
Neuron-Specific Enolase (NSE) as a Biomarker for Autistic Spectrum Disease (ASD)


Abstract
1. Introduction
2. Materials and Methods
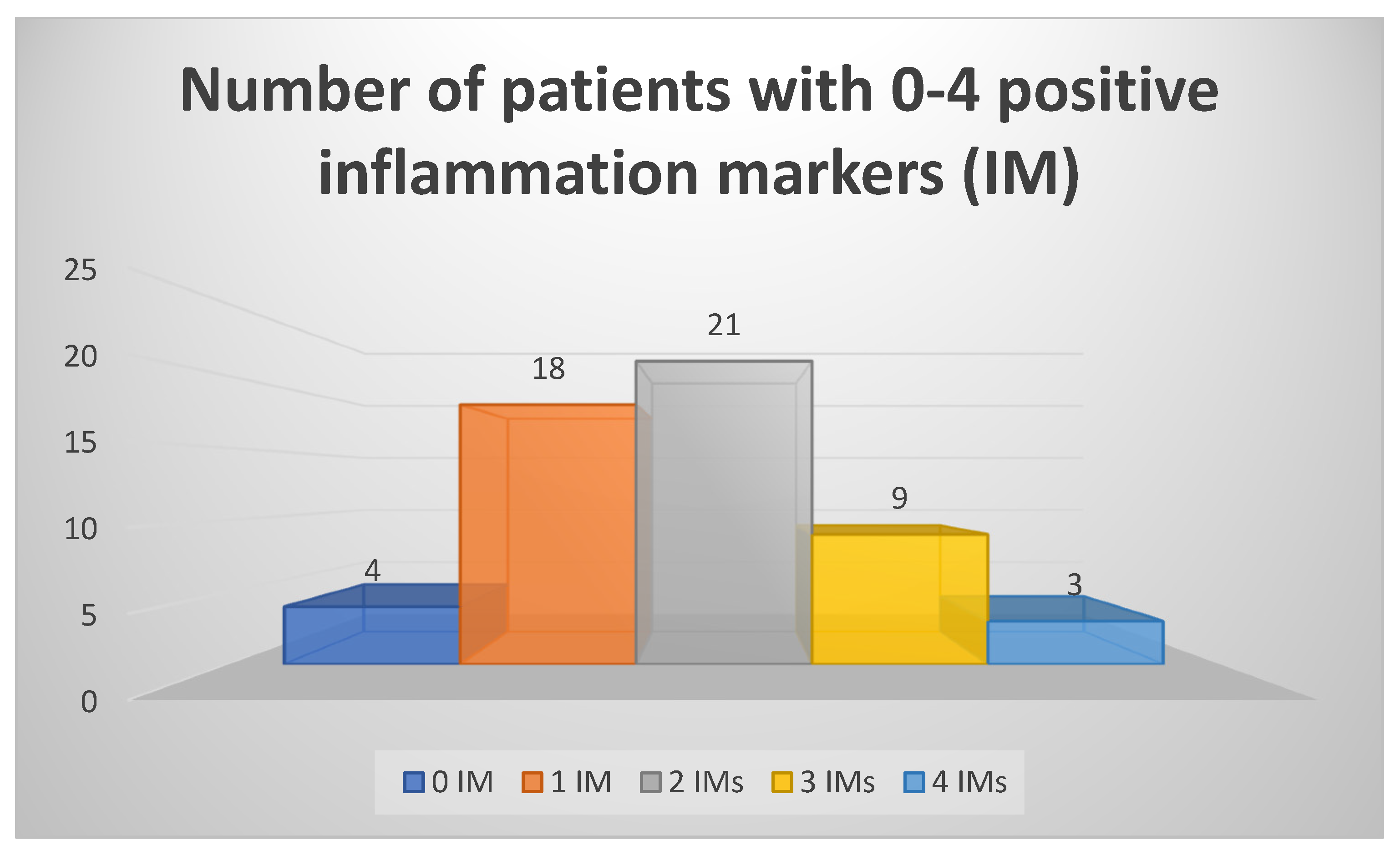
3. Results
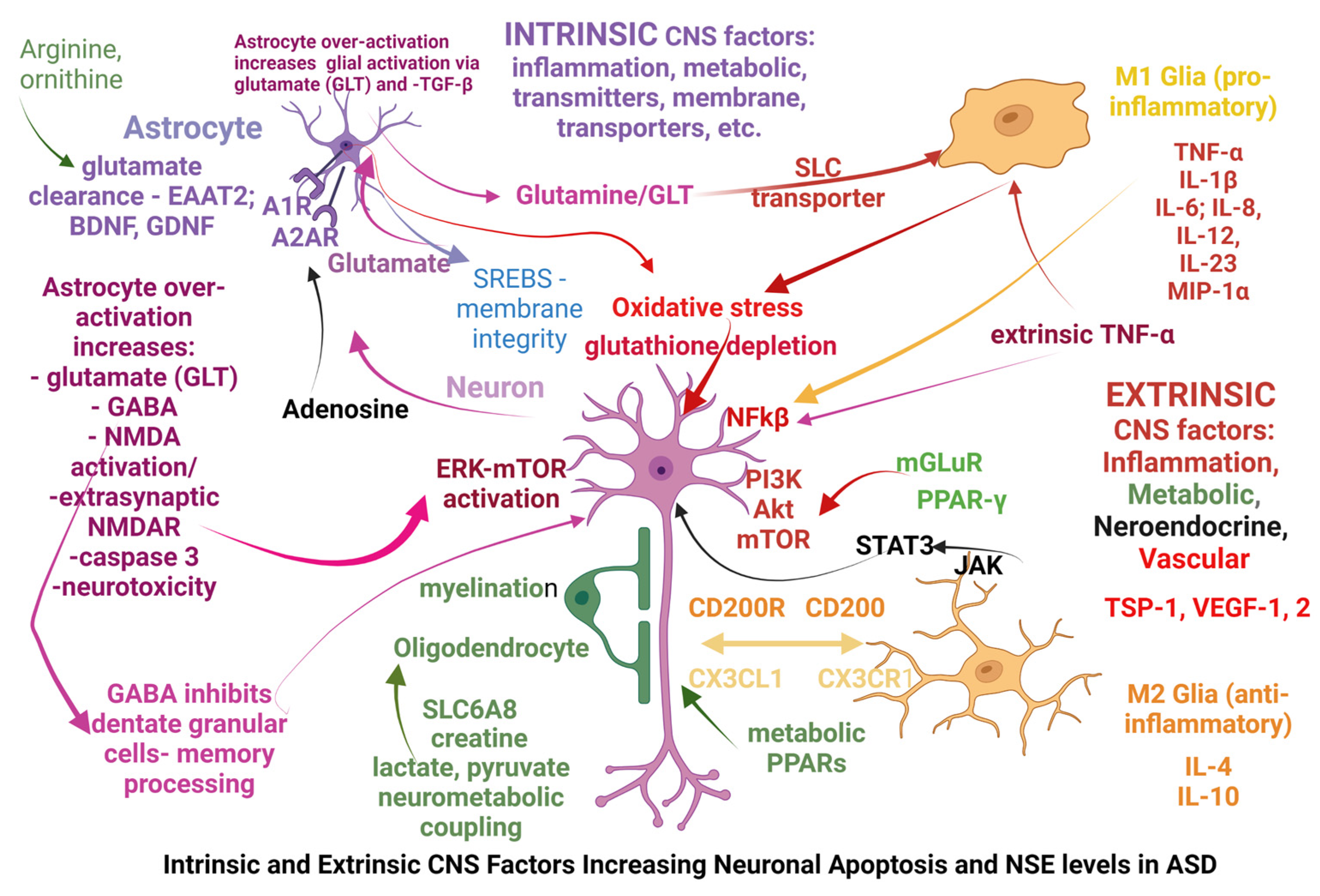
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kanner, L. Autistic disturbances of affective contact. Nerv. Child 1943, 2, 217–250. [Google Scholar]
- Wong, R.S.Y. Neuroinflammation in autism spectrum disorders: Potential target for mesenchymal stem cell-based therapy. Egypt. J. Neurol. Psychiatry Neurosurg. 2022, 58, 91. [Google Scholar] [CrossRef]
- Gesundheit, B.; Ashwood, P.; Keating, A.; Naor, D.; Melamed, M.; Rosenzweig, J.P. Therapeutic properties of mesenchymal stem cells for autism spectrum disorders. Med. Hypotheses 2015, 84, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Dawson, G.; Sun, J.M.; Davlantis, K.S.; Murias, M.; Franz, L.; Troy, J.; Simmons, R.; Sabatos-DeVito, M.; Durham, R.; Kurtzberg, J. Autologous Cord Blood Infusions Are Safe and Feasible in Young Children with Autism Spectrum Disorder: Results of a Single-Center Phase I Open-Label Trial. Stem Cells Transl. Med. 2017, 6, 1332–1339. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.M.; Dawson, G.; Franz, L.; Howard, J.; McLaughlin, C.; Kistler, B.; Waters-Pick, B.; Meadows, N.; Troy, J.; Kurtzberg, J. Infusion of human umbilical cord tissue mesenchymal stromal cells in children with autism spectrum disorder. Stem Cells Transl. Med. 2020, 9, 1137–1146. [Google Scholar] [CrossRef]
- Murias, M.; Major, S.; Compton, S.; Buttinger, J.; Sun, J.M.; Kurtzberg, J.; Dawson, G. Electrophysiological Biomarkers Predict Clinical Improvement in an Open-Label Trial Assessing Efficacy of Autologous Umbilical Cord Blood for Treatment of Autism. Stem Cells Transl. Med. 2018, 7, 783–791. [Google Scholar] [CrossRef]
- Gopalarethinam, J.; Nair, A.P.; Iyer, M.; Vellingiri, B.; Subramaniam, M.D. Advantages of mesenchymal stem cell over the other stem cells. Acta Histochem. 2023, 125, 152041. [Google Scholar] [CrossRef]
- Croen, L.A.; Braunschweig, D.; Haapanen, L.; Yoshida, C.K.; Fireman, B.; Grether, J.K.; Kharrazi, M.; Hansen, R.L.; Ashwood, P.; Van de Water, J. Maternal Mid-Pregnancy Autoantibodies to Fetal Brain Protein: The Early Markers for Autism Study. Biol. Psychiatry 2008, 64, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Braunschweig, D.; Duncanson, P.; Boyce, R.; Hansen, R.; Ashwood, P.; Pessah, I.N.; Hertz-Picciotto, I.; Van de Water, J. Behavioral Correlates of Maternal Antibody Status Among Children with Autism. J. Autism Dev. Disord. 2011, 42, 1435–1445. [Google Scholar] [CrossRef]
- Li, X.; Chauhan, A.; Sheikh, A.M.; Patil, S.; Chauhan, V.; Li, X.-M.; Ji, L.; Brown, T.; Malik, M. Elevated immune response in the brain of autistic patients. J. Neuroimmunol. 2009, 207, 111–116. [Google Scholar] [CrossRef]
