
Molecular Mechanism of Sirtuin 1 Inhibition by Human Immunodeficiency Virus 1 Tat Protein

 , , ,
, , ,  , and
, and
Abstract
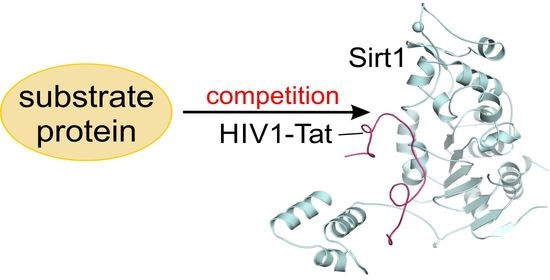
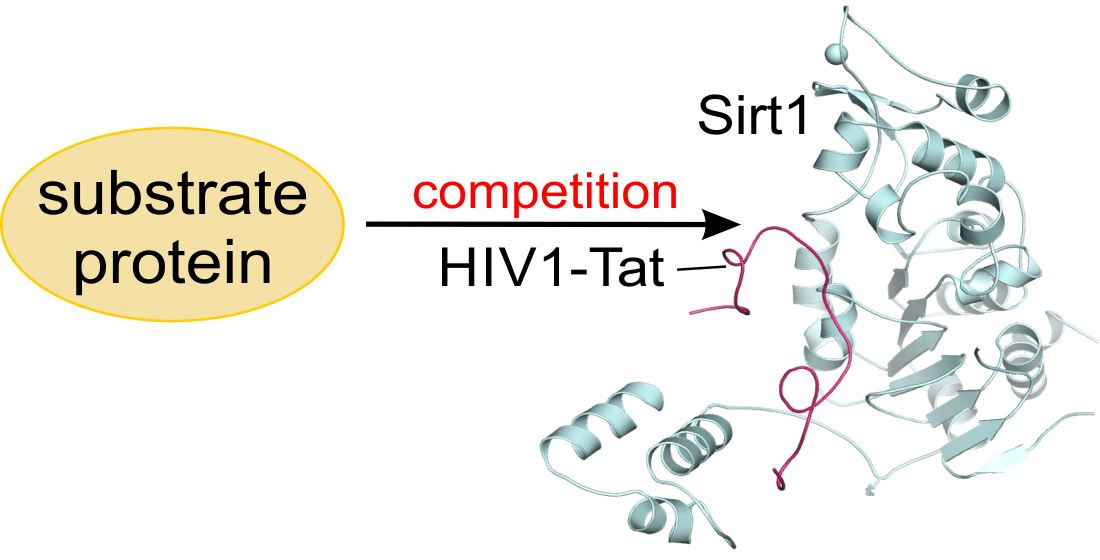
1. Introduction
2. Results
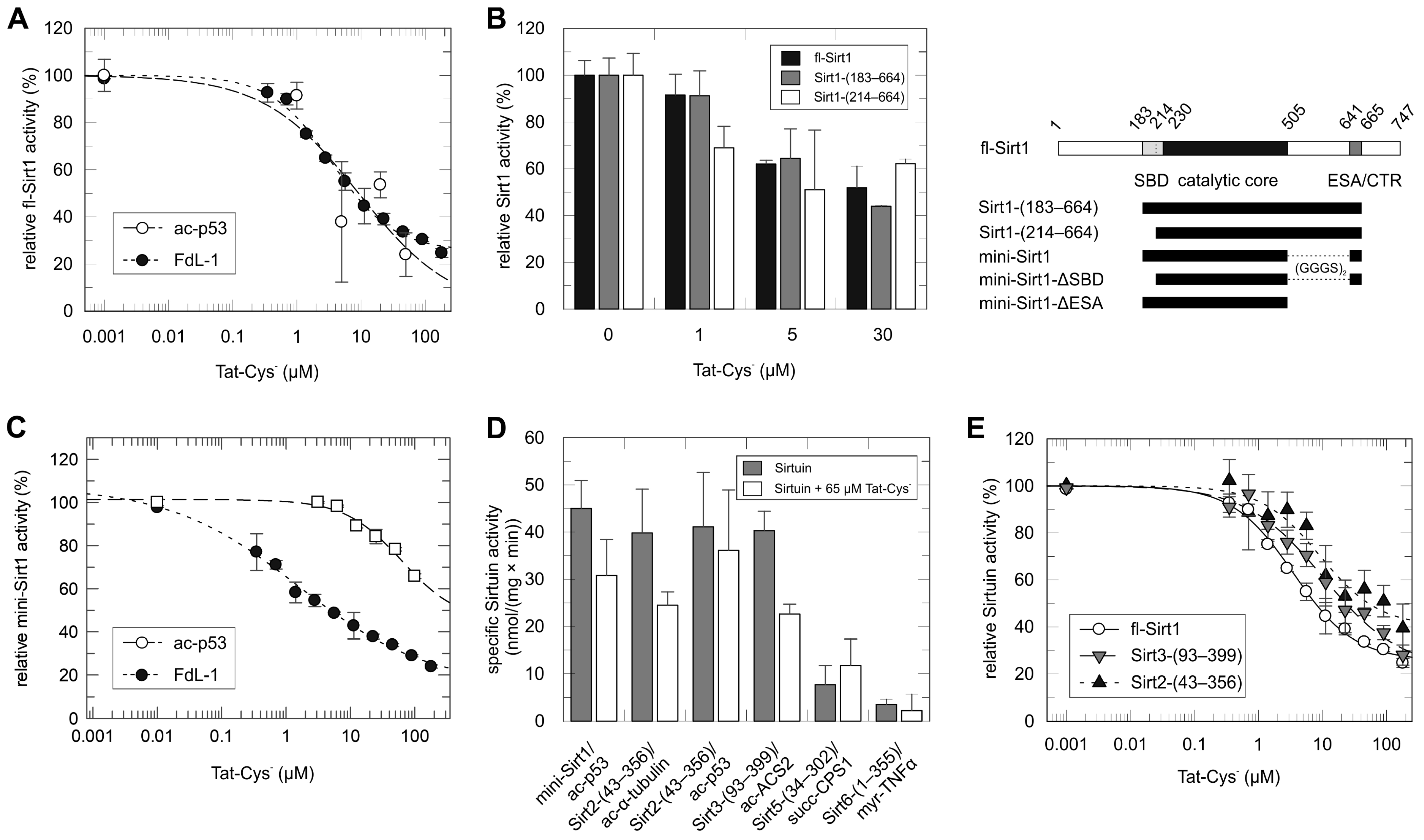
2.1. Tat Inhibits Directly the Catalytic Core of Sirt1 and Also Those of Other Sirtuin Isoforms
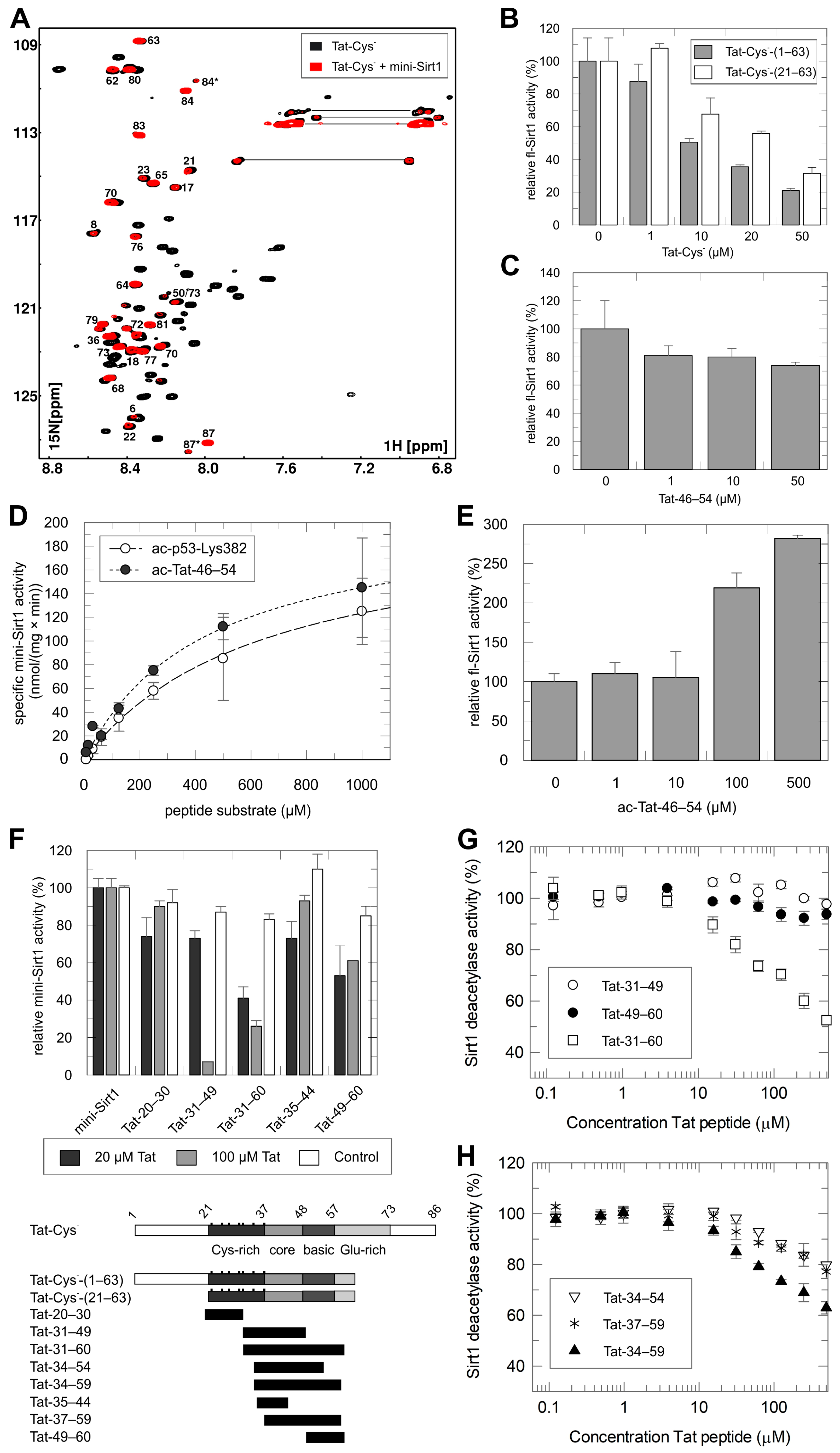
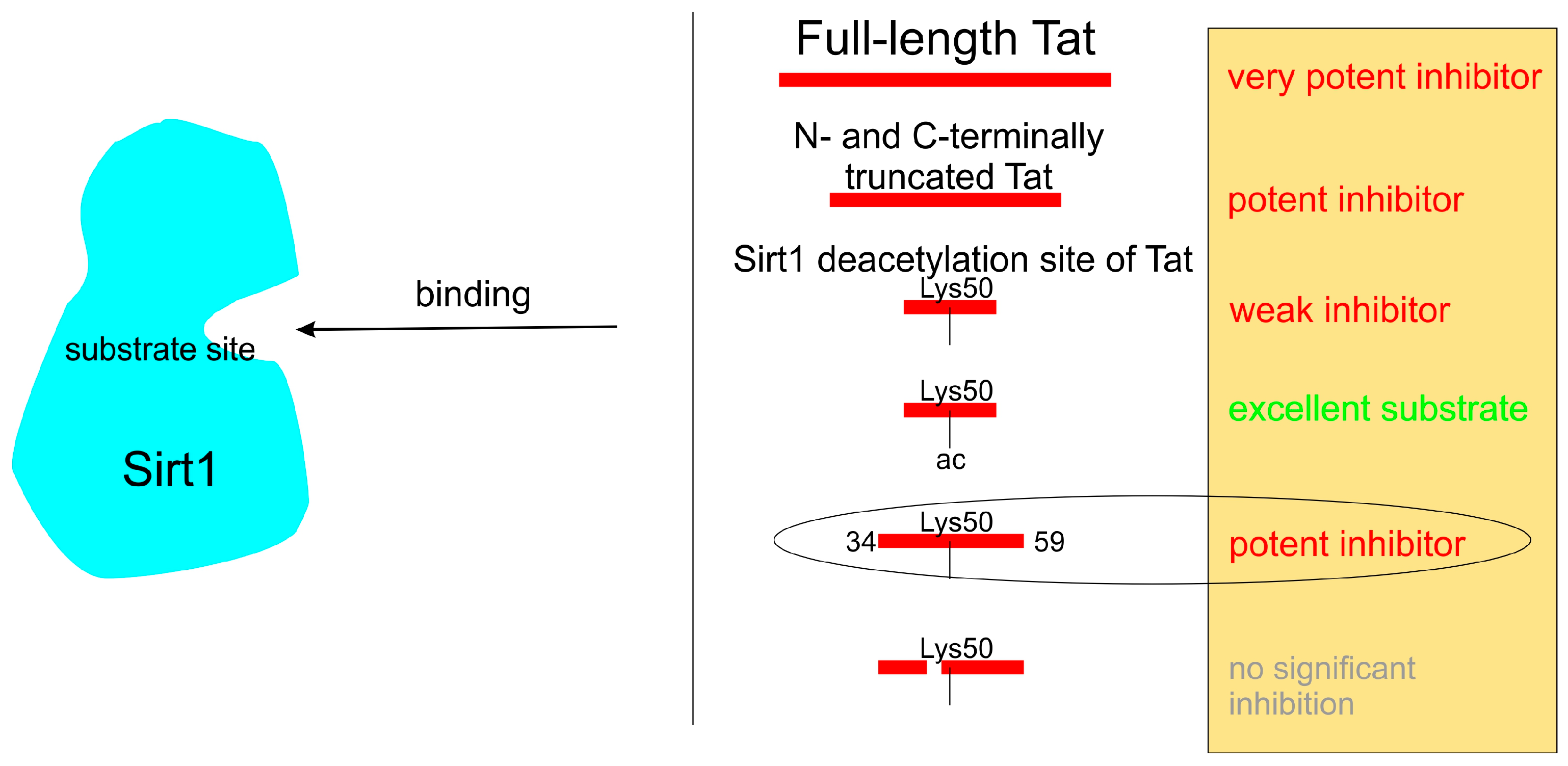
2.2. Mapping of the Tat Region Inhibiting Sirt1
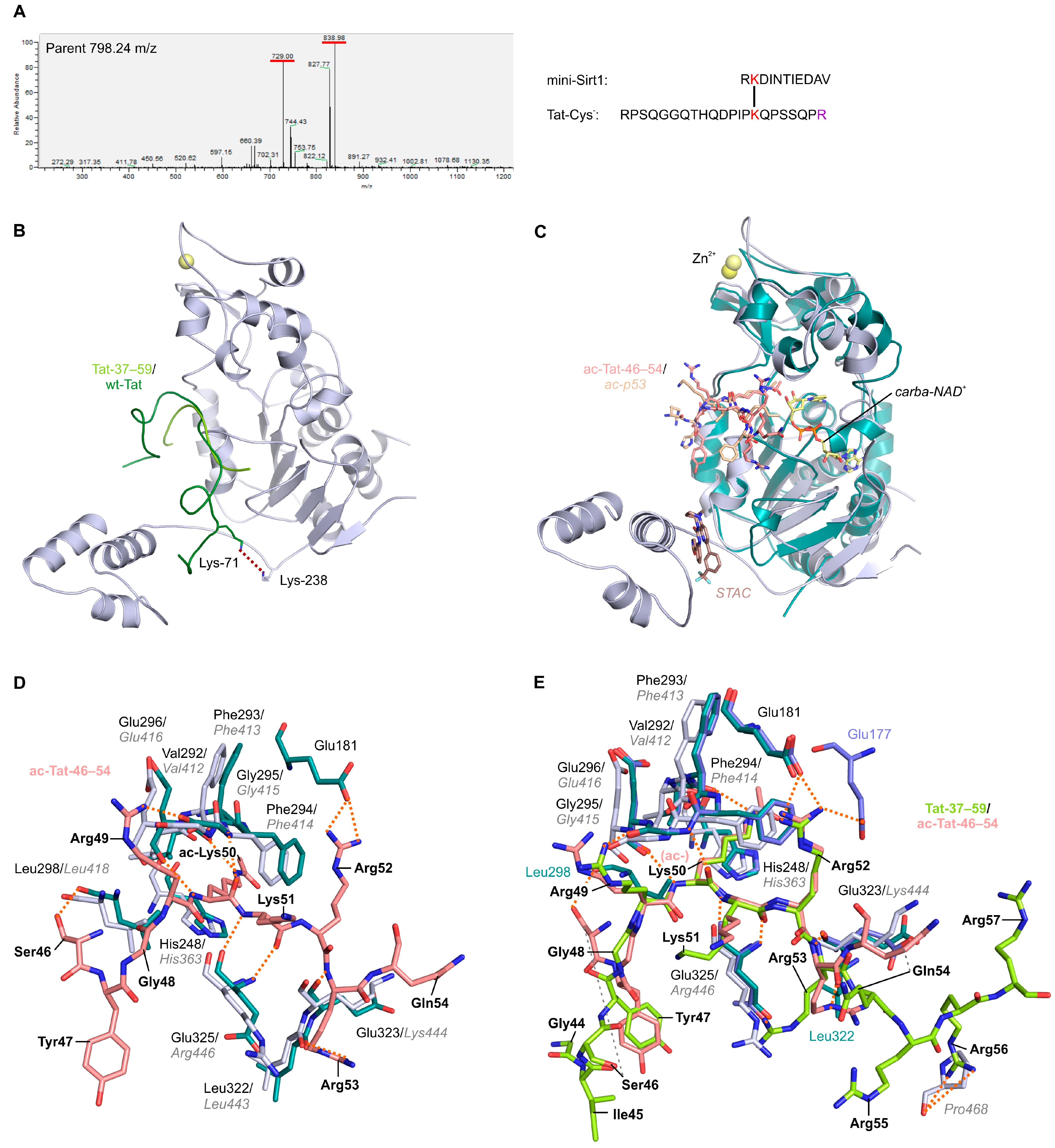
2.3. Structural Basis of Tat-Dependent Sirtuin Inhibition
3. Discussion
4. Conclusions
5. Methods
5.1. Chemicals
5.2. Expression and Purification of Proteins
5.3. NMR Spectroscopy
5.4. Peptide- and FdL-Based Sirtuin Activity Assays
5.5. Crosslinking and Mass Spectrometric Analysis
5.6. Crystallization and Structure Determination of Sirtuin Complexes
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AIDS | acquired immunodeficiency syndrome |
| ARM | arginine-rich motif |
| AROS | active regulator of Sirt1 |
| Dbc1 | deleted in breast cancer 1 |
| DSSO | disuccinimidyl sulfoxide |
| ESA | essential for Sirt1 activity |
| FdL | Fluor-de-Lys |
| fl-Sirt1 | full-length sirtuin 1 |
| GDH | glutamate dehydrogenase |
| HIC1 | hypermethylated in cancer 1 |
| HIV1 | human immunodeficiency virus 1 |
