

Exercise Improves the Coordination of the Mitochondrial Unfolded Protein Response and Mitophagy in Aging Skeletal Muscle

Abstract

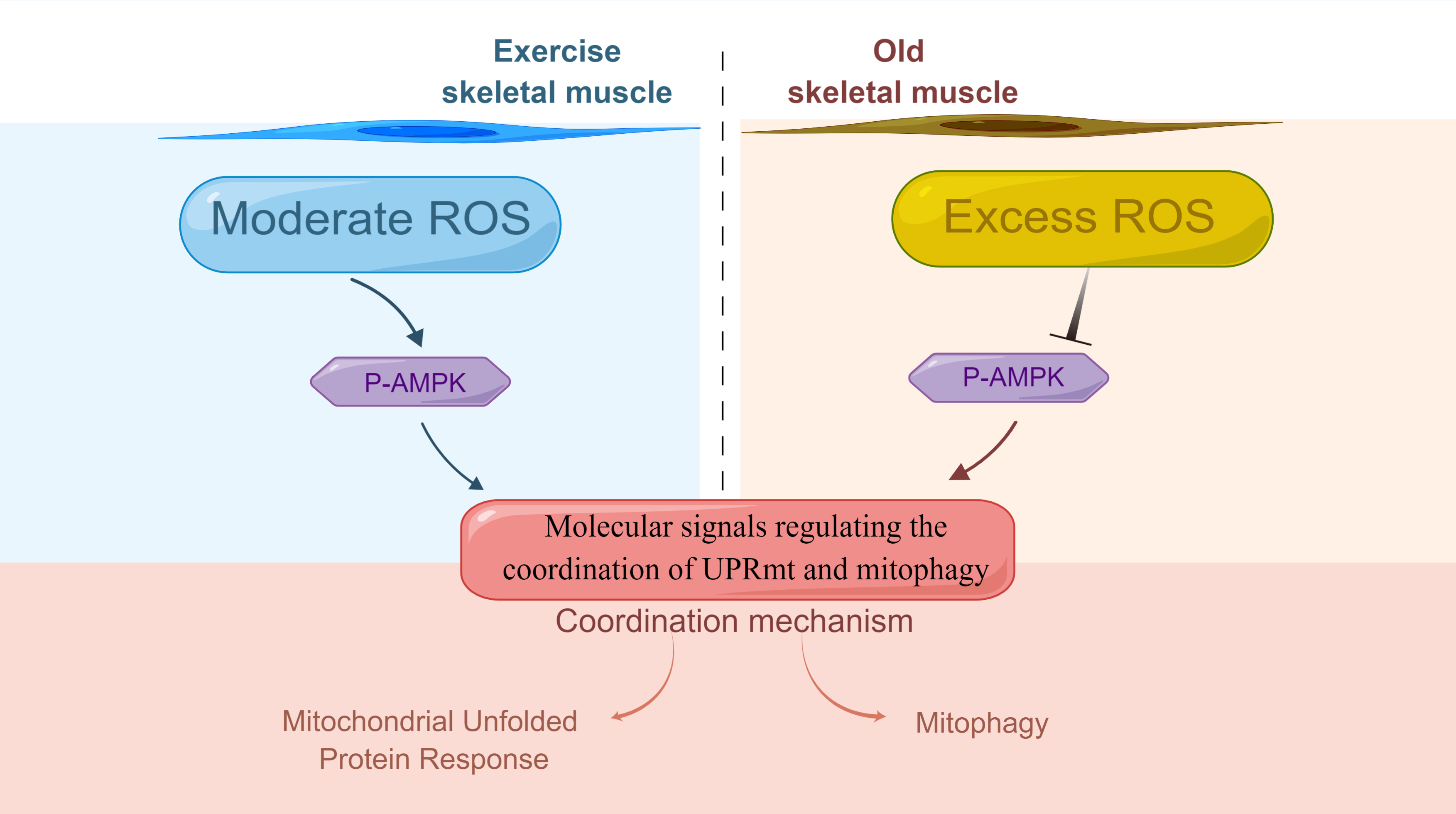{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. MQC
2.1. UPRmt
2.2. Mitophagy
3. Coordination Mechanism of the UPRmt and Mitophagy
3.1. Relationship between the UPRmt and Mitophagy
3.2. Molecular Signaling Regulates the Coordination of the UPRmt and Mitophagy
3.2.1. FUNDC1
3.2.2. CHCHD2
3.2.3. SIRT1
3.2.4. SIRT3
3.2.5. eIF2α
3.2.6. MOTS-c
3.2.7. FGF21
4. MQC Dysfunction in Aging Skeletal Muscle
4.1. UPRmt Abnormalities in Aging Skeletal Muscle
4.2. Mitophagy Aberrations in Aging Skeletal Muscle
4.3. The Coordination Mechanism in Aging Skeletal Muscles Is Impaired
5. Exercise Maintains Mitochondrial Health in Aging Skeletal Muscle by Enhancing the UPRmt and Mitophagy
5.1. Exercise and the UPRmt
5.2. Exercise and Mitophagy
5.3. Exercise Promotes the Coordination Mechanism of the UPRmt and Mitophagy
6. Exercise Induces ROS to Regulate the Coordination Mechanism of the UPRmt and Mitophagy
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carter, H.N.; Chen, C.C.; Hood, D.A. Mitochondria, muscle health, and exercise with advancing age. Physiology 2015, 30, 208–223. [Google Scholar] [CrossRef] [PubMed]
- Mrowka, R.; Westphal, A. Skeletal muscle in the fight against chronic diseases. Acta Physiol. 2018, 223, e13086. [Google Scholar] [CrossRef] [PubMed]
- Dennison, E.M.; Sayer, A.A.; Cooper, C. Epidemiology of sarcopenia and insight into possible therapeutic targets. Nat. Rev. Rheumatol. 2017, 13, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Wu, J.; Yang, Z.; Wang, X.; Zhang, D.; Ma, J. Mitochondrial quality control in cardiac ischemia/reperfusion injury: New insights into mechanisms and implications. Cell Biol. Toxicol. 2022, 39, 33–51. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Fan, Y.B.; Tao, X.H.; Pan, W.L.; Wu, Y.X.; Wang, X.H.; He, Y.Q.; Xiao, W.F.; Li, Y.S. Mitochondrial Quality Control in Sarcopenia: Updated Overview of Mechanisms and Interventions. Aging Dis. 2021, 12, 2016–2030. [Google Scholar] [CrossRef]
- Zhang, Y.; Oliveira, A.N.; Hood, D.A. The intersection of exercise and aging on mitochondrial protein quality control. Exp. Gerontol. 2020, 131, 110824. [Google Scholar] [CrossRef]
- Hood, D.A.; Memme, J.M.; Oliveira, A.N.; Triolo, M. Maintenance of Skeletal Muscle Mitochondria in Health, Exercise, and Aging. Annu. Rev. Physiol. 2019, 81, 19–41. [Google Scholar] [CrossRef]
- Slavin, M.B.; Memme, J.M.; Oliveira, A.N.; Moradi, N.; Hood, D.A. Regulatory networks coordinating mitochondrial quality control in skeletal muscle. Am. J. Physiol. Cell Physiol. 2022, 322, C913–C926. [Google Scholar] [CrossRef]
- Xin, N.; Durieux, J.; Yang, C.; Wolff, S.; Kim, H.E.; Dillin, A. The UPRmt preserves mitochondrial import to extend lifespan. J. Cell Biol. 2022, 221, e202201071. [Google Scholar] [CrossRef]
