. Graph neural network approaches for drug-target interactions. Curr. Opin. Struct. Biol. 2022, 73, 102327. [Google Scholar] [CrossRef]
- Wang, J.; Liu, X.; Shen, S.; Deng, L.; Liu, H. DeepDDS: Deep graph neural network with attention mechanism to predict synergistic drug combinations. Brief. Bioinform. 2022, 23, bbab390. [Google Scholar] [CrossRef]
- Zhou, J.; Cui, G.; Hu, S.; Zhang, Z.; Yang, C.; Liu, Z.; Wang, L.; Li, C.; Sun, M. Graph neural networks: A review of methods and applications. AI Open 2020, 1, 57–81. [Google Scholar] [CrossRef]
- Ding, Y.; Wang, F.; Lei, X.; Liao, B.; Wu, F.X. Deep belief network-Based Matrix Factorization Model for MicroRNA-Disease Associations Prediction. Evol. Bioinform. Online 2020, 16, 1176934320919707. [Google Scholar] [CrossRef]
- Chen, G.; Tsoi, A.; Xu, H.; Zheng, W.J. Predict effective drug combination by deep belief network and ontology fingerprints. J. Biomed. Inform. 2018, 85, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yan, C.C.; Zhang, X.; You, Z.-H. Long non-coding RNAs and complex diseases: From experimental results to computational models. Brief. Bioinform. 2017, 18, 558–576. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Shi, H.; Wang, Z.; Zhang, C.; Liu, L.; Wang, L.; He, W.; Hao, D.; Liu, S.; Zhou, M. Inferring novel lncRNA–disease associations based on a random walk model of a lncRNA functional similarity network. Mol. Biosyst. 2014, 10, 2074–2081. [Google Scholar] [CrossRef] [PubMed]
- Yin, N.; Ma, W.; Pei, J.; Ouyang, Q.; Tang, C.; Lai, L. Synergistic and Antagonistic Drug Combinations Depend on Network Topology. PLoS ONE 2014, 9, e93960. [Google Scholar] [CrossRef]
- Kaschek, D.; Sharanek, A.; Guillouzo, A.; Timmer, J.; Weaver, R.J. A dynamic mathematical model of bile acid clearance in HepaRG cells. Toxicol. Sci. 2018, 161, 48–57. [Google Scholar] [CrossRef]
- Cohen, A.A.; Geva-Zatorsky, N.; Eden, E.; Frenkel-Morgenstern, M.; Issaeva, I.; Sigal, A.; Milo, R.; Cohen-Saidon, C.; Liron, Y.; Kam, Z.; et al. Dynamic Proteomics of Individual Cancer Cells in Response to a Drug. Science 2008, 322, 1511–1516. [Google Scholar] [CrossRef]
- Geva-Zatorsky, N.; Dekel, E.; Cohen, A.A.; Danon, T.; Cohen, L.; Alon, U. Protein Dynamics in Drug Combinations: A Linear Superposition of Individual-Drug Responses. Cell 2010, 140, 643–651. [Google Scholar] [CrossRef]
- Wong, P.K.; Yu, F.; Shahangian, A.; Cheng, G.; Sun, R.; Ho, C.-M. Closed-loop control of cellular functions using combinatory drugs guided by a stochastic search algorithm. Proc. Natl. Acad. Sci. USA 2008, 105, 5105–5110. [Google Scholar] [CrossRef]
- Zinner, R.G.; Barrett, B.L.; Popova, E.; Damien, P.; Volgin, A.Y.; Gelovani, J.G.; Lotan, R.; Tran, H.T.; Pisano, C.; Mills, G.B.; et al. Algorithmic guided screening of drug combinations of arbitrary size for activity against cancer cells. Mol. Cancer Ther. 2009, 8, 521–532. [Google Scholar] [CrossRef]
- Feala, J.D.; Cortes, J.; Duxbury, P.M.; Piermarocchi, C.; McCulloch, A.D.; Paternostro, G. Systems approaches and algorithms for discovery of combinatorial therapies. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Julkunen, H.; Cichonska, A.; Gautam, P.; Szedmak, S.; Douat, J.; Pahikkala, T.; Aittokallio, T.; Rousu, J. Leveraging multi-way interactions for systematic prediction of pre-clinical drug combination effects. Nat. Commun. 2020, 11, 6136. [Google Scholar] [CrossRef] [PubMed]
- Madani, A.; Krause, B.; Greene, E.R.; Subramanian, S.; Mohr, B.P.; Holton, J.M.; Olmos, J.L., Jr.; Xiong, C.; Sun, Z.Z.; Socher, R.; et al. Large language models generate functional protein sequences across diverse families. Nat. Biotechnol. 2023, 41, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Juhi, A.; Pipil, N.; Santra, S.; Mondal, S.; Behera, J.K.; Mondal, H. The Capability of ChatGPT in Predicting and Explaining Common Drug-Drug Interactions. Cureus 2023, 15, e36272. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Data Type | Database | URL | Latest Update | Description |
|---|---|---|---|---|
| Synergistic Drug Combination | DrugComb [21] | https://drugcomb.fimm.fi/ (accessed on 13 July 2023) | 2021 | Synergistic Drug Combination mainly contains data on the response of cancer cell lines to or combinations of drugs. |
| DrugCombDB [22] | http://drugcombdb.denglab.org/ (accessed on 13 July 2023) | 2019 | ||
| NCI-ALMANAC [23] | https://dtp.cancer.gov/ncialmanac (accessed on 13 July 2023) | 2017 | ||
| SYNERGxDB [24] | https://www.synergxdb.ca/ (accessed on 13 July 2023) | 2019 | ||
| Bioactivity resources | ChEMBL [25] | https://www.ebi.ac.uk/chembl/ (accessed on 13 July 2023) | 2023 | Bioactivity resources mainly contain the biological activity data of small molecules, drug targets, enzymes and proteins. |