- Morgan, J.T.; Chana, G.; Pardo, C.A.; Achim, C.; Semendeferi, K.; Buckwalter, J.; Courchesne, E.; Everall, I.P. Microglial Activation and Increased Microglial Density Observed in the Dorsolateral Prefrontal Cortex in Autism. Biol. Psychiatry 2010, 68, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Sugihara, G.; Ouchi, Y.; Nakamura, K.; Futatsubashi, M.; Takebayashi, K.; Yoshihara, Y.; Omata, K.; Matsumoto, K.; Tsuchiya, K.J.; et al. Microglial Activation in Young Adults with Autism Spectrum Disorder. JAMA Psychiatry 2013, 70, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Usui, N.; Kobayashi, H.; Shimada, S. Neuroinflammation and Oxidative Stress in the Pathogenesis of Autism Spectrum Disorder. Int. J. Mol. Sci. 2023, 24, 5487. [Google Scholar] [CrossRef]
- Vargas, D.L.; Nascimbene, C.; Krishnan, C.; Zimmerman, A.W.; Pardo, C.A. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 2005, 57, 67–81. [Google Scholar] [CrossRef]
- Molloy, C.A.; Morrow, A.L.; Meinzen-Derr, J.; Schleifer, K.; Dienger, K.; Manning-Courtney, P.; Altaye, M.; Wills-Karp, M. Elevated cytokine levels in children with autism spectrum disorder. J. Neuroimmunol. 2006, 172, 198–205. [Google Scholar] [CrossRef]
- Gesundheit, B.; Rosenzweig, J.P.; Naor, D.; Lerer, B.; Zachor, D.A.; Procházka, V.; Melamed, M.; Kristt, D.A.; Steinberg, A.; Shulman, C.; et al. Immunological and autoimmune considerations of Autism Spectrum Disorders. J. Autoimmun. 2013, 44, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hughes, H.; Moreno, R.J.; Ashwood, P. Innate immune dysfunction and neuroinflammation in autism spectrum disorder (ASD). Brain Behav. Immun. 2023, 108, 245–254. [Google Scholar] [CrossRef]
- Laurence, J.A.; Fatemi, S.H. Glial fibrillary acidic protein is elevated in superior frontal, parietal and cerebellar cortices of autistic subjects. Cerebellum 2005, 4, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Malik, M.; Tauqeer, Z.; Sheikh, A.M.; Wen, G.; Nagori, A.; Yang, K.; Brown, W.T.; Li, X. NF-κB Signaling in the Brain of Autistic Subjects. Mediat. Inflamm. 2011, 2011, 785265, Erratum in Mediat. Inflamm. 2013, 2013, 691975. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, K.K. Glial fibrillary acidic protein: From intermediate filament assembly and gliosis to neurobiomarker. Trends Neurosci. 2015, 38, 364–374. [Google Scholar] [CrossRef]
- Vakilzadeh, G.; Martinez-Cerdeño, V. Pathology and Astrocytes in Autism. Neuropsychiatr. Dis. Treat. 2023, 19, 841–850. [Google Scholar] [CrossRef]
- Gesundheit, B.; Zisman, P.D.; Hochbaum, L.; Posen, Y.; Steinberg, A.; Friedman, G.; Ravkin, H.D.; Rubin, E.; Faktor, O.; Ellis, R. Autism spectrum disorder diagnosis using a new panel of immune- and inflammatory-related serum biomarkers: A case-control multicenter study. Front. Pediatr. 2023, 11, 967954. [Google Scholar] [CrossRef] [PubMed]
- Levin, A.R.; Naples, A.J.; Scheffler, A.W.; Webb, S.J.; Shic, F.; Sugar, C.A.; Murias, M.; Bernier, R.A.; Chawarska, K.; Dawson, G.; et al. Day-to-Day Test-Retest Reliability of EEG Profiles in Children with Autism Spectrum Disorder and Typical Development. Front. Integr. Neurosci. 2020, 14, 21. [Google Scholar] [CrossRef] [PubMed]
- Webb, S.J.; Naples, A.J.; Levin, A.R.; Hellemann, G.; Borland, H.; Benton, J.; Carlos, C.; McAllister, T.; Santhosh, M.; Seow, H.; et al. The Autism Biomarkers Consortium for Clinical Trials: Initial Evaluation of a Battery of Candidate EEG Biomarkers. Am. J. Psychiatry 2023, 180, 41–49, Erratum in Am. J. Psychiatry 2023, 180, 145. [Google Scholar] [CrossRef]