| NAD+ | nicotinamide adenine dinucleotide |
| NAM | nicotinamide |
| PCAF | p300/CBP-associated factor |
| PMSF | phenylmethylsulfonyl fluoride |
| p-TEFb | positive-acting transcription elongation factor b |
| SBD | STAC-binding domain |
| STAC | sirtuin-activating compound |
| TAR | transactivation response |
| Tat | transactivator of transcription |
References
- Morris, B.J. Seven sirtuins for seven deadly diseases of aging. Free. Radic. Biol. Med. 2013, 56, 133–171. [Google Scholar] [CrossRef] [PubMed]
- Klatzmann, D.; Champagne, E.; Chamaret, S.; Gruest, J.; Guetard, D.; Hercend, T.; Gluckman, J.C.; Montagnier, L. T-lymphocyte T4 molecule behaves as the receptor for human retrovirus LAV. Nature 1984, 312, 767–768. [Google Scholar] [CrossRef] [PubMed]
- Barre-Sinoussi, F.; Chermann, J.C.; Rey, F.; Nugeyre, M.T.; Chamaret, S.; Gruest, J.; Dauguet, C.; Axler-Blin, C.; Vezinet-Brun, F.; Rouzioux, C.; et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 1983, 220, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Dayton, A.I.; Sodroski, J.G.; Rosen, C.A.; Goh, W.C.; Haseltine, W.A. The trans-activator gene of the human T cell lymphotropic virus type III is required for replication. Cell 1986, 44, 941–947. [Google Scholar] [CrossRef] [PubMed]