- Roca-Portoles, A.; Tait, S.W.G. Mitochondrial quality control: From molecule to organelle. Cell Mol. Life Sci. 2021, 78, 3853–3866. [Google Scholar] [CrossRef]
- Andreux, P.A.; Houtkooper, R.H.; Auwerx, J. Pharmacological approaches to restore mitochondrial function. Nat. Rev. Drug Discov. 2013, 12, 465–483. [Google Scholar] [CrossRef]
- Urbina-Varela, R.; Castillo, N.; Videla, L.A.; Del Campo, A. Impact of Mitophagy and Mitochondrial Unfolded Protein Response as New Adaptive Mechanisms Underlying Old Pathologies: Sarcopenia and Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2020, 21, 7704. [Google Scholar] [CrossRef]
- Cordeiro, A.V.; Peruca, G.F.; Braga, R.R.; Brícola, R.S.; Lenhare, L.; Silva, V.R.R.; Anaruma, C.P.; Katashima, C.K.; Crisol, B.M.; Barbosa, L.T.; et al. High-intensity exercise training induces mitonuclear imbalance and activates the mitochondrial unfolded protein response in the skeletal muscle of aged mice. GeroScience 2021, 43, 1513–1518. [Google Scholar] [CrossRef]
- Leduc-Gaudet, J.P.; Reynaud, O.; Hussain, S.N.; Gouspillou, G. Parkin overexpression protects from ageing-related loss of muscle mass and strength. J. Physiol. 2019, 597, 1975–1991. [Google Scholar] [CrossRef]
- Wang, S.; Chen, Y.; Li, X.; Zhang, W.; Liu, Z.; Wu, M.; Pan, Q.; Liu, H. Emerging role of transcription factor EB in mitochondrial quality control. Biomed. Pharm. 2020, 128, 110272. [Google Scholar] [CrossRef]
- Zhu, H.; Toan, S.; Mui, D.; Zhou, H. Mitochondrial quality surveillance as a therapeutic target in myocardial infarction. Acta Physiol. 2021, 231, e13590. [Google Scholar] [CrossRef]
- Katiyar, A.; Fujimoto, M.; Tan, K.; Kurashima, A.; Srivastava, P.; Okada, M.; Takii, R.; Nakai, A. HSF1 is required for induction of mitochondrial chaperones during the mitochondrial unfolded protein response. FEBS Open Bio. 2020, 10, 1135–1148. [Google Scholar] [CrossRef]
- Horibe, T.; Hoogenraad, N.J. The chop gene contains an element for the positive regulation of the mitochondrial unfolded protein response. PLoS ONE 2007, 2, e835. [Google Scholar] [CrossRef]
- Fawcett, T.W.; Martindale, J.L.; Guyton, K.Z.; Hai, T.; Holbrook, N.J. Complexes containing activating transcription factor (ATF)/cAMP-responsive-element-binding protein (CREB) interact with the CCAAT/enhancer-binding protein (C/EBP)-ATF composite site to regulate Gadd153 expression during the stress response. Biochem. J. 1999, 339, 135–141. [Google Scholar] [CrossRef]
- Yamazaki, T.; Ohmi, A.; Kurumaya, H.; Kato, K.; Abe, T.; Yamamoto, H.; Nakanishi, N.; Okuyama, R.; Umemura, M.; Kaise, T.; et al. Regulation of the human CHOP gene promoter by the stress response transcription factor ATF5 via the AARE1 site in human hepatoma HepG2 cells. Life Sci. 2010, 87, 294–301. [Google Scholar] [CrossRef]
