
Phosphorylation Mimetic of Myosin Regulatory Light Chain Mitigates Cardiomyopathy-Induced Myofilament Impairment in Mouse Models of RCM and DCM

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Transgenic Mice
2.2. Recombinant RLC Proteins—Cloning, Expression, and Purification
2.3. Mechanical and ATP Turnover Measurements in Skinned Left Ventricular Papillary Muscle (LVPM) Fibers from Transgenic Tg-RLC and Tg-ELC Mice
2.3.1. Preparation of Skinned LVPM Fibers
2.3.2. Depletion of Endogenous RLC from LVPM and Reconstitution with Recombinant RLC Proteins
2.3.3. Steady-State Force Development and Ca2+-Sensitivity of Force in Reconstituted Muscle Strips
2.3.4. Mant-ATP Chase Experiments
2.4. Treatment of Skinned Muscle Strips with Omecamtiv Mecarbil (OM)
2.5. Statistical Analysis
3. Results
3.1. Effect of Phosphomimetic S15D-RLC on Force Measurements in LVPM Fibers from Tg-ELC Mouse Model of RCM
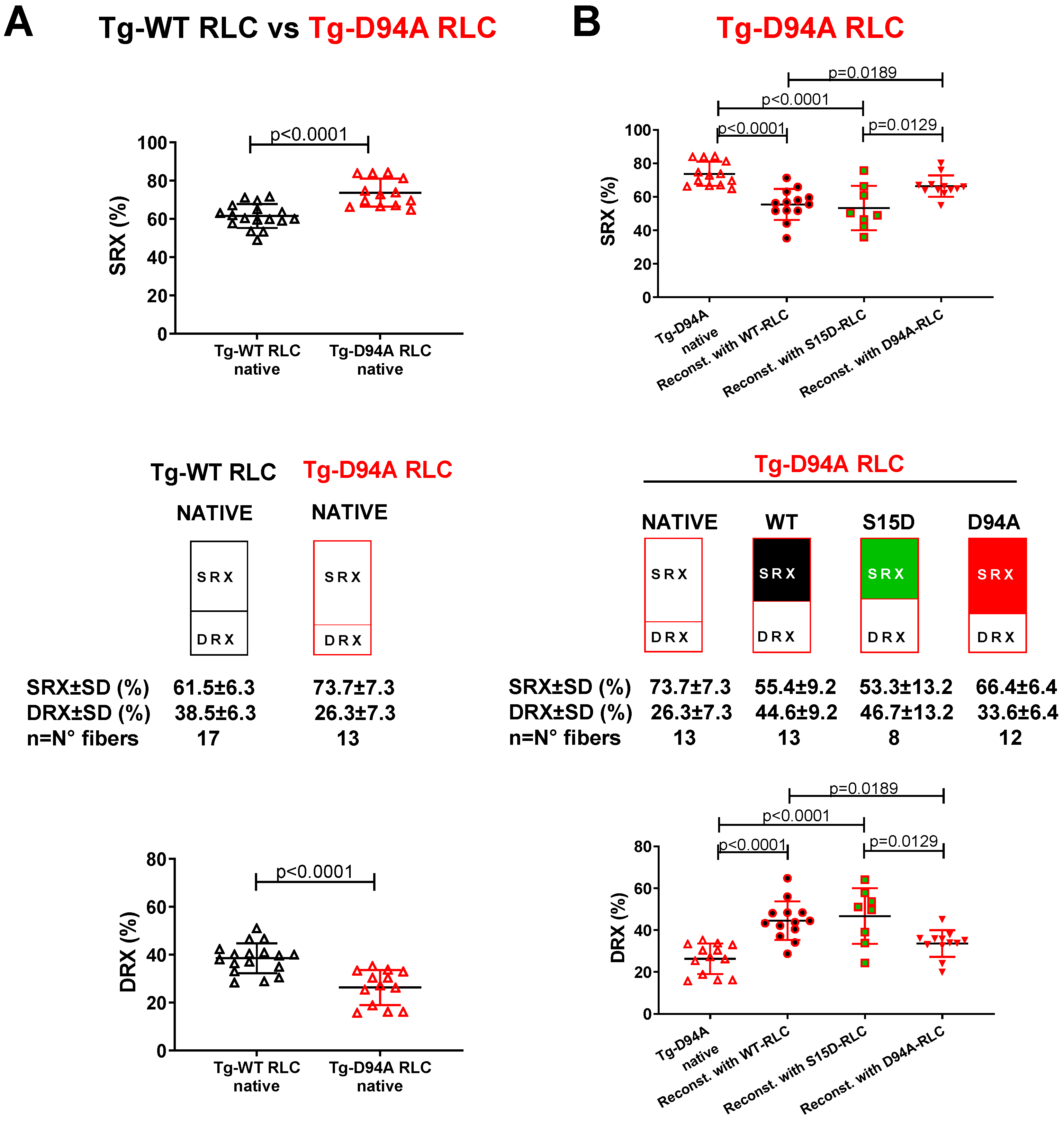
3.2. Effect of Phosphomimetic S15D-RLC on the Super-Relaxed State in LVPM Fibers from Tg-ELC and Tg-RLC Mouse Modes of RCM and DCM
3.2.1. Transgenic ELC Mice
3.2.2. Transgenic RLC Mice
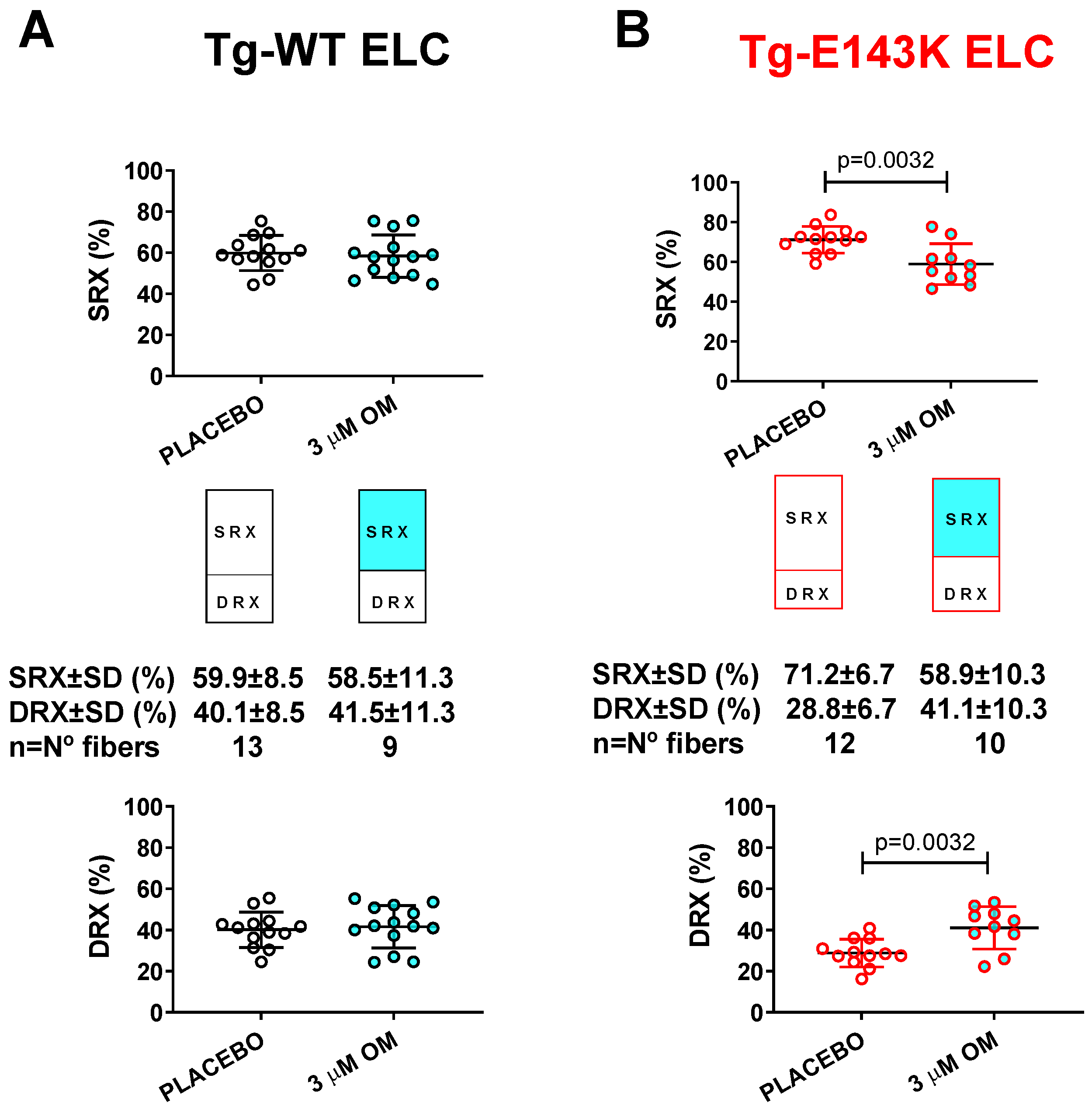
3.3. Treatment of RCM and DCM Mice with Omecamtiv Mecarbil (OM)
3.3.1. Steady-State Force and Force–pCa Relationship
3.3.2. SRX↔DRX Equilibrium in LVPM Fibers from RCM and DCM Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Geeves, M.A.; Holmes, K.C. The molecular mechanism of muscle contraction. Adv. Protein Chem. 2005, 71, 161–193. [Google Scholar] [PubMed]
- Tobacman, L.S. Thin filament-mediated regulation of cardiac contraction. Annu. Rev. Physiol. 1996, 58, 447–481. [Google Scholar] [CrossRef] [PubMed]
- McKillop, D.F.; Geeves, M.A. Regulation of the interaction between actin and myosin subfragment 1: Evidence for three states of the thin filament. Biophys. J. 1993, 65, 693–701. [Google Scholar] [CrossRef]
- Brunello, E.; Fusi, L.; Ghisleni, A.; Park-Holohan, S.J.; Ovejero, J.G.; Narayanan, T.; Irving, M. Myosin filament-based regulation of the dynamics of contraction in heart muscle. Proc. Natl. Acad. Sci. USA 2020, 117, 8177–8186. [Google Scholar] [CrossRef]