- Clark, E.; Nava, B.; Caputi, M. Tat is a multifunctional viral protein that modulates cellular gene expression and functions. Oncotarget 2017, 8, 27569–27581. [Google Scholar] [CrossRef]
- Wong, K.; Sharma, A.; Awasthi, S.; Matlock, E.F.; Rogers, L.; Van Lint, C.; Skiest, D.J.; Burns, D.K.; Harrod, R. HIV-1 Tat interactions with p300 and PCAF transcriptional coactivators inhibit histone acetylation and neurotrophin signaling through CREB. J. Biol. Chem. 2005, 280, 9390–9399. [Google Scholar] [CrossRef]
- Kwon, H.S.; Ott, M. The ups and downs of SIRT1. Trends Biochem. Sci. 2008, 33, 517–525. [Google Scholar] [CrossRef]
- Pagans, S.; Pedal, A.; North, B.J.; Kaehlcke, K.; Marshall, B.L.; Dorr, A.; Hetzer-Egger, C.; Henklein, P.; Frye, R.; McBurney, M.W.; et al. SIRT1 regulates HIV transcription via Tat deacetylation. PLoS Biol. 2005, 3, e41. [Google Scholar] [CrossRef]
- Kwon, H.S.; Brent, M.M.; Getachew, R.; Jayakumar, P.; Chen, L.F.; Schnolzer, M.; McBurney, M.W.; Marmorstein, R.; Greene, W.C.; Ott, M. Human immunodeficiency virus type 1 Tat protein inhibits the SIRT1 deacetylase and induces T cell hyperactivation. Cell Host Microbe 2008, 3, 158–167. [Google Scholar] [CrossRef]
- Thakur, B.K.; Chandra, A.; Dittrich, T.; Welte, K.; Chandra, P. Inhibition of SIRT1 by HIV-1 viral protein Tat results in activation of p53 pathway. Biochem. Biophys. Res. Commun. 2012, 424, 245–250. [Google Scholar] [CrossRef]