- Uddin, L.Q.; Dajani, D.R.; Voorhies, W.; Bednarz, H.; Kana, R.K. Progress and roadblocks in the search for brain-based biomarkers of autism and attention-deficit/hyperactivity disorder. Transl. Psychiatry 2017, 7, e1218. [Google Scholar] [CrossRef]
- Sussman, D.; Leung, R.C.; Vogan, V.M.; Lee, W.; Trelle, S.; Lin, S.; Cassel, D.B.; Chakravarty, M.M.; Lerch, J.P.; Anagnostou, E.; et al. The autism puzzle: Diffuse but not pervasive neuroanatomical abnormalities in children with ASD. Neuroimage Clin. 2015, 8, 170–179. [Google Scholar] [CrossRef]
- Khundrakpam, B.S.; Lewis, J.D.; Kostopoulos, P.; Carbonell, F.; Evans, A.C. Cortical thickness abnormalities in autism spectrum disorders through late childhood, adolescence, and adulthood: A large-scale MRI study. Cereb. Cortex 2017, 27, 1721–1731. [Google Scholar] [CrossRef]
- Zielinski, B.A.; Prigge, M.B.D.; Nielsen, J.A.; Froehlich, A.L.; Abildskov, T.J.; Anderson, J.S.; Fletcher, P.T.; Zygmunt, K.M.; Travers, B.G.; Lange, N.; et al. Longitudinal changes in cortical thickness in autism and typical development. Brain 2014, 137, 1799–1812. [Google Scholar] [CrossRef]
- Schumann, C.M.; Hamstra, J.; Goodlin-Jones, B.L.; Lotspeich, L.J.; Kwon, H.; Buonocore, M.H.; Lammers, C.R.; Reiss, A.L.; Amaral, D.G. The amygdala is enlarged in children but not adolescents with autism, the hippocampus is enlarged at all ages. J. Neurosci. 2004, 24, 6392–6401. [Google Scholar] [CrossRef] [PubMed]
- Nordahl, C.W.; Scholz, R.; Yang, X.; Buonocore, M.H.; Simon, T.; Rogers, S.; Amaral, D.G. Increased rate of amygdala growth in children aged 2 to 4 years with autism spectrum disorders. Arch. Gen. Psychiatry 2015, 69, 53–61. [Google Scholar] [CrossRef]
- Groen, W.; Teluij, M.; Buitelaar, J.; Tendolkar, I. Amygdala and hippocampus enlargement during adolescence in autism. J. Am. Acad. Child Adolesc. Psychiatry 2010, 49, 552–560. [Google Scholar] [PubMed]
- Van Rooij, D.; Anagnostou, E.; Arango, C.; Auzias, G.; Behrmann, M.; Busatto, G.F.; Calderoni, S.; Daly, E.; Deruelle, C.; Di Martino, A.; et al. Cortical and subcortical brain morphometry differences between patients with autism spectrum disorder and healthy individuals across the lifespan: Tesults from the ENIGMA ASD working group. Am. J. Psychiatry 2018, 175, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Rylaarsdam, L.E.; Guemez-Gamboa, A. Genetic Causes and Modifiers of Autism Spectrum Disorder. Front. Cell. Neurosci. 2019, 13, 385. [Google Scholar] [CrossRef] [PubMed]
- Wiśniowiecka-Kowalnik, B.; Nowakowska, B.A. Genetics and epigenetics of autism spectrum disorder—Current evidence in the field. J. Appl. Genet. 2019, 60, 37–47. [Google Scholar] [CrossRef]
- Genovese, A.; Butler, M.G. The Autism Spectrum: Behavioral, Psychiatric and Genetic Associations. Genes 2023, 14, 677. [Google Scholar] [CrossRef]
- Havdahl, A.; Niarchou, M.; Starnawska, A.; Uddin, M.; van der Merwe, C.; Warrier, V. Genetic contributions to autism spectrum disorder. Psychol. Med. 2021, 51, 2260–2273. [Google Scholar] [CrossRef]