- Wang, Y.; Yuan, C.C.; Kazmierczak, K.; Szczesna-Cordary, D.; Burghardt, T.P. Single cardiac ventricular myosins are autonomous motors. Open Biol. 2018, 8, 170240. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, J.P.; Debold, E.P.; Ahmad, F.; Armstrong, A.; Frederico, A.; Conner, D.A.; Mende, U.; Lohse, M.J.; Warshaw, D.; Seidman, C.E.; et al. Cardiac myosin missense mutations cause dilated cardiomyopathy in mouse models and depress molecular motor function. Proc. Natl. Acad. Sci. USA 2006, 103, 14525–14530. [Google Scholar] [CrossRef]
- Alcalai, R.; Seidman, J.G.; Seidman, C.E. Genetic Basis of Hypertrophic Cardiomyopathy: From Bench to the Clinics. J. Cardiovasc. Electrophysiol. 2008, 19, 104–110. [Google Scholar] [CrossRef]
- Burghardt, T.P.; Sikkink, L.A. Regulatory light chain mutants linked to heart disease modify the cardiac myosin lever arm. Biochemistry 2013, 52, 1249–1259. [Google Scholar] [CrossRef]
- Huang, W.; Liang, J.; Yuan, C.C.; Kazmierczak, K.; Zhou, Z.; Morales, A.; McBride, K.L.; Fitzgerald-Butt, S.M.; Hershberger, R.E.; Szczesna-Cordary, D. Novel familial dilated cardiomyopathy mutation in MYL2 affects the structure and function of myosin regulatory light chain. FEBS J. 2015, 282, 2379–2393. [Google Scholar] [CrossRef]
- Huang, W.; Szczesna-Cordary, D. Molecular mechanisms of cardiomyopathy phenotypes associated with myosin light chain mutations. J. Muscle Res. Cell Motil. 2015, 36, 433–445. [Google Scholar] [CrossRef]
- Muthu, P.; Kazmierczak, K.; Jones, M.; Szczesna-Cordary, D. The effect of myosin RLC phosphorylation in normal and cardiomyopathic mouse hearts. J. Cell. Mol. Med. 2012, 16, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.C.; Muthu, P.; Kazmierczak, K.; Liang, J.; Huang, W.; Irving, T.C.; Kanashiro-Takeuchi, R.M.; Hare, J.M.; Szczesna-Cordary, D. Constitutive phosphorylation of cardiac myosin regulatory light chain prevents development of hypertrophic cardiomyopathy in mice. Proc. Natl. Acad. Sci. USA 2015, 112, E4138–E4146. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Kazmierczak, K.; Liang, J.; Sitbon, Y.H.; Szczesna-Cordary, D. Phosphomimetic-mediated in vitro rescue of hypertrophic cardiomyopathy linked to R58Q mutation in myosin regulatory light chain. FEBS J. 2019, 286, 151–168. [Google Scholar] [CrossRef] [PubMed]
- Sitbon, Y.H.; Kazmierczak, K.; Liang, J.; Yadav, S.; Veerasammy, M.; Kanashiro-Takeuchi, R.M.; Szczesna-Cordary, D. Ablation of the N terminus of cardiac essential light chain promotes the super-relaxed state of myosin and counteracts hypercontractility in hypertrophic cardiomyopathy mutant mice. FEBS J. 2020, 287, 3989–4004. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, H.L.; Bowman, B.F.; Stull, J.T. Myosin light chain phosphorylation in vertebrate striated muscle: Regulation and function. Am. J. Physiol. 1993, 264 Pt 1, C1085–C1095. [Google Scholar] [CrossRef] [PubMed]