- Fiorese, C.J.; Schulz, A.M.; Lin, Y.F.; Rosin, N.; Pellegrino, M.W.; Haynes, C.M. The Transcription Factor ATF5 Mediates a Mammalian Mitochondrial UPR. Curr. Biol. 2016, 26, 2037–2043. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Wang, J.; Levichkin, I.V.; Stasinopoulos, S.; Ryan, M.T.; Hoogenraad, N.J. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002, 21, 4411–4419. [Google Scholar] [CrossRef] [PubMed]
- Papa, L.; Germain, D. Estrogen receptor mediates a distinct mitochondrial unfolded protein response. J. Cell Sci. 2011, 124, 1396–1402. [Google Scholar] [CrossRef] [PubMed]
- Rangarajan, P.; Karthikeyan, A.; Lu, J.; Ling, E.A.; Dheen, S.T. Sirtuin 3 regulates Foxo3a-mediated antioxidant pathway in microglia. Neuroscience 2015, 311, 398–414. [Google Scholar] [CrossRef] [PubMed]
- Shpilka, T.; Haynes, C.M. The mitochondrial UPR: Mechanisms, physiological functions and implications in ageing. Nat. Rev. Mol. Cell Bio. 2018, 19, 109–120. [Google Scholar] [CrossRef]
- Queliconi, B.B.; Kojima, W.; Kimura, M.; Imai, K.; Udagawa, C.; Motono, C.; Hirokawa, T.; Tashiro, S.; Caaveiro, J.M.M.; Tsumoto, K.; et al. Unfolding is the driving force for mitochondrial import and degradation of the Parkinson’s disease-related protein DJ-1. J. Cell Sci. 2021, 134, jcs258653. [Google Scholar] [CrossRef]
- Montava-Garriga, L.; Ganley, I.G. Outstanding questions in mitophagy: What we do and do not know. J. Mol. Biol. 2020, 432, 206–230. [Google Scholar] [CrossRef]
- Gustafsson, Å.B.; Dorn, G.W., 2nd. Evolving and expanding the roles of mitophagy as a homeostatic and pathogenic process. Physiol. Rev. 2019, 99, 853–892. [Google Scholar] [CrossRef]
- Miyazaki, N.; Shiratori, R.; Oshima, T.; Zhang, Z.; Valencia, R.; Kranrod, J.; Fang, L.; Seubert, J.M.; Ito, K.; Aoki, S. PINK1-dependent and Parkin-independent mitophagy is involved in reprogramming of glycometabolism in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2022, 625, 167–173. [Google Scholar] [CrossRef]
- Esteban-Martínez, L.; Boya, P. BNIP3L/NIX-dependent mitophagy regulates cell differentiation via metabolic reprogramming. Autophagy 2018, 14, 915–917. [Google Scholar] [CrossRef]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef]
- McWilliams, T.G.; Prescott, A.R.; Allen, G.F.; Tamjar, J.; Munson, M.J.; Thomson, C.; Muqit, M.M.; Ganley, I.G. mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J. Cell Biol. 2016, 214, 333–345. [Google Scholar] [CrossRef]
- Sun, N.; Yun, J.; Liu, J.; Malide, D.; Liu, C.; Rovira, I.I.; Holmström, K.M.; Fergusson, M.M.; Yoo, Y.H.; Combs, C.A.; et al. Measuring In Vivo Mitophagy. Mol. Cell 2015, 60, 685–696. [Google Scholar] [CrossRef]
- Stotland, A.; Gottlieb, R.A. α-MHC MitoTimer mouse: In vivo mitochondrial turnover model reveals remarkable mitochondrial heterogeneity in the heart. J. Mol. Cell Cardiol. 2016, 90, 53–58. [Google Scholar] [CrossRef]
- Wang, Y.; Jasper, H.; Toan, S.; Muid, D.; Chang, X.; Zhou, H. Mitophagy coordinates the mitochondrial unfolded protein response to attenuate inflammation-mediated myocardial injury. Redox Biol. 2021, 45, 102049. [Google Scholar] [CrossRef]
- Han, K.; Hassanzadeh, S.; Singh, K.; Menazza, S.; Nguyen, T.T.; Stevens, M.V.; Nguyen, A.; San, H.; Anderson, S.A.; Lin, Y.; et al. Parkin regulation of CHOP modulates susceptibility to cardiac endoplasmic reticulum stress. Sci. Rep. 2017, 7, 2093. [Google Scholar] [CrossRef]
- Memme, J.M.; Oliveira, A.N.; Hood, D.A. p53 regulates skeletal muscle mitophagy and mitochondrial quality control following denervation-induced muscle disuse. J. Biol. Chem. 2022, 298, 101540. [Google Scholar] [CrossRef]
- Lee, H.; Ha, T.Y.; Jung, C.H.; Nirmala, F.S.; Park, S.Y.; Huh, Y.H.; Ahn, J. Mitochondrial dysfunction in skeletal muscle contributes to the development of acute insulin resistance in mice. J. Cachexia Sarcopenia Muscle 2021, 12, 1925–1939. [Google Scholar] [CrossRef]
- Jin, S.M.; Youle, R.J. The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/Parkin-mediated mitophagy of polarized mitochondria. Autophagy 2013, 9, 1750–1757. [Google Scholar] [CrossRef]
- Berry, B.J.; Nieves, T.O.; Wojtovich, A.P. Decreased Mitochondrial Membrane Potential Activates the Mitochondrial Unfolded Protein Response. Micropublication Biol. 2021, 2021. [Google Scholar] [CrossRef]
- Quiles, J.M.; Gustafsson, Å.B. Mitochondrial Quality Control and Cellular Proteostasis: Two Sides of the Same Coin. Front. Physiol. 2020, 11, 515. [Google Scholar] [CrossRef] [PubMed]
- Hauffe, R.; Rath, M.; Schell, M.; Ritter, K.; Kappert, K.; Deubel, S.; Ott, C.; Jähnert, M.; Jonas, W.; Schürmann, A.; et al. HSP60 reduction protects against diet-induced obesity by modulating energy metabolism in adipose tissue. Mol. Metab. 2021, 53, 101276. [Google Scholar] [CrossRef] [PubMed]
- Burbulla, L.F.; Fitzgerald, J.C.; Stegen, K.; Westermeier, J.; Thost, A.K.; Kato, H.; Mokranjac, D.; Sauerwald, J.; Martins, L.M.; Woitalla, D.; et al. Mitochondrial proteolytic stress induced by loss of mortalin function is rescued by Parkin and PINK1. Cell Death Dis. 2014, 5, e1180. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.E.; Andrews, L.A.; Burman, J.L.; Lin, W.Y.; Pallanck, L.J. PINK1-Parkin pathway activity is regulated by degradation of PINK1 in the mitochondrial matrix. PLoS Genet. 2014, 10, e1004279. [Google Scholar] [CrossRef]
- Xu, Z.; Fu, T.; Guo, Q.; Zhou, D.; Sun, W.; Zhou, Z.; Chen, X.; Zhang, J.; Liu, L.; Xiao, L.; et al. Disuse-associated loss of the protease LONP1 in muscle impairs mitochondrial function and causes reduced skeletal muscle mass and strength. Nat. Commun. 2022, 13, 894. [Google Scholar] [CrossRef]
- Gitschlag, B.L.; Kirby, C.S.; Samuels, D.C.; Gangula, R.D.; Mallal, S.A.; Patel, M.R. Homeostatic Responses Regulate Selfish Mitochondrial Genome Dynamics in C. elegans. Cell Metab. 2016, 24, 91–103. [Google Scholar] [CrossRef]
- Cooper, J.F.; Machiela, E.; Dues, D.J.; Spielbauer, K.K.; Senchuk, M.M.; Van Raamsdonk, J.M. Activation of the mitochondrial unfolded protein response promotes longevity and dopamine neuron survival in Parkinson’s disease models. Sci. Rep. 2017, 7, 16441. [Google Scholar] [CrossRