



Identification of BACE-1 Inhibitors against Alzheimer’s Disease through E-Pharmacophore-Based Virtual Screening and Molecular Dynamics Simulation Studies: An Insilco Approach


Abstract
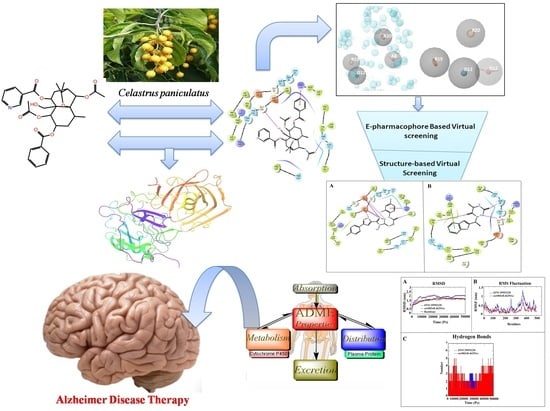
1. Introduction
2. Materials and Methods
2.1. Protein Preparation and Grid Generation
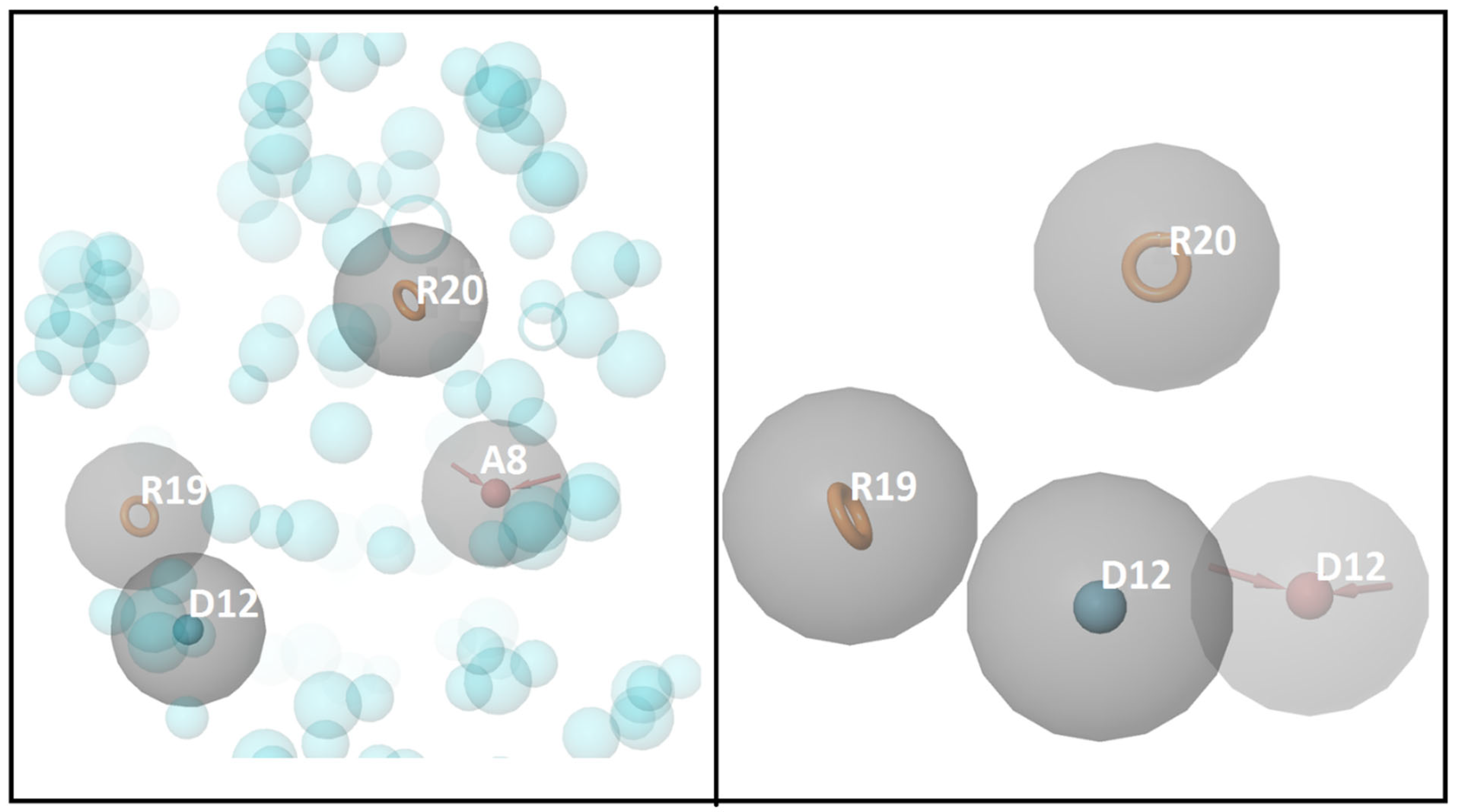
2.2. Molecular Docking and E-Pharmacophore Modeling
2.3. E-Pharmacophore-Based Virtual Screening
2.4. Structure-Based Virtual Screening
2.5. Enrichment Calculations
2.6. Binding Free Energy
2.7. Molecular Dynamic Simulation (MDS)
2.8. Density Functional Theory (DFT)
2.9. ADME Prediction
3. Results and Discussion
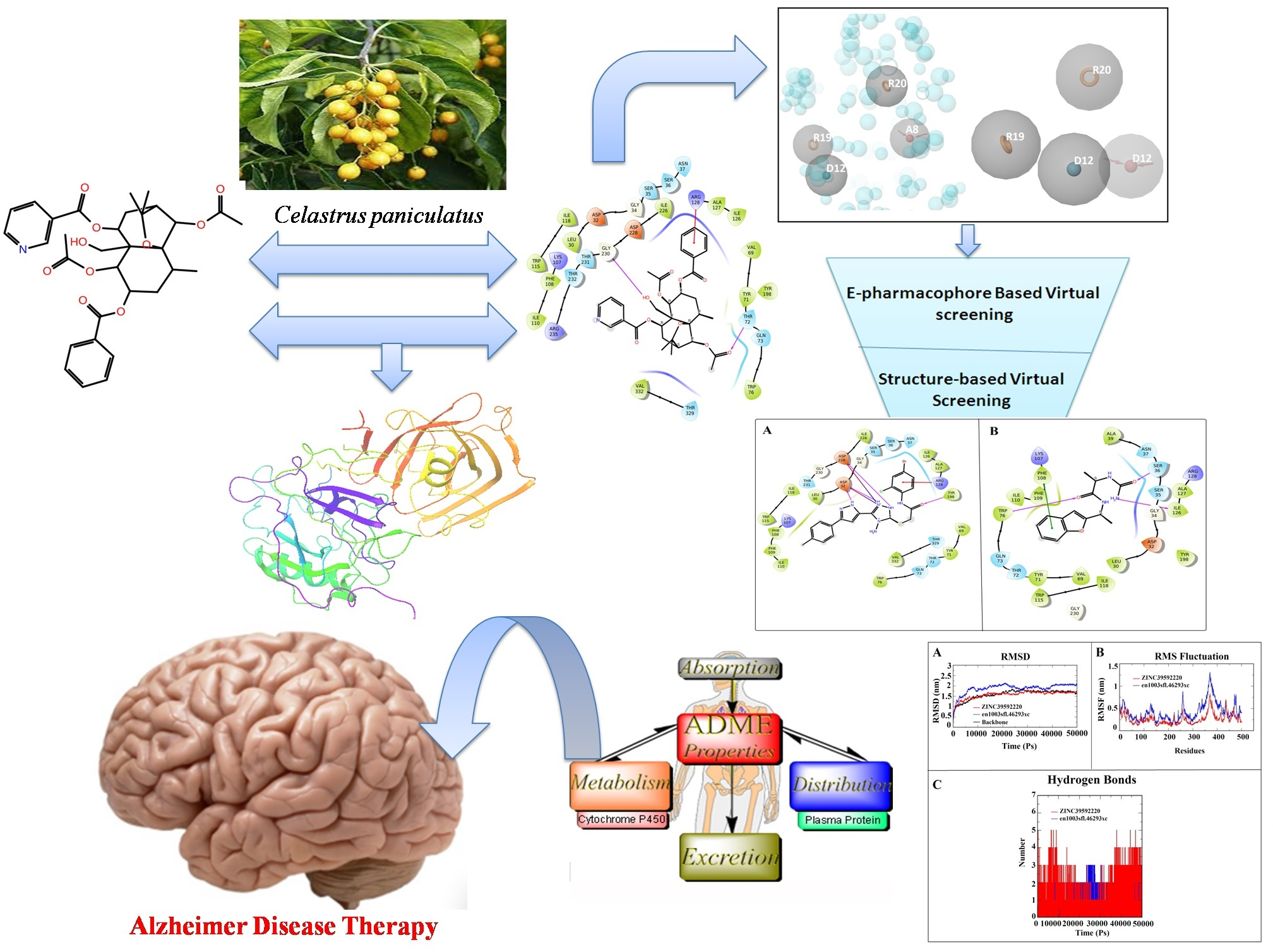
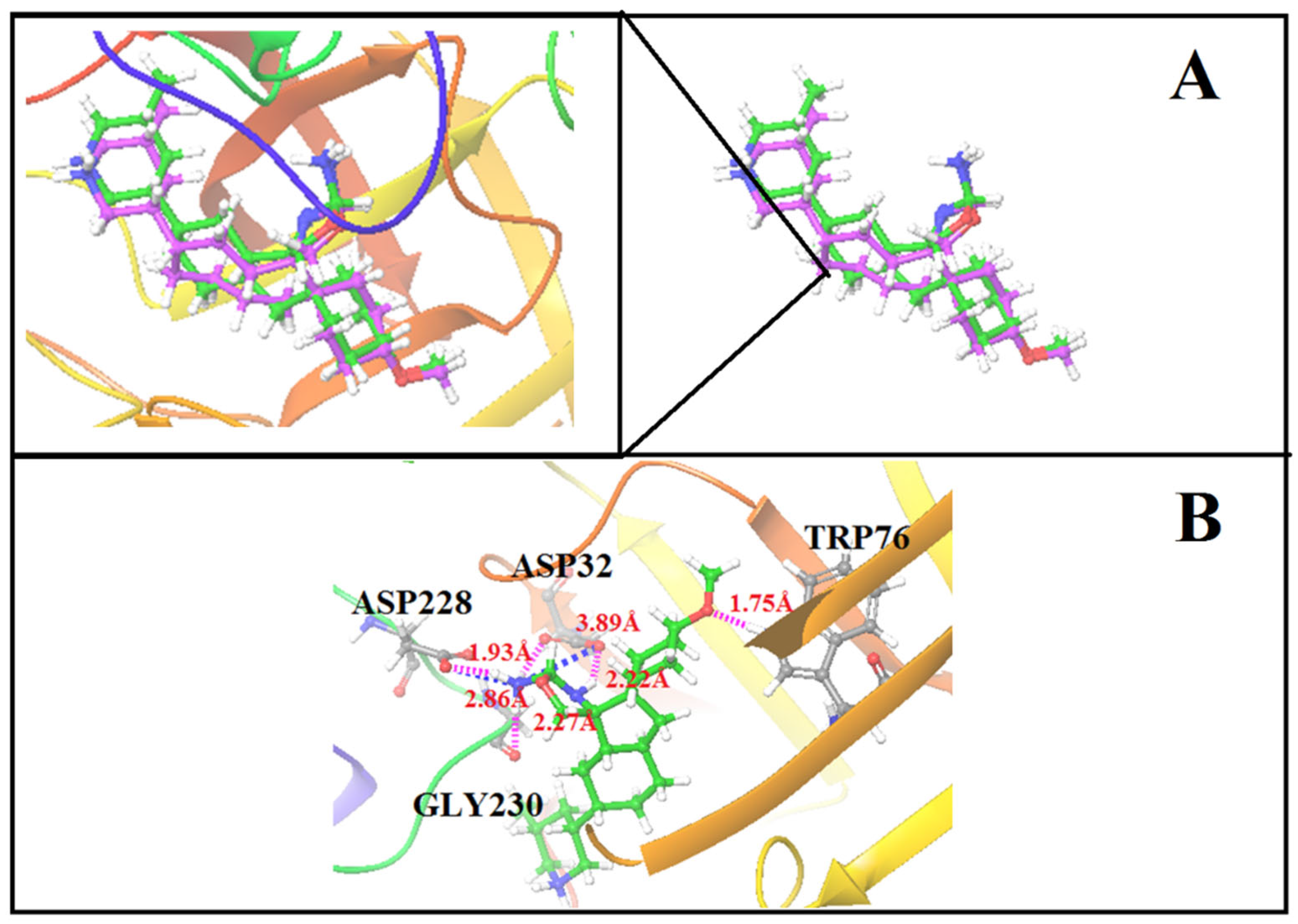
3.1. Molecular Docking and E-Pharmacophore
3.2. E-Pharmacophore-Based Virtual Screening
3.3. Enrichment Calculations
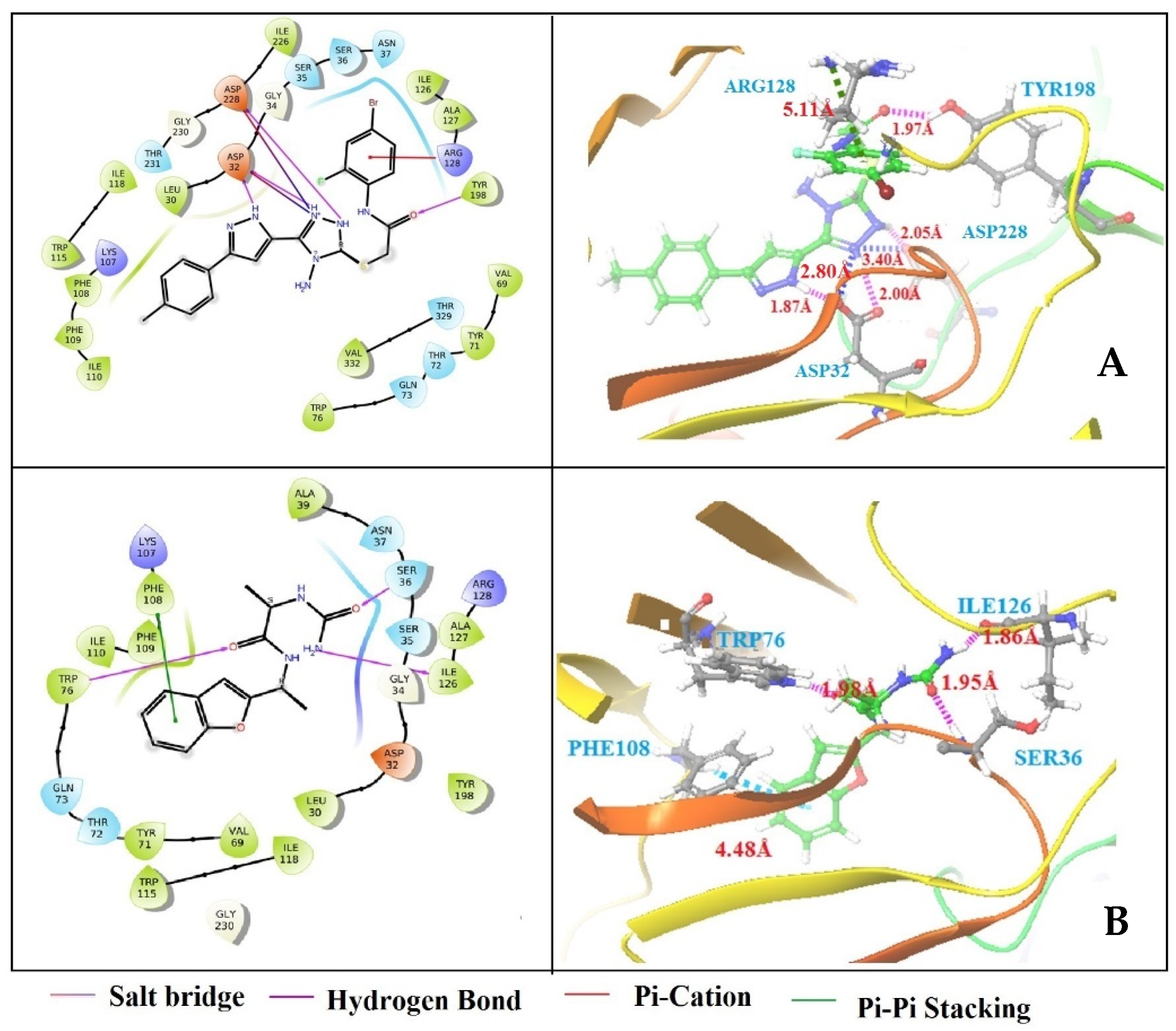
3.4. Structure-Based Virtual Screening
3.5. Binding Free Energy
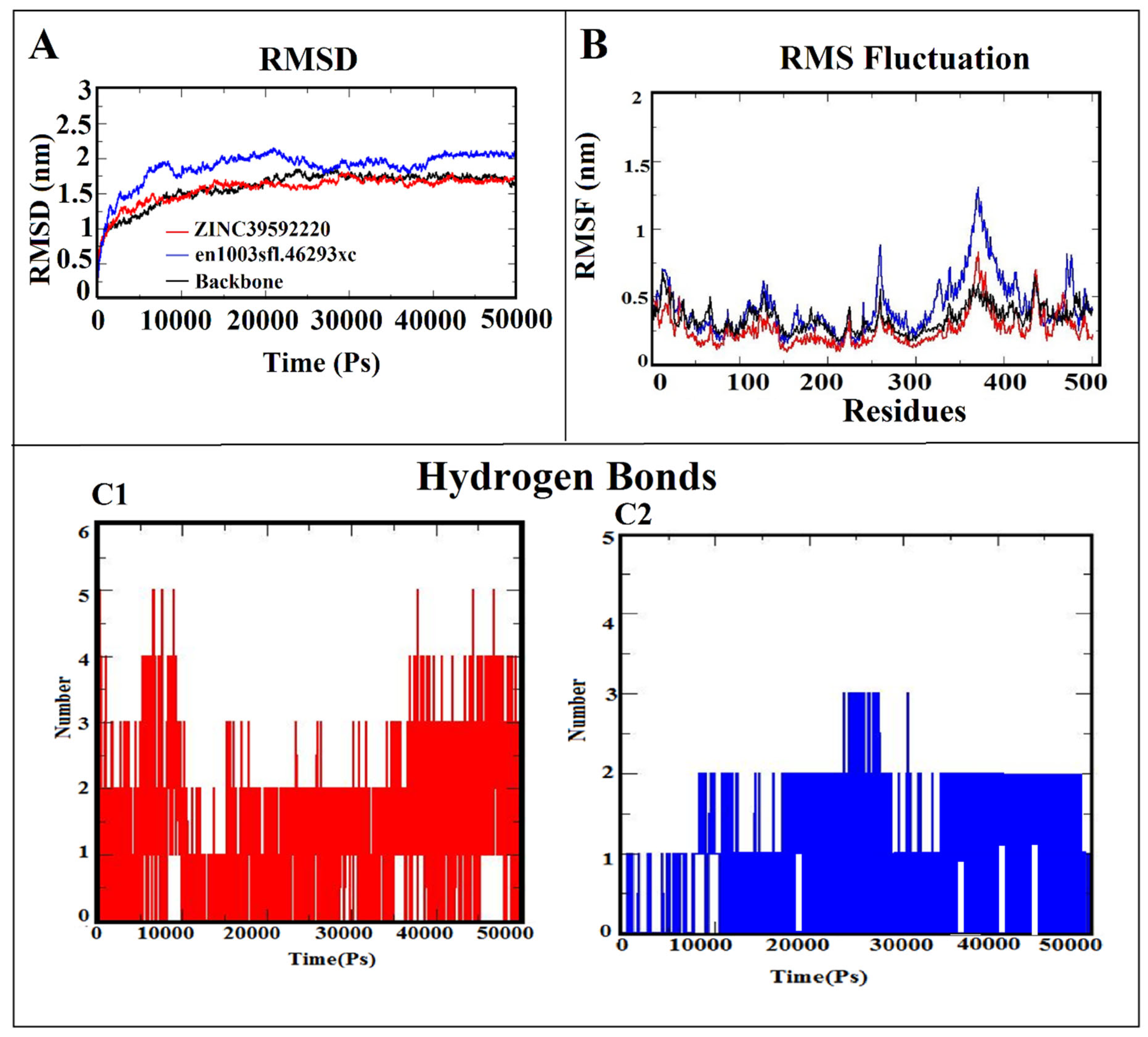
3.6. Molecular Dynamic Simulation (MDS)
3.7. MM-PBSA Calculation
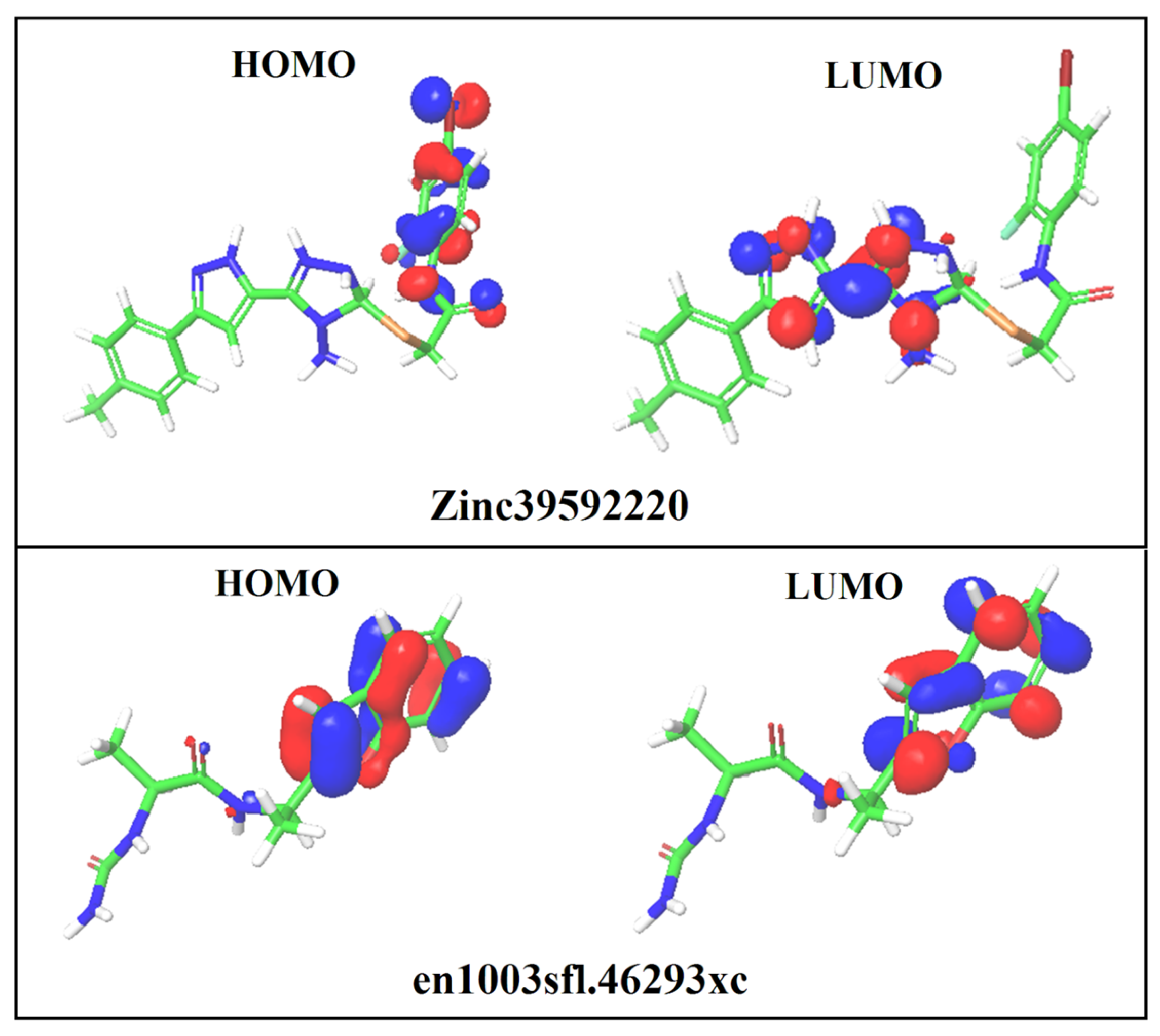
3.8. DFT Analysis
3.9. ADME Properties
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hussain, I.; Powell, D.J.; Howlett, D.R.; Chapma, G.A.; Gilmour, L.; Murdock, P.R.; Tew, D.G.; Meek, T.D.; Chapman, C.; Schneider, K.; et al. ASP1 (BACE2) cleaves the amyloid precursor protein at the beta-secretase site. Mol. Cell. Neurosci. 2000, 16, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Sait, A.; Angeli, C.; Doig, A.J.; Day, P.J.R. Viral Involvement in Alzheimer’s Disease. ACS Chem. Neurosci. 2021, 12, 1049–1060. [Google Scholar] [CrossRef]