- Kamm, K.E.; Stull, J.T. The function of myosin and myosin light chain kinase phosphorylation in smooth muscle. Annu. Rev. Pharmacol. Toxicol. 1985, 25, 593–620. [Google Scholar] [CrossRef]
- Kamm, K.E.; Stull, J.T. Signaling to Myosin Regulatory Light Chain in Sarcomeres. J. Biol. Chem. 2011, 286, 9941–9947. [Google Scholar] [CrossRef]
- Seguchi, O.; Takashima, S.; Yamazaki, S.; Asakura, M.; Asano, Y.; Shintani, Y.; Wakeno, M.; Minamino, T.; Kondo, H.; Furukawa, H.; et al. A cardiac myosin light chain kinase regulates sarcomere assembly in the vertebrate heart. J. Clin. Investig. 2007, 117, 2812–2824. [Google Scholar] [CrossRef]
- Terry, M.; Walker, D.D.; Ferrari, M.B. Protein phosphatase activity is necessary for myofibrillogenesis. Cell Biochem. Biophys. 2006, 45, 265–278. [Google Scholar] [CrossRef]
- Chang, A.N.; Battiprolu, P.K.; Cowley, P.M.; Chen, G.; Gerard, R.D.; Pinto, J.R.; Hill, J.A.; Baker, A.J.; Kamm, K.E.; Stull, J.T. Constitutive phosphorylation of cardiac myosin regulatory light chain in vivo. J. Biol. Chem. 2015, 290, 10703–10716. [Google Scholar] [CrossRef]
- Kazmierczak, K.; Liang, J.; Gomez-Guevara, M.; Szczesna-Cordary, D. Functional comparison of phosphomimetic S15D and T160D mutants of myosin regulatory light chain exchanged in cardiac muscle preparations of HCM and WT mice. Front. Cardiovasc. Med. 2022, 9, 988066. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Yuan, C.C.; Kazmierczak, K.; Liang, J.; Huang, W.; Takeuchi, L.M.; Kanashiro-Takeuchi, R.M.; Szczesna-Cordary, D. Therapeutic potential of AAV9-S15D-RLC gene delivery in humanized MYL2 mouse model of HCM. J. Mol. Med. 2019, 97, 1033–1047. [Google Scholar] [CrossRef]
- Brodehl, A.; Gerull, B. Genetic Insights into Primary Restrictive Cardiomyopathy. J. Clin. Med. 2022, 11, 2094. [Google Scholar] [CrossRef] [PubMed]
- Olson, T.M.; Karst, M.L.; Whitby, F.G.; Driscoll, D.J. Myosin Light Chain Mutation Causes Autosomal Recessive Cardiomyopathy With Mid-Cavitary Hypertrophy and Restrictive Physiology. Circulation 2002, 105, 2337–2340. [Google Scholar] [CrossRef] [PubMed]