- Sanders, B.D.; Jackson, B.; Marmorstein, R. Structural basis for sirtuin function: What we know and what we don’t. Biochim. Biophys. Acta 2010, 1804, 1604–1616. [Google Scholar] [CrossRef] [PubMed]
- Moniot, S.; Weyand, M.; Steegborn, C. Structures, substrates, and regulators of Mammalian sirtuins—Opportunities and challenges for drug development. Front. Pharmacol. 2012, 3, 16. [Google Scholar] [CrossRef] [PubMed]
- Rauh, D.; Fischer, F.; Gertz, M.; Lakshminarasimhan, M.; Bergbrede, T.; Aladini, F.; Kambach, C.; Becker, C.F.W.; Zerweck, J.; Schutkowski, M.; et al. An acetylome peptide microarray reveals specificities and deacetylation substrates for all human sirtuin isoforms. Nat. Commun. 2013, 4, 2327. [Google Scholar] [CrossRef] [PubMed]
- Moniot, S.; Schutkowski, M.; Steegborn, C. Crystal structure analysis of human Sirt2 and its ADP-ribose complex. J. Struct. Biol. 2013, 182, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Wei, W.; Jiang, Y.; Peng, H.; Cai, J.; Mao, C.; Dai, H.; Choy, W.; Bemis, J.E.; Jirousek, M.R.; et al. Crystal structures of human SIRT3 displaying substrate-induced conformational changes. J. Biol. Chem. 2009, 284, 24394–24405. [Google Scholar] [CrossRef] [PubMed]