- Winblad, B.; Amouyel, P.; Andrieu, S.; Ballard, C.; Brayne, C.; Brodaty, H.; Cedazo-Minguez, A.; Dubois, B.; Edvardsson, D.; Feldman, H.; et al. Defeating Alzheimer’s disease and other dementias: A priority for European science and society. Lancet Neurol. 2016, 15, 455–532. [Google Scholar] [CrossRef] [PubMed]
- Jannat, S.; Balupuri, A.; Ali, M.Y.; Hong, S.S.; Choi, C.W.; Choi, Y.H.; Ku, J.M.; Kim, W.J.; Leem, J.Y.; Kim, J.E.; et al. Inhibition of β-site amyloid precursor protein cleaving enzyme 1 and cholinesterases by pterosins via a specific structure-activity relationship with a strong BBB permeability. Exp. Mol. Med. 2019, 51, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Choi, R.J.; Roy, A.; Jung, H.J.; Ali, M.Y.; Min, B.S.; Park, C.H.; Yokozawa, T.; Fan, T.P.; Choi, J.S.; Jung, H.A. BACE1 molecular docking and anti-Alzheimer’s disease activities of ginsenosides. J. Ethnopharmacol. 2016, 190, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yang, M.W.; Li, X.W.; Yue, J.W.; Chen, J.Z.; Yang, M.W.; Huang, X.; Zhu, L.L.; Hong, F.F.; Yang, S.L. Therapeutic effects of natural drugs on Alzheimer’s disease. Front. Pharmacol. 2019, 10, 1355. [Google Scholar] [CrossRef]
- Li, Y.; Sun, H.; Chen, Z.; Xu, H.; Bu, G.; Zheng, H. Implications of GABAergic Neurotransmission in Alzheimer’s Disease. Front. Aging Neurosci. 2016, 8, 31. [Google Scholar] [CrossRef]