- Caleshu, C.; Sakhuja, R.; Nussbaum, R.L.; Schiller, N.B.; Ursell, P.C.; Eng, C.; De Marco, T.; McGlothlin, D.; Burchard, E.G.; Rame, J.E. Furthering the link between the sarcomere and primary cardiomyopathies: Restrictive cardiomyopathy associated with multiple mutations in genes previously associated with hypertrophic or dilated cardiomyopathy. Am. J. Med. Genet. A 2011, 155, 2229–2235. [Google Scholar] [CrossRef]
- Yuan, C.C.; Kazmierczak, K.; Liang, J.; Kanashiro-Takeuchi, R.; Irving, T.C.; Gomes, A.V.; Wang, Y.; Burghardt, T.P.; Szczesna-Cordary, D. Hypercontractile mutant of ventricular myosin essential light chain leads to disruption of sarcomeric structure and function and results in restrictive cardiomyopathy in mice. Cardiovasc. Res. 2017, 113, 1124–1136. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Hedges, D.J.; Morales, A. Dilated cardiomyopathy: The complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 2013, 10, 531–547. [Google Scholar] [CrossRef]
- Yuan, C.C.; Kazmierczak, K.; Liang, J.; Zhou, Z.; Yadav, S.; Gomes, A.V.; Irving, T.C.; Szczesna-Cordary, D. Sarcomeric perturbations of myosin motors lead to dilated cardiomyopathy in genetically modified MYL2 mice. Proc. Natl. Acad. Sci. USA 2018, 115, E2338–E2347. [Google Scholar] [CrossRef]
- Mamidi, R.; Li, J.; Gresham Kenneth, S.; Verma, S.; Doh Chang, Y.; Li, A.; Lal, S.; dos Remedios Cristobal, G.; Stelzer Julian, E. Dose-Dependent Effects of the Myosin Activator Omecamtiv Mecarbil on Cross-Bridge Behavior and Force Generation in Failing Human Myocardium. Circ. Heart Fail. 2017, 10, e004257. [Google Scholar] [CrossRef]
- Teerlink, J.R.; Felker, G.M.; McMurray, J.J.; Ponikowski, P.; Metra, M.; Filippatos, G.S.; Ezekowitz, J.A.; Dickstein, K.; Cleland, J.G.; Kim, J.B.; et al. Acute Treatment With Omecamtiv Mecarbil to Increase Contractility in Acute Heart Failure: The ATOMIC-AHF Study. J. Am. Coll. Cardiol. 2016, 67, 1444–1455. [Google Scholar] [CrossRef]
- Sitbon, Y.H.; Diaz, F.; Kazmierczak, K.; Liang, J.; Wangpaichitr, M.; Szczesna-Cordary, D. Cardiomyopathic mutations in essential light chain reveal mechanisms regulating the super relaxed state of myosin. J. Gen. Physiol. 2021, 153, e202012801. [Google Scholar] [CrossRef]
- Szczesna, D.; Ghosh, D.; Li, Q.; Gomes, A.V.; Guzman, G.; Arana, C.; Zhi, G.; Stull, J.T.; Potter, J.D. Familial hypertrophic cardiomyopathy mutations in the regulatory light chains of myosin affect their structure, Ca2+ binding, and phosphorylation. J. Biol. Chem. 2001, 276, 7086–7092. [Google Scholar] [CrossRef]