- Sauve, A.A.; Wolberger, C.; Schramm, V.L.; Boeke, J.D. The biochemistry of sirtuins. Annu. Rev. Biochem. 2006, 75, 435–465. [Google Scholar] [CrossRef] [PubMed]
- Schutkowski, M.; Fischer, F.; Roessler, C.; Steegborn, C. New assays and approaches for discovery and design of Sirtuin modulators. Expert Opin. Drug Discov. 2014, 9, 183–199. [Google Scholar] [CrossRef]
- Tennen, R.I.; Berber, E.; Chua, K.F. Functional dissection of SIRT6: Identification of domains that regulate histone deacetylase activity and chromatin localization. Mech. Ageing Dev. 2010, 131, 185–192. [Google Scholar] [CrossRef]
- Gertz, M.; Steegborn, C. Using mitochondrial sirtuins as drug targets: Disease implications and available compounds. Cell. Mol. Life Sci. 2016, 73, 2871–2896. [Google Scholar] [CrossRef]
- Kang, H.; Suh, J.Y.; Jung, Y.S.; Jung, J.W.; Kim, M.K.; Chung, J.H. Peptide switch is essential for Sirt1 deacetylase activity. Mol. Cell 2011, 44, 203–213. [Google Scholar] [CrossRef]
- Lakshminarasimhan, M.; Curth, U.; Moniot, S.; Mosalaganti, S.; Raunser, S.; Steegborn, C. Molecular architecture of the human protein deacetylase Sirt1 and its regulation by AROS and resveratrol. Biosci. Rep. 2013, 33, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Sinclair, D.A.; Ellis, J.L.; Steegborn, C. Sirtuin activators and inhibitors: Promises, achievements, and challenges. Pharmacol. Ther. 2018, 188, 140–154. [Google Scholar] [CrossRef] [PubMed]