- Pandey, S.N.; Rangra, N.K.; Singh, S.; Arora, S.; Gupta, V. Evolving Role of Natural Products from Traditional Medicinal Herbs in the Treatment of Alzheimer’s Disease. ACS Chem. Neurosci. 2021, 12, 2718–2728. [Google Scholar] [CrossRef]
- Liu, H.; Han, Y.; Wang, T.; Zhang, H.; Xu, Q.; Yuan, J.; Li, Z. Targeting microglia for therapy of Parkinson’s disease by using biomimetic ultra-small nanoparticles. J. Am. Chem. Soc. 2020, 142, 21730–21742. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.K.; Tailang, M.; Kumar, S.; Chandrasekaran, B.; Alghazwani, Y.; Chandramoorthy, H.C.; Kumar, A.; Deshpande, H.; Wal, P.; Balamurugan, M.; et al. Recognition of therapeutical potentials of Alchornea laxiflora (Benth.) Pax & K. Hoffm., an underexplored medicinal herb: A systematic review. Front. Pharmacol. 2022, 13, 958453. [Google Scholar] [CrossRef]
- Mohandas, E.; Rajmohan, V.; Raghunath, B. Neurobiology of Alzheimer’s disease. Indian J. Psychiatry 2009, 51, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Ayaz, M.; Sadiq, A.; Junaid, M.; Ullah, F.; Ovais, M.; Ullah, I.; Ahmed, J.; Shahid, M. Flavonoids as prospective neuroprotectants and their therapeutic propensity in aging associated neurological disorders. Front. Aging Neurosci. 2019, 11, 155. [Google Scholar] [CrossRef] [PubMed]
- Malamas, M.S.; Erdei, J.; Gunawan, I.; Turner, J.; Hu, Y.; Wagner, E.; Fan, K.; Chopra, R.; Olland, A.; Bard, J.; et al. Design and synthesis of 5,5’-disubstituted aminohydantoins as potent and selective human beta-secretase (BACE1) inhibitors. J. Med. Chem. 2010, 53, 1146–1158. [Google Scholar] [CrossRef]