- Muthu, P.; Liang, J.; Schmidt, W.; Moore, J.R.; Szczesna-Cordary, D. In Vitro Rescue Study of a Malignant Familial Hypertrophic Cardiomyopathy Phenotype by Pseudo-Phosphorylation of Myosin Regulatory Light Chain. Arch. Biochem. Biophys. 2014, 552–553, 29–39. [Google Scholar] [CrossRef]
- Pant, K.; Watt, J.; Greenberg, M.; Jones, M.; Szczesna-Cordary, D.; Moore, J.R. Removal of the cardiac myosin regulatory light chain increases isometric force production. FASEB J. 2009, 23, 3571–3580. [Google Scholar] [CrossRef]
- Yuan, C.C.; Kazmierczak, K.; Liang, J.; Ma, W.; Irving, T.C.; Szczesna-Cordary, D. Molecular basis of force-pCa relation in MYL2 cardiomyopathy mice: Role of the super-relaxed state of myosin. Proc. Natl. Acad. Sci. USA 2022, 119, e2110328119. [Google Scholar] [CrossRef] [PubMed]
- Hooijman, P.; Stewart, M.A.; Cooke, R. A new state of cardiac Myosin with very slow ATP turnover: A potential cardioprotective mechanism in the heart. Biophys. J. 2011, 100, 1969–1976. [Google Scholar] [CrossRef] [PubMed]
- Szczesna-Cordary, D.; Guzman, G.; Ng, S.S.; Zhao, J. Familial hypertrophic cardiomyopathy-linked alterations in Ca2+ binding of human cardiac myosin regulatory light chain affect cardiac muscle contraction. J. Biol. Chem. 2004, 279, 3535–3542. [Google Scholar] [CrossRef] [PubMed]
- Muthu, P.; Huang, W.; Kazmierczak, K.; Szczesna-Cordary, D. Functional Consequences of Mutations in the Myosin Regulatory Light Chain Associated with Hypertrophic Cardiomyopathy. In Cardiomyopathies—From Basic Research to Clinical Management; Veselka, J., Ed.; InTech: Rijeka, Croatia, 2012; Chapet 17; pp. 383–408. [Google Scholar]
- Greenberg, M.J.; Kazmierczak, K.; Szczesna-Cordary, D.; Moore, J.R. Cardiomyopathy-linked myosin regulatory light chain mutations disrupt myosin strain-dependent biochemistry. Proc. Natl. Acad. Sci. USA 2010, 107, 17403–17408. [Google Scholar] [CrossRef] [PubMed]
- Pulcastro, H.C.; Awinda, P.O.; Breithaupt, J.J.; Tanner, B.C. Effects of myosin light chain phosphorylation on length-dependent myosin kinetics in skinned rat myocardium. Arch. Biochem. Biophys. 2016, 601, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Alamo, L.; Ware, J.S.; Pinto, A.; Gillilan, R.E.; Seidman, J.G.; Seidman, C.E.; Padron, R. Effects of myosin variants on interacting-heads motif explain distinct hypertrophic and dilated cardiomyopathy phenotypes. Elife 2017, 6, e24634. [Google Scholar] [CrossRef]
- Woodhead, J.L.; Zhao, F.Q.; Craig, R.; Egelman, E.H.; Alamo, L.; Padrón, R. Atomic model of a myosin filament in the relaxed state. Nature 2005, 436, 1195–1199. [Google Scholar] [CrossRef] [PubMed]
- Schmid, M.; Toepfer, C.N. Cardiac myosin super relaxation (SRX): A perspective on fundamental biology, human disease and therapeutics. Biol. Open 2021, 10, bio057646. [Google Scholar] [CrossRef]
- Malik, F.I.; Hartman, J.J.; Elias, K.A.; Morgan, B.P.; Rodriguez, H.; Brejc, K.; Anderson, R.L.; Sueoka, S.H.; Lee, K.H.; Finer, J.T.; et al. Cardiac myosin activation: A potential therapeutic approach for systolic heart failure. Science 2011, 331, 1439–1443. [Google Scholar] [CrossRef]
- Teerlink, J.R.; Diaz, R.; Felker, G.M.; McMurray, J.J.V.; Metra, M.; Solomon, S.D.; Adams, K.F.; Anand, I.; Arias-Mendoza, A.; Biering-Sørensen, T.; et al. Cardiac Myosin Activation with Omecamtiv Mecarbil in Systolic Heart Failure. N. Engl. J. Med. 2021, 384, 105–116. [Google Scholar] [CrossRef]
- Woody, M.S.; Greenberg, M.J.; Barua, B.; Winkelmann, D.A.; Goldman, Y.E.; Ostap, E.M. Positive cardiac inotrope omecamtiv mecarbil activates muscle despite suppressing the myosin working stroke. Nat. Commun. 2018, 9, 3838. [Google Scholar] [CrossRef]
- Mamidi, R.; Holmes, J.B.; Doh, C.Y.; Dominic, K.L.; Madugula, N.; Stelzer, J.E. cMyBPC phosphorylation modulates the effect of omecamtiv mecarbil on myocardial force generation. J. Gen. Physiol. 2021, 153, e202012816. [Google Scholar] [CrossRef]
- Abraham, T.P.; Jones, M.; Kazmierczak, K.; Liang, H.-Y.; Pinheiro, A.C.; Wagg, C.S.; Lopaschuk, G.D.; Szczesna-Cordary, D. Diastolic dysfunction in familial hypertrophic cardiomyopathy transgenic model mice. Cardiovasc. Res. 2009, 82, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Kazmierczak, K.; Xu, Y.; Jones, M.; Guzman, G.; Hernandez, O.M.; Kerrick, W.G.L.; Szczesna-Cordary, D. The Role of the N-Terminus of the Myosin Essential Light Chain in Cardiac Muscle Contraction. J. Mol. Biol. 2009, 387, 706–725. [Google Scholar] [CrossRef]
- Wang, Y.; Ajtai, K.; Burghardt, T.P. Ventricular myosin modifies in vitro step-size when phosphorylated. J. Mol. Cell. Cardiol. 2014, 72, 231–237. [Google Scholar] [CrossRef]