- Mujtaba, S.; He, Y.; Zeng, L.; Farooq, A.; Carlson, J.E.; Ott, M.; Verdin, E.; Zhou, M.M. Structural basis of lysine-acetylated HIV-1 Tat recognition by PCAF bromodomain. Mol. Cell 2002, 9, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Kaehlcke, K.; Dorr, A.; Hetzer-Egger, C.; Kiermer, V.; Henklein, P.; Schnoelzer, M.; Loret, E.; Cole, P.A.; Verdin, E.; Ott, M. Acetylation of Tat defines a cyclinT1-independent step in HIV transactivation. Mol. Cell 2003, 12, 167–176. [Google Scholar] [CrossRef]
- Boehm, M. Arbeiten zur Strukturaufklärung Immunologisch Relevanter Proteine: Bet v 1 und HIV-1 Tat. Ph.D. Thesis, University of Bayreuth, Bayreuth, Germany, 1998. [Google Scholar]
- Kaeberlein, M.; McDonagh, T.; Heltweg, B.; Hixon, J.; Westman, E.A.; Caldwell, S.D.; Napper, A.; Curtis, R.; DiStefano, P.S.; Fields, S.; et al. Substrate-specific activation of sirtuins by resveratrol. J. Biol. Chem. 2005, 280, 17038–17045. [Google Scholar] [CrossRef]
- Dai, H.; Case, A.W.; Riera, T.V.; Considine, T.; Lee, J.E.; Hamuro, Y.; Zhao, H.; Jiang, Y.; Sweitzer, S.M.; Pietrak, B.; et al. Crystallographic structure of a small molecule SIRT1 activator-enzyme complex. Nat. Commun. 2015, 6, 7645. [Google Scholar] [CrossRef]
- Yuan, H.; Marmorstein, R. Structural basis for sirtuin activity and inhibition. J. Biol. Chem. 2012, 287, 42428–42435. [Google Scholar] [CrossRef]
- Tahirov, T.H.; Babayeva, N.D.; Varzavand, K.; Cooper, J.J.; Sedore, S.C.; Price, D.H. Crystal structure of HIV-1 Tat complexed with human P-TEFb. Nature 2010, 465, 747–751. [Google Scholar] [CrossRef]
- Blazek, D.; Peterlin, B.M. Tat-SIRT1 tango. Mol. Cell 2008, 29, 539–540. [Google Scholar] [CrossRef]
- Smith, K.M.; Himiari, Z.; Tsimbalyuk, S.; Forwood, J.K. Structural Basis for Importin-alpha Binding of the Human Immunodeficiency Virus Tat. Sci. Rep. 2017, 7, 1650. [Google Scholar] [CrossRef]
- Schlicker, C.; Boanca, G.; Lakshminarasimhan, M.; Steegborn, C. Structure-based Development of Novel Sirtuin Inhibitors. Aging 2011, 3, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Suenkel, B.; Steegborn, C. Recombinant Preparation, Biochemical Analysis, and Structure Determination of Sirtuin Family Histone/Protein Deacylases. Methods Enzymol. 2016, 573, 183–208. [Google Scholar] [PubMed]
- You, W.; Rotili, D.; Li, T.M.; Kambach, C.; Meleshin, M.; Schutkowski, M.; Chua, K.F.; Mai, A.; Steegborn, C. Structural Basis of Sirtuin 6 Activation by Synthetic Small Molecules. Angew. Chem. Int. Ed. 2017, 56, 1007–1011. [Google Scholar] [CrossRef] [PubMed]
- Gurka, S. Diploma Thesis, University of Bayreuth, Bayreuth, Germany, 2002.