| DrugBank [26] | https://www.drugbank.com (accessed on 14 July 2023) | 2023 | ||
| PubChem [27] | https://pubchem.ncbi.nlm.nih.gov (accessed on 14 July 2023) | 2023 | ||
| Gene expression | GEO [28] | https://www.ncbi.nlm.nih.gov/geo/ (accessed on 14 July 2023) | 2013 | It mainly includes gene expression data with or without perturbation, gene methylation data and some interaction data. |
| CMap [29] | https://clue.io (accessed on 14 July 2023) | 2021 | ||
| LINCS | https://lincsproject.org/ (accessed on 14 July 2023) | 2022 | ||
| Toxicity effects resources | SIDER [30] | http://sideeffects.embl.de/ (accessed on 15 July 2023) | 2015 | Toxicity effects resources mainly contain information about drugs and targets, as well as information on side effects. |
| TOXRIC [31] | https://toxric.bioinforai.tech/ (accessed on 15 July 2023) | 2022 | ||
| Tox21BodyMap [32] | https://sandbox.ntp.niehs.nih.gov/bodymap/ (accessed on 15 July 2023) | 2020 | ||
| Pathways resources | Reactome [33] | https://reactome.org/ (accessed on 15 July 2023) | 2023 | Pathways resources mainly contain various biological pathways that facilitate drug combination prediction. |
| Pathbank [34] | https://pathbank.org/ (accessed on 15 July 2023) | 2020 | ||
| KEGG Pathways [35] | https://www.kegg.jp/kegg/pathway.html (accessed on 15 July 2023) | 2023 | ||
| Interactions resources | TTD [36] | https://db.idrblab.net/ttd/ (accessed on 16 July 2023) | 2023 | It mainly contains information about drug targets and targeted drugs that interact with them. |
| Bingding DB [37] | http://www.bindingdb.org (accessed on 16 July 2023) | 2023 | ||
| HPRD [38] | http://www.hprd.org/ (accessed on 16 July 2023) | 2008 | ||
| STRING [39] | https://string-db.org/ (accessed on 16 July 2023) | 2021 | ||
| STITCH [40] | http://stitch.embl.de/ (accessed on 16 July 2023) | 2016 |
| Methods | Algorithms | URL | Characteristics | Reference |
|---|---|---|---|---|
| Traditional machine learning | Support vector machine | — | Do well in identifying subtle patterns in complex data sets; poor interpretability; run slowly on large data sets. | [58] |
| Decision tree | https://github.com/Lianlian-Wu/ForSyn (accessed on 18 July 2023) | Display visually; easy to over fit; accuracy may decrease when processing data with complex relationships. | [59] | |
| Gradient boosting | — | Do well in handling nonlinear relationships and high dimensional data; easy to over fit; hyperparameters tuning is complex. | [60] | |
| Deep learning methods | Feedforward neural network | — | Do well in handling nonlinear relationships and high dimensional data; easy to over fit; poor interpretability; processing large data takes a long time | [61] |
| Autoencoder | https://github.com/qiaoliuhub/drug_combination (accessed on 19 July 2023) | Feature learning ability is strong; poor interpretability. | [62] | |
| Graph convolutional network | https://github.com/Sinwang404/DeepDDS/tree/master (accessed on 19 July 2023) | Being able to capture the relationship and topological information between the nodes in the graph, poor interpretability, and robustness is of concern. | [63] | |
| Deep belief network | Perform well in supervised study; easy to over fit; poor interpretability. | [64] | ||
| Mathematical methods | Network analysis | — | Be able to capture complex interactions; good interpretability. | [65] |
| Dynamic mathematical model | — | Be able to simulate drug reaction more accurately; poor interpretability. | [15] | |
| Search algorithms | Breadth first search algorithm | — | Be able to consider a large amount of potential drug-target interactions; robustness is of concern. | [66] |
| Systems biology methods | Signature-based model | https://tanlab.ucdenver.edu/kMap(accessed on 20 July 2023) | Understand drug action mechanisms and influencing factors more comprehensively; high requirements on data quality. | [67] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, Y.; Ren, H.; Lan, L.; Li, Y.; Huang, T. Review of Predicting Synergistic Drug Combinations. Life 2023, 13, 1878. https://doi.org/10.3390/life13091878
Pan Y, Ren H, Lan L, Li Y, Huang T. Review of Predicting Synergistic Drug Combinations. Life. 2023; 13(9):1878. https://doi.org/10.3390/life13091878
Chicago/Turabian StylePan, Yichen, Haotian Ren, Liang Lan, Yixue Li, and Tao Huang. 2023. "Review of Predicting Synergistic Drug Combinations" Life 13, no. 9: 1878. https://doi.org/10.3390/life13091878
APA StylePan, Y., Ren, H., Lan, L., Li, Y., & Huang, T. (2023). Review of Predicting Synergistic Drug Combinations. Life, 13(9), 1878. https://doi.org/10.3390/life13091878