- Yu, H.; Chakravorty, S.; Song, W.; Ferenczi, M.A. Phosphorylation of the regulatory light chain of myosin in striated muscle: Methodological perspectives. Eur. Biophys. J. 2016, 45, 779–805. [Google Scholar] [CrossRef] [PubMed]
- Toepfer, C.N.; Garfinkel, A.C.; Venturini, G.; Wakimoto, H.; Repetti, G.; Alamo, L.; Sharma, A.; Agarwal, R.; Ewoldt, J.F.; Cloonan, P.; et al. Myosin Sequestration Regulates Sarcomere Function, Cardiomyocyte Energetics, and Metabolism, Informing the Pathogenesis of Hypertrophic Cardiomyopathy. Circulation 2020, 141, 828–842. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Force Parameter/ Recombinant RLC-Reconstituted LVPM | LVPM from Tg-WT ELC Mice | LVPM from Tg-E143K Mice | ||
|---|---|---|---|---|
| WT | S15D | WT | S15D | |
| N° fibers | 8 | 7 | 9 | 10 |
| Fmax (kN/m2) ± SD | 21.56 ± 4.64 | 34.25 ± 9.66 ** | 23.56 ± 4.42 | 28.58 ± 1.95 |
| pCa50 ± SD | 5.32 ± 0.1 | 5.41 ± 0.06 * | 5.51 ± 0.09 | 5.51 ± 0.09 |
| nH ± SD | 2.03 ± 0.74 | 1.87 ± 0.41 | 2.2 ± 0.61 | 1.73 ± 0.24 * |
| SRX Parameter/Recombinant RLC-Reconstituted LVPM | LVPM from Tg-WT ELC Mice | LVPM from Tg-E143K ELC Mice | ||||
|---|---|---|---|---|---|---|
| Native | WT | S15D | Native | WT | S15D | |
| N° fibers | 14 | 6 | 7 | 13 | 11 | 10 |
| DRX (%) ± SD | 38.9 ± 8.2 | 35.1 ± 14.2 | 37.2 ± 18.8 | 29.4 ± 3.7 ## | 32.3 ± 11.9 | 44 ± 12.8 * |
| SRX (%) ± SD | 61.1 ± 8.2 | 64.9 ± 14.2 | 62.8 ± 18.8 | 70.6 ± 3.7 ## | 67.8 ± 11.9 | 56 ± 12.8 * |
| T1 (s) ± SD | 4.4 ± 2.4 | 6.6 ± 4.9 | 5.3 ± 2.5 | 4.2 ± 2.5 | 4.8 ± 3.6 | 7 ± 5.2 |
| T2 (s) ± SD | 129.8 ± 67 | 125.3 ± 81.3 | 173.4 ± 167.7 | 126.5 ± 81.2 | 92.2 ± 64 | 136.4 ± 116.5 |
| SRX Parameter/Recombinant RLC-Reconstituted LVPM | LVPM from Tg-D94A RLC Mice | |||
|---|---|---|---|---|
| Native | WT | S15D | D94A | |
| No. fibers | 13 | 13 | 8 | 12 |
| DRX ± SD | 26.3 ± 7.3 | 44.9 ± 10.2 ****,^ | 46.7 ± 13.2 ****,^ | 33.6 ± 6.4 |
| SRX ± SD | 73.7 ± 7.3 | 55.1 ± 10.2 ****,^ | 53.3 ± 13.2 ****,^ | 66.4 ± 6.4 |
| T1 ± SD | 6.5 ± 3.3 | 4.9 ± 2.8 | 5.7 ± 5.1 | 2.7 ± 1.7 * |
| T2 ± SD | 161.8 ± 149 | 136.9 ± 103.1 | 117.7 ± 130.8 | 67.4 ± 77.3 |
| Force Parameter/ OM- vs. Placebo-Treated Fibers | LVPM from Tg-WT ELC Mice | LVPM from Tg-E143K ELC Mice | LVPM from Tg-WT RLC Mice | LVPM from Tg-D94A RLC Mice | ||||
|---|---|---|---|---|---|---|---|---|
| OM | Placebo | OM | Placebo | OM | Placebo | OM | Placebo | |
| No. fibers | 6 | 4 | 6 | 4 | 8 | 4 | 6 | 4 |