- Koesterich, J.; An, J.-Y.; Inoue, F.; Sohota, A.; Ahituv, N.; Sanders, S.J.; Kreimer, A. Characterization of De Novo Promoter Variants in Autism Spectrum Disorder with Massively Parallel Reporter Assays. Int. J. Mol. Sci. 2023, 24, 3509. [Google Scholar] [CrossRef]
- Anney, R.; Klei, L.; Pinto, D.; Regan, R.; Conroy, J.; Magalhaes, T.R.; Correia, C.; Abrahams, B.S.; Sykes, N.; Pagnamenta, A.T.; et al. A genome-wide scan for common alleles affecting risk for autism. Hum. Mol. Genet. 2010, 19, 4072–4082. [Google Scholar] [CrossRef]
- Pugsley, K.; Scherer, S.W.; Bellgrove, M.A.; Hawi, Z. Environmental exposures associated with elevated risk for autism spectrum disorder may augment the burden of deleterious de novo mutations among probands. Mol. Psychiatry 2021, 27, 710–730. [Google Scholar] [CrossRef]
- Apte, M.; Kumar, A. Correlation of mutated gene and signalling pathways in ASD. IBRO Neurosci. Rep. 2023, 14, 384–392. [Google Scholar] [CrossRef]
- Chow, M.L.; Pramparo, T.; Winn, M.E.; Barnes, C.C.; Li, H.-R.; Weiss, L.; Fan, J.-B.; Murray, S.; April, C.; Belinson, H.; et al. Age-Dependent Brain Gene Expression and Copy Number Anomalies in Autism Suggest Distinct Pathological Processes at Young Versus Mature Ages. PLoS Genet. 2012, 8, e1002592. [Google Scholar] [CrossRef]
- Huang, Z.-X.; Chen, Y.; Guo, H.-R.; Chen, G.-F. Systematic Review and Bioinformatic Analysis of microRNA Expression in Autism Spectrum Disorder Identifies Pathways Associated with Cancer, Metabolism, Cell Signaling, and Cell Adhesion. Front. Psychiatry 2021, 12, 630876. [Google Scholar] [CrossRef]
- Vasu, M.M.; Anitha, A.; Thanseem, I.; Suzuki, K.; Yamada, K.; Takahashi, T.; Wakuda, T.; Iwata, K.; Tsujii, M.; Sugiyama, T.; et al. Serum microRNA profiles in children with autism. Mol. Autism 2014, 5, 40. [Google Scholar] [CrossRef]
- Abu-Elneel, K.; Liu, T.; Gazzaniga, F.S.; Nishimura, Y.; Wall, D.P.; Geschwind, D.H.; Lao, K.; Kosik, K.S. Heterogeneous dysregulation of microRNAs across the autism spectrum. Neurogenetics 2008, 9, 153–161. [Google Scholar] [CrossRef]
- Kichukova, T.M.; Popov, N.T.; Ivanov, I.S.; Vachev, T.I. Profiling of Circulating Serum MicroRNAs in Children with Autism Spectrum Disorder using Stem-loop qRT-PCR Assay. Folia Medica 2017, 59, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Cortese, S.; Solmi, M.; Michelini, G.; Bellato, A.; Blanner, C.; Canozzi, A.; Eudave, L.; Farhat, L.C.; Højlund, M.; Köhler-Forsberg, O.; et al. Candidate diagnostic biomarkers for neurodevelopmental disorders in children and adolescents: A systematic review. World Psychiatry 2023, 22, 129–149. [Google Scholar] [CrossRef]
- Parellada, M.; Andreu-Bernabeu, Á.; Burdeus, M.; Cáceres, A.S.J.; Urbiola, E.; Carpenter, L.L.; Kraguljac, N.V.; McDonald, W.M.; Nemeroff, C.B.; Rodriguez, C.I.; et al. In Search of Biomarkers to Guide Interventions in Autism Spectrum Disorder: A Systematic Review. Am. J. Psychiatry 2023, 180, 23–40. [Google Scholar] [CrossRef] [PubMed]
- Ansel, A.; Posen, Y.; Ellis, R.; Deutsch, L.; Zisman, P.D.; Gesundheit, B. Biomarkers for Autism Spectrum Disorders (ASD): A Meta-analysis. Rambam Maimonides Med. J. 2019, 10, e0021. [Google Scholar] [CrossRef]
- Hollestein, V.; Poelmans, G.; Forde, N.J.; Beckmann, C.F.; Ecker, C.; Mann, C.; Schäfer, T.; Moessnang, C.; Baumeister, S.; Banaschewski, T.; et al. Excitatory/inhibitory imbalance in autism: The role of glutamate and GABA gene-sets in symptoms and cortical brain structure. Transl. Psychiatry 2023, 13, 18. [Google Scholar] [CrossRef] [PubMed]
- French, L.; Paus, T. A FreeSurfer view of the cortical transcriptome generated from the Allen Human Brain Atlas. Front. Neurosci. 2015, 9, 323. [Google Scholar] [CrossRef] [PubMed]
- Hawrylycz, M.J.; Lein, E.S.; Guillozet-Bongaarts, A.L.; Shen, E.H.; Ng, L.; Miller, J.A.; van de Lagemaat, L.N.; Smith, K.A.; Ebbert, A.; Riley, Z.L.; et al. An anatomically comprehen-sive atlas of the adult human brain transcriptome. Nature 2012, 489, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Buch, A.M.; Vértes, P.E.; Seidlitz, J.; Kim, S.H.; Grosenick, L.; Liston, C. Molecular and network-level mechanisms explaining individual differences in autism spectrum disorder. Nat. Neurosci. 2023, 26, 650–663. [Google Scholar] [CrossRef] [PubMed]
- Hewitson, L.; Mathews, J.A.; Devlin, M.; Schutte, C.; Lee, J.; German, D.C. Blood biomarker discovery for autism spectrum disorder: A proteomic analysis. PLoS ONE 2021, 16, e0246581. [Google Scholar] [CrossRef] [PubMed]
- Araghi-Niknam, M.; Fatemi, S.H. Levels of Bcl-2 and P53 are altered in superior frontal and cerebellar cortices of autistic subjects. Cell. Mol. Neurobiol. 2003, 23, 945–952. [Google Scholar] [CrossRef]
- Ramirez-Celis, A.; Edmiston, E.; Schauer, J.; Vu, T.; Van de Water, J. Peptides of neuron specific enolase as potential ASD biomarkers: From discovery to epitope mapping. Brain Behav. Immun. 2019, 84, 200–208. [Google Scholar] [CrossRef]
- Delpire, E.; Ben-Ari, Y. A Wholistic View of How Bumetanide Attenuates Autism Spectrum Disorders. Cells 2022, 11, 2419. [Google Scholar] [CrossRef]
- Dogan, M.; Company, I.H.M.; Albayrak, Y.; Erbas, O. Torasemide Improves the Propionic Acid-Induced Autism in Rats: A Histopathological and Imaging Study. Alpha Psychiatry 2023, 24, 22–31. [Google Scholar] [CrossRef]
- Singh, R.; Kisku, A.; Kungumaraj, H.; Nagaraj, V.; Pal, A.; Kumar, S.; Sulakhiya, K. Autism Spectrum Disorders: A Recent Update on Targeting Inflammatory Pathways with Natural Anti-Inflammatory Agents. Biomedicines 2023, 11, 115. [Google Scholar] [CrossRef]
- Courchesne, E.; Campbell, K.; Solso, S. Brain growth across the life span in autism: Age-specific changes in anatomical pathology. Brain Res. 2011, 1380, 138–145. [Google Scholar] [CrossRef]
- Rodin, R.E.; Dou, Y.; Kwon, M.; Sherman, M.A.; D’gama, A.M.; Doan, R.N.; Rento, L.M.; Girskis, K.M.; Bohrson, C.L.; Kim, S.N.; et al. The landscape of somatic mutation in cerebral cortex of autistic and neurotypical individuals revealed by ultra-deep whole-genome sequencing. Nat. Neurosci. 2021, 24, 176–185. [Google Scholar] [CrossRef]
- Price, M.Z.; Price, R.L. Extended-Release Viloxazine Compared with Atomoxetine for Attention Deficit Hyperactivity Disorder. CNS Drugs 2023, 37, 655–660. [Google Scholar] [CrossRef]
- Yu, C.; Garcia-Olivares, J.; Candler, S.; Schwabe, S.; Maletic, V. New Insights into the Mechanism of Action of Viloxazine: Serotonin and Norepinephrine Modulating Properties. J. Exp. Pharmacol. 2020, 12, 285–300. [Google Scholar] [CrossRef]
- Hazlett, H.C.; Poe, M.D.; Gerig, G.; Styner, M.; Chappell, C.; Smith, R.G.; Vachet, C.; Piven, J. Early Brain Overgrowth in Autism Associated with an Increase in Cortical Surface Area Before Age 2 Years. Arch. Gen. Psychiatry 2011, 68, 467–476. [Google Scholar] [CrossRef]
- Tang, G.; Gudsnuk, K.; Kuo, S.-H.; Cotrina, M.L.; Rosoklija, G.; Sosunov, A.; Sonders, M.S.; Kanter, E.; Castagna, C.; Yamamoto, A.; et al. Loss of mTOR-Dependent Macroautophagy Causes Autistic-like Synaptic Pruning Deficits. Neuron 2014, 83, 1131–1143, Erratum in Neuron 2014, 83, 1482. [Google Scholar] [CrossRef]
- Kim, H.-J.; Cho, M.-H.; Shim, W.H.; Kim, J.K.; Jeon, E.-Y.; Kim, D.-H.; Yoon, S.-Y. Deficient autophagy in microglia impairs synaptic pruning and causes social behavioral defects. Mol. Psychiatry 2016, 22, 1576–1584. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, H.; Hale, V.A.; Dolcet, X.; Davies, A. NF-κB signalling regulates the growth of neural processes in the developing PNS and CNS. Development 2005, 132, 1713–1726. [Google Scholar] [CrossRef]
- Liao, X.; Li, Y. Nuclear Factor Kappa B in Autism Spectrum Disorder: A Systematic Review. Pharmacol. Res. 2020, 159, 104918. [Google Scholar] [CrossRef]
- Afridi, R.; Kim, J.-H.; Rahman, H.; Suk, K. Metabolic Regulation of Glial Phenotypes: Implications in Neuron–Glia Interactions and Neurological Disorders. Front. Cell. Neurosci. 2020, 14, 20. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, S.; Jungalwala, F.B. Receptor for advanced glycation end products (RAGE) mediates neuronal differentiation and neurite outgrowth. J. Neurosci. Res. 2007, 86, 1254–1266. [Google Scholar] [CrossRef]
- Juranek, J.; Mukherjee, K.; Kordas, B.; Załęcki, M.; Korytko, A.; Zglejc-Waszak, K.; Szuszkiewicz, J.; Banach, M. Role of RAGE in the Pathogenesis of Neurological Disorders. Neurosci. Bull. 2022, 38, 1248–1262. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Mehan, S.; Narula, A.S. Therapeutic modulation of JAK-STAT, mTOR, and PPAR-γ signaling in neurological dysfunctions. J. Mol. Med. 2022, 101, 9–49. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.R.; Batra, G.; Saini, L.; Sharma, S.; Mishra, A.; Singla, R.; Singh, A.; Singh, R.S.; Jain, A.; Bansal, S.; et al. Valproic acid and Propionic acid modulated mechanical pathways associated with Autism Spectrum Disorder at prenatal and neonatal exposure. CNS Neurol. Disord.-Drug Targets 2022, 21, 399–408. [Google Scholar] [CrossRef]
- Foley, K.A.; MacFabe, D.F.; Vaz, A.; Ossenkopp, K.; Kavaliers, M. Sexually dimorphic effects of prenatal exposure to propionic acid and lipopolysaccharide on social behavior in neonatal, adolescent, and adult rats: Implications for autism spectrum disorders. Int. J. Dev. Neurosci. 2014, 39, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, N.; Yang, J.-J.; Zhao, D.-M.; Chen, B.; Zhang, G.-Q.; Chen, S.; Cao, R.-F.; Yu, H.; Zhao, C.-Y.; et al. Probiotics and fructo-oligosaccharide intervention modulate the microbiota-gut brain axis to improve autism spectrum reducing also the hyper-serotonergic state and the dopamine metabolism disorder. Pharmacol. Res. 2020, 157, 104784. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Han, S.; Yu, L.; Du, L.; You, Y.; Chen, J.; Wang, M.; Wu, S.; Li, S.; Sun, X.; et al. Jia Wei Xiao Yao San ameliorates chronic stress-induced depression-like behaviors in mice by regulating the gut microbiome and brain metabolome in relation to purine metabolism. Phytomedicine 2022, 98, 153940. [Google Scholar] [CrossRef]
- Liang, Y.; Xiao, Z.; Ke, X.; Yao, P.; Chen, Y.; Lin, L.; Lu, J. Urinary Metabonomic Profiling Discriminates Between Children with Autism and Their Healthy Siblings. Experiment 2020, 26, e926634-1–e926634-8. [Google Scholar] [CrossRef]
- Amin, N.; Liu, J.; Bonnechere, B.; MahmoudianDehkordi, S.; Arnold, M.; Batra, R.; Chiou, Y.-J.; Fernandes, M.; Ikram, M.A.; Kraaij, R.; et al. Interplay of Metabolome and Gut Microbiome in Individuals with Major Depressive Disorder vs Control Individuals. JAMA Psychiatry 2023, 80, 597. [Google Scholar] [CrossRef]
- Aatsinki, A.-K.; Lahti, L.; Uusitupa, H.-M.; Munukka, E.; Keskitalo, A.; Nolvi, S.; O’Mahony, S.; Pietilä, S.; Elo, L.L.; Eerola, E.; et al. Gut microbiota composition is associated with temperament traits in infants. Brain Behav. Immun. 2019, 80, 849–858. [Google Scholar] [CrossRef]
- Thomas, S.D.; Jha, N.K.; Ojha, S.; Sadek, B. mTOR Signaling Disruption and Its Association with the Development of Autism Spectrum Disorder. Molecules 2023, 28, 1889. [Google Scholar] [CrossRef]
- Stancioiu, F. Effect of Fatty Acids and Antioxidants on Glucose Tolerance. Acta Endocrinol. (Bucharest) 2007, 3, 391–404. [Google Scholar] [CrossRef]
- Agostinho, P.; Madeira, D.; Dias, L.; Simões, A.P.; Cunha, R.A.; Canas, P.M. Purinergic signaling orchestrating neuron-glia communication. Pharmacol. Res. 2020, 162, 105253. [Google Scholar] [CrossRef]
- Van Deijk, A.-L.F.; Camargo, N.; Timmerman, J.; Heistek, T.; Brouwers, J.F.; Mogavero, F.; Mansvelder, H.D.; Smit, A.B.; Verheijen, M.H. Astrocyte lipid metabolism is critical for synapse development and function in vivo. Glia 2017, 65, 670–682. [Google Scholar] [CrossRef] [PubMed]
- Sato, A. mTOR, a Potential Target to Treat Autism Spectrum Disorder. CNS Neurol. Disord.-Drug Targets 2016, 15, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Durankuş, F.; Budak, K.; Albayrak, Y.; Sever, I.H.; Özkul, B.; Uyanıkgil, Y.; Albayrak, N.; Erbas, O. Atorvastatin Improves the Propionic Acid-Induced Autism in Rats: The Roles of Sphingosine-1-Phosphate and Anti-inflammatory Action. Cureus 2023, 15, e36870. [Google Scholar] [CrossRef]
- Śmiałek, D.; Jóźwiak, S.; Kotulska, K. Safety of Sirolimus in Patients with Tuberous Sclerosis Complex under Two Years of Age—A Bicenter Retrospective Study. J. Clin. Med. 2023, 12, 365. [Google Scholar] [CrossRef] [PubMed]