- Huang, H.J.; Leem, C.C.; Chen, C.Y. In silico design of BACE1 inhibitor for Alzheimer’s disease by traditional Chinese medicine. Biomed. Res. Int. 2014, 2014, 741703. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Osswald, H.L. BACE1 (β-secretase) inhibitors for the treatment of Alzheimer’s disease. Chem. Soc. Rev. 2014, 43, 6765–6813. [Google Scholar] [CrossRef] [PubMed]
- Balakumar, C.; Ramesh, M.; Tham, C.L.; Khathi, S.P.; Kozielski, F.; Srinivasulu, C.; Hampannavar, G.A.; Sayyad, N.; Soliman, M.E.; Karpoormath, R. Ligand- and structure-based in silico studies to identify kinesin spindle protein (KSP) inhibitors as potential anticancer agents. J. Biomol. Struct. Dyn. 2018, 36, 3687–3704. [Google Scholar] [CrossRef]
- Balakumar, C.; Lamba, P.; Kishore, D.P.; Narayana, B.L.; Rao, K.V.; Rajwinder, K.; Rao, A.R.; Shireesha, B.; Narsaiah, B. Synthesis, anti-inflammatory evaluation and docking studies of some new fluorinated fused quinazolines. Eur. J. Med. Chem. 2010, 45, 4904–4913. [Google Scholar] [CrossRef]
- Ramachandran, B.; Srinivasadesikan, V.; Chou, T.M.; Jeyakanthan, J.; Lee, S.L. Atomistic simulation on flavonoids derivatives as potential inhibitors of bacterial gyrase of Staphylococcus aureus. J. Biomol. Struct. Dyn. 2020, 40, 4314–4327. [Google Scholar] [CrossRef]