| Fmax (kN/m2) ± SD | 66.4 ± 5.1 *** | 48.3 ± 4.5 | 56.8 ± 4.8 * | 47.8 ± 3.2 | 55.8 ± 5.7 | 51.7 ± 2 | 64.5 ± 7.8 ** | 46.9 ± 8.1 |
| pCa50 ± SD | 5.9 ± 0.1 ** | 5.5 ± 0.04 | 5.7 ± 0.1 | 5.6 ± 0.03 | 5.9 ± 0.1 *** | 5.6 ± 0.1 | 5.8 ± 0.1 * | 5.6 ± 0.1 |
| nH ± SD | 1.2 ± 0.1 * | 1.5 ± 0.2 | 1.8 ± 0.2 * | 2.2 ± 0.1 | 1.5 ± 0.2 ** | 2 ± 0.2 | 1.9 ± 0.3 * | 2.2 ± 0.2 |
| SRX Parameter/ OM- vs. Placebo-Treated Fibers | LVPM from Tg-WT ELC Mice | LVPM from Tg-E143K ELC Mice | LVPM from Tg-WT RLC Mice | LVPM from Tg-D94A RLC Mice | ||||
|---|---|---|---|---|---|---|---|---|
| OM | Placebo | OM | Placebo | OM | Placebo | OM | Placebo | |
| No. fibers | 14 | 13 | 10 | 12 | 12 | 14 | 11 | 12 |
| DRX ± SD | 41.6 ± 10.4 | 40.1 ± 8.5 | 41.1 ± 10.3 ** | 28.8 ± 6.7 | 39.3 ± 13.4 | 35.9 ± 8.5 | 47.2 ± 15.3 **** | 22.7 ± 6.2 |
| SRX ± SD | 58.4 ± 10.4 | 59.9 ± 8.5 | 58.9 ± 10.3 ** | 71.2 ± 6.7 | 60.7 ± 13.4 | 64.1 ± 8.5 | 52.8 ± 15.3 **** | 77.3 ± 6.2 |
| T1 ± SD | 3.7 ± 3.4 | 3.2 ± 1.8 | 3.7 ± 2.4 | 4.2 ± 2.7 | 4.2 ± 3 | 5.6 ± 3.8 | 3.7 ± 1.9 * | 6.9 ± 4.1 |
| T2 ± SD | 101.4 ± 152.5 | 69 ± 38.5 | 89.3 ± 78.9 | 112.9 ± 94.3 | 123.8 ± 86.8 | 119.6 ± 122.4 | 83.1 ± 65.6 * | 230.6 ± 213.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kazmierczak, K.; Liang, J.; Maura, L.G.; Scott, N.K.; Szczesna-Cordary, D. Phosphorylation Mimetic of Myosin Regulatory Light Chain Mitigates Cardiomyopathy-Induced Myofilament Impairment in Mouse Models of RCM and DCM. Life 2023, 13, 1463. https://doi.org/10.3390/life13071463
Kazmierczak K, Liang J, Maura LG, Scott NK, Szczesna-Cordary D. Phosphorylation Mimetic of Myosin Regulatory Light Chain Mitigates Cardiomyopathy-Induced Myofilament Impairment in Mouse Models of RCM and DCM. Life. 2023; 13(7):1463. https://doi.org/10.3390/life13071463
Chicago/Turabian StyleKazmierczak, Katarzyna, Jingsheng Liang, Luis G. Maura, Natissa K. Scott, and Danuta Szczesna-Cordary. 2023. "Phosphorylation Mimetic of Myosin Regulatory Light Chain Mitigates Cardiomyopathy-Induced Myofilament Impairment in Mouse Models of RCM and DCM" Life 13, no. 7: 1463. https://doi.org/10.3390/life13071463
APA StyleKazmierczak, K., Liang, J., Maura, L. G., Scott, N. K., & Szczesna-Cordary, D. (2023). Phosphorylation Mimetic of Myosin Regulatory Light Chain Mitigates Cardiomyopathy-Induced Myofilament Impairment in Mouse Models of RCM and DCM. Life, 13(7), 1463. https://doi.org/10.3390/life13071463