- Khera, R.; Mehan, S.; Bhalla, S.; Kumar, S.; Alshammari, A.; Alharbi, M.; Sadhu, S.S. Guggulsterone Mediated JAK/STAT and PPAR-Gamma Modulation Prevents Neurobehavioral and Neurochemical Abnormalities in Propionic Acid-Induced Experimental Model of Autism. Molecules 2022, 27, 889. [Google Scholar] [CrossRef]
- Sharma, A.; Bhalla, S.; Mehan, S. PI3K/AKT/mTOR signalling inhibitor chrysophanol ameliorates neurobehavioural and neurochemical defects in propionic acid-induced experimental model of autism in adult rats. Metab. Brain Dis. 2022, 37, 1909–1929. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.U.H.; Allende-Vega, N.; Gitenay, D.; Gerbal-Chaloin, S.; Gondeau, C.; Vo, D.-N.; Belkahla, S.; Orecchioni, S.; Talarico, G.; Bertolini, F.; et al. The PDK1 Inhibitor Dichloroacetate Controls Cholesterol Homeostasis Through the ERK5/MEF2 Pathway. Sci. Rep. 2017, 7, 10654. [Google Scholar] [CrossRef]
- Khalaf, K.; Tornese, P.; Cocco, A.; Albanese, A. Tauroursodeoxycholic acid: A potential therapeutic tool in neurodegenerative diseases. Transl. Neurodegener. 2022, 11, 33. [Google Scholar] [CrossRef]
- Nabetani, M.; Mukai, T.; Taguchi, A. Cell Therapies for Autism Spectrum Disorder Based on New Pathophysiology: A Review. Cell Transplant. 2023, 32, 9636897231163217. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| ESR | γ-Globulins | Ferritin | CRP | Eosinophils | TNF-α | α-2 Globulins | NSE | |
|---|---|---|---|---|---|---|---|---|
| Normal laboratory range | <16 mm/h | 11.1–18.8% | 5.3–99.9 ng/mL | <0.1 mg/dL | 0–4.5% | <8.1 pg/mL | <11.8% | <16.3 ng/mL |
| Patients tested (n=) | 55 | 55 | 51 | 51 | 55 | 52 | 55 | 41 |
| Number (n=); percentage (%) of abnormal values | 11; 20% | 15; 27.3% | 14; 27.4% | 15; 29.4% | 18; 32.7% | 36; 69.2% | 39; 70.9% | 40; 97.5% |
| Mean ± SD | 11 ± 9.9 | N/A | N/A | N/A | N/A | 9.22 ± 3.98 | 13.1 ± 4.39 | 24.0 ± 10.4 |
| Number (n=) of Lower/Higher values | 11 High | 15 Low | 10 Low/4 High | 15 High | 18 High | 36 High | 39 High | 40 High |
| Low MCV, Low MHC | High RBC, Low MCV (Thalassemia) | Low Na (<138 mmol/L) | High MCV | Urea | Liver Function Tests | Homocysteine | IGF-1; hGH | |
|---|---|---|---|---|---|---|---|---|
| Number, percentage | 1/55; (1.81%) | 2/55; (3.63%) | 4/55; (7.27%) | 5/55; (9.09%) | 5/55; (9.09%) | 6/55; (10.9%) | 5/30; (16.6%) | 2/10; (20%) |
| Mean ± SD | Low/Low | High/Low | Low | High | High | High | 4 Low, 1 High | Low |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stancioiu, F.; Bogdan, R.; Dumitrescu, R. Neuron-Specific Enolase (NSE) as a Biomarker for Autistic Spectrum Disease (ASD). Life 2023, 13, 1736. https://doi.org/10.3390/life13081736
Stancioiu F, Bogdan R, Dumitrescu R. Neuron-Specific Enolase (NSE) as a Biomarker for Autistic Spectrum Disease (ASD). Life. 2023; 13(8):1736. https://doi.org/10.3390/life13081736
Chicago/Turabian StyleStancioiu, Felician, Raluca Bogdan, and Radu Dumitrescu. 2023. "Neuron-Specific Enolase (NSE) as a Biomarker for Autistic Spectrum Disease (ASD)" Life 13, no. 8: 1736. https://doi.org/10.3390/life13081736
APA StyleStancioiu, F., Bogdan, R., & Dumitrescu, R. (2023). Neuron-Specific Enolase (NSE) as a Biomarker for Autistic Spectrum Disease (ASD). Life, 13(8), 1736. https://doi.org/10.3390/life13081736