- Muthumanickam, S.; Indhumathi, T.; Boomi, P.; Balajee, R.; Jeyakanthan, J.; Anand, K.; Ravikumar, S.; Kumar, P.; Sudha, A.; Jiang, X. In silico approach of naringin as potent phosphatase and tensin homolog (PTEN) protein agonist against prostate cancer. J. Biomol. Struct Dyn. 2022, 40, 1629–1638. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R.; Roitberg, A.E. Design of e-pharmacophore models using compound fragments for the trans-sialidase of Trypanosoma cruzi: Screening for novel inhibitor scaffolds. J. Mol. Graph. Model. 2013, 45, 84–97. [Google Scholar] [CrossRef]
- Sudheer, K.K.; Pradeep, N.; Sandeep, S.; Hema, K.; Chiranjeevi, P.; Amineni, U. Inhibitor design against JNK1through e-pharmacophore modeling docking and molecular dynamics simulations. J. Recept. Signal Transduct. Res. 2016, 36, 558–571. [Google Scholar] [CrossRef]
- Palakurti, R.D.; Sriram, D.; Yogeeswari, P.; Vadrevu, R. Multiple e-Pharmacophore modeling combined with high-throughput virtual screening and docking to identify potential inhibitors of β-secretase (BACE1). Mol. Inform. 2013, 32, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Sindhu, T.; Srinivasan, P. Identification of potential dual agonists of FXR and TGR5 using e-pharmacophore based virtual screening. Mol. BioSyst. 2015, 11, 1305–1318. [Google Scholar] [CrossRef]
- Sankar, M.; Ramachandran, B.; Pandi, B.; Mutharasappan, N.; Ramasamy, V.; Prabu, P.G.; Shanmugaraj, G.; Wang, Y.; Muniyandai, B.; Rathinasamy, S.; et al. In silico screening of natural phytocompounds towards identification of potential lead compounds to treat COVID-19. Front. Mol. Biosci. 2021, 8, 637122. [Google Scholar] [CrossRef] [PubMed]
- Muthumanickam, S.; Kamaladevi, A.; Boomi, P.; Gowrishankar, G.; Pandian, S.K. Indian ethnomedicinal phytochemicals as promising inhibitors of rna-binding domain of SARS-CoV-2 nucleocapsid phosphoprotein: An in-silico study. Front. Mol. Biosci. 2021, 8, 637329. [Google Scholar] [CrossRef]
- Muthumanickam, S.; Langeswaran, K.; Sangavi, J.; Boomi, P. Screening of inhibitors as potential remedial against Ebolavirus infection: Pharmacophore-based approach. J. Biomol. Struct. Dyn. 2021, 39, 395–408. [Google Scholar] [CrossRef]
- Ramar, M.K.; Chidambaram, K.; Chandrasekaran, B.; Kandasamy, R. Standardization, in-silico and in-vivo safety assessment of methanol extract of Ziziphus mauritiana Lam leaves. Regul. Toxicol. Pharmacol. 2022, 131, 105144. [Google Scholar] [CrossRef]
- Alqahtani, A.M.; Chidambaram, K.; Pino-Figueroa, A.; Chandrasekaran, B.; Dhanaraj, P.; Venkatesan, K. Curcumin-Celecoxib: A synergistic and rationale combination chemotherapy for breast cancer. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 1916–1927. [Google Scholar] [CrossRef]
- Nagamani, S.; Muthusamy, K.; Marshal, J.J. E-pharmacophore filtering and molecular dynamics simulation studies in the discovery of potent drug-like molecules for chronic kidney disease. J. Biomol. Struct. Dyn. 2016, 34, 2233–2250. [Google Scholar] [CrossRef]
- Liu, S.; Fu, R.; Cheng, X.; Chen, S.P.; Zhou, L.H. Exploring the binding of BACE-1 inhibitors using comparative binding energy analysis (COMBINE). BMC Struct. Biol. 2012, 12, 21. [Google Scholar] [CrossRef]