- Smith, B.C.; Hallows, W.C.; Denu, J.M. A continuous microplate assay for sirtuins and nicotinamide-producing enzymes. Anal. Biochem. 2009, 394, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Sparta, K.M.; Krug, M.; Heinemann, U.; Mueller, U.; Weiss, M.S. XDSAPP2.0. J. Appl. Cryst. 2016, 49, 1085–1092. [Google Scholar] [CrossRef]
- Winter, G.; Waterman, D.G.; Parkhurst, J.M.; Brewster, A.S.; Gildea, R.J.; Gerstel, M.; Fuentes-Montero, L.; Vollmar, M.; Michels-Clark, T.; Young, I.D.; et al. DIALS: Implementation and evaluation of a new integration package. Acta Crystallogr. Sect. D Struct. Biol. Crystallogr. 2018, 74, 85–97. [Google Scholar] [CrossRef]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr. Sect. D 2004, 60, 2126–2132. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sirt3/Tat-37–59 | Sirt3/ac-Tat-46–54 | |
|---|---|---|
| Resolution (a) | 54.24–1.95 (2.02–1.95) | 39.36–1.65 (1.71–1.65) |
| Space group | P21 | C2221 |
| Unit cell constants | a = 54.5 Å, b = 78.1 Å, c = 76.6 Å α = 90.0°, β = 96.1°, γ = 90.0° | a = 78.7 Å, b = 127.8 Å, c = 77.6 Å α = β = γ = 90.0° |
| Unique reflections (a) | 46,174 (4615) | 47,332 (4698) |
| Multiplicity (a) | 5.5 (5.5) | 11.2 (11.2) |
| Completeness (a) | 98.8% (98.9%) | 99.9% (99.9%) |
| I/σ(I) (a) | 12.0 (0.6) | 13.9 (0.9) |
| Rmeas (a) | 29.9% (160.4%) | 9.5% (22.6%) |
| CC1/2 (a) | 0.95 (0.45) | 1.00 (0.47) |
| Protein atoms | 4563 | 2244 |
| Ligand atoms | 2 | 1 |
| Solvent atoms | 472 | 287 |
| Rcryst/Rfree | 22.7%/27.6% | 18.1%/20.9% |
| Average B-factors | ||
| Protein | 38.6 | 35.6 |
| Ligands | 25.9 | 26.9 |
| Solvent | 41.3 | 47.1 |
| RMSD bond lengths | 0.002 Å | 0.017 Å |
| RMSD bond angles | 0.520° | 1.380° |
| Ramachandran | ||
| Favored | 97.2% | 98.9% |
| Allowed | 2.8% | 1.1% |
| Outliers | 0.0% | 0.0% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adolph, R.S.; Beck, E.; Schweimer, K.; Di Fonzo, A.; Weyand, M.; Rösch, P.; Wöhrl, B.M.; Steegborn, C. Molecular Mechanism of Sirtuin 1 Inhibition by Human Immunodeficiency Virus 1 Tat Protein. Life 2023, 13, 949. https://doi.org/10.3390/life13040949
Adolph RS, Beck E, Schweimer K, Di Fonzo A, Weyand M, Rösch P, Wöhrl BM, Steegborn C. Molecular Mechanism of Sirtuin 1 Inhibition by Human Immunodeficiency Virus 1 Tat Protein. Life. 2023; 13(4):949. https://doi.org/10.3390/life13040949
Chicago/Turabian StyleAdolph, Ramona S., Eileen Beck, Kristian Schweimer, Andrea Di Fonzo, Michael Weyand, Paul Rösch, Birgitta M. Wöhrl, and Clemens Steegborn. 2023. "Molecular Mechanism of Sirtuin 1 Inhibition by Human Immunodeficiency Virus 1 Tat Protein" Life 13, no. 4: 949. https://doi.org/10.3390/life13040949
APA StyleAdolph, R. S., Beck, E., Schweimer, K., Di Fonzo, A., Weyand, M., Rösch, P., Wöhrl, B. M., & Steegborn, C. (2023). Molecular Mechanism of Sirtuin 1 Inhibition by Human Immunodeficiency Virus 1 Tat Protein. Life, 13(4), 949. https://doi.org/10.3390/life13040949