- Mahmud, S.; Paul, G.K.; Biswas, S.; Afrose, S.; Mita, M.A.; Hasan, M.R.; Shimu, M.S.S.; Hossain, A.; Promi, M.M.; Ema, F.K.; et al. Prospective Role of Peptide-Based Antiviral Therapy Against the Main Protease of SARS-CoV-2. Front. Mol. Biosci. 2021, 8, 628585. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Tech. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No | Compound | Site Score | Vector Score | Volume Score | Fitness Score |
|---|---|---|---|---|---|
| 1 | ZINC39592220 | 0.824 | 0.837 | 0.474 | 1.624 |
| 2 | LEG 18146402 | 0.567 | 0.832 | 0.359 | 1.575 |
| 3 | SYN 19994146 | 0.434 | 0.847 | 0.277 | 1.571 |
| 4 | F0513-0458 | 0.924 | 0.957 | 0.345 | 1.481 |
| 5 | LEG 18141414 | 0.840 | 0.815 | 0.387 | 1.432 |
| 6 | 10236 | 0.499 | 0.740 | 0.254 | 1.413 |
| 7 | en1003sfl.46293xc | 0.818 | 0.855 | 0.299 | 1.399 |
| S. No | Compound ID | XP Docking Score (kcal/mol) | Glide Energy (kcal/mol) | ∆G Bind kcal/mol |
|---|---|---|---|---|
| 1 | ZINC39592220 | −8.182 | −59.425 | −58.803 |
| 2 | en1003sfl.46293xc | −7.184 | −55.284 | −56. 951 |
| 3 | LEG 18146402 | −6.933 | −49.358 | −45.351 |
| 4 | SYN 19994146 | −6.581 | −50.369 | −48.259 |
| 5 | F0513-0458 | −6.358 | −48.259 | −50.258 |
| 6 | LEG 18141414 | −5.982 | −42.357 | −50.592 |
| S. No | Databases | Compound ID | ∆G Bind kcal/mol |
|---|---|---|---|
| 1 | ZINC | ZINC39592220 | −58.803 |
| 2 | Enamine | en1003sfl.46293xc | −56.951 |
| S. No | Compound ID | HOMO (eV) | LUMO (eV) | EHOMO–ELUMO(eV) | Solvation Energy (kcal/mol) |
|---|---|---|---|---|---|
| 1 | ZINC39592220 | −0.237 | −0.094 | 0.143 | −25.40 |
| 2 | en1003sfl.46293xc | −0.217 | −0.026 | −0.191 | −20.29 |
| S. No | Compounds ID | MW | HBD | HBA | %HOA | Qplog Po/w | QPP Caco |
|---|---|---|---|---|---|---|---|
| 1 | ZINC39592220 | 288 | 3 | 5 | 93.01 | −1.204 | −4.877 |
| 2 | en1003sfl.46293xc | 255.35 | 5 | 9 | 75.360 | −2.001 | −3.305 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chidambaram, K. Identification of BACE-1 Inhibitors against Alzheimer’s Disease through E-Pharmacophore-Based Virtual Screening and Molecular Dynamics Simulation Studies: An Insilco Approach. Life 2023, 13, 952. https://doi.org/10.3390/life13040952
Chidambaram K. Identification of BACE-1 Inhibitors against Alzheimer’s Disease through E-Pharmacophore-Based Virtual Screening and Molecular Dynamics Simulation Studies: An Insilco Approach. Life. 2023; 13(4):952. https://doi.org/10.3390/life13040952
Chicago/Turabian StyleChidambaram, Kumarappan. 2023. "Identification of BACE-1 Inhibitors against Alzheimer’s Disease through E-Pharmacophore-Based Virtual Screening and Molecular Dynamics Simulation Studies: An Insilco Approach" Life 13, no. 4: 952. https://doi.org/10.3390/life13040952
APA StyleChidambaram, K. (2023). Identification of BACE-1 Inhibitors against Alzheimer’s Disease through E-Pharmacophore-Based Virtual Screening and Molecular Dynamics Simulation Studies: An Insilco Approach. Life, 13(4), 952. https://doi.org/10.3390/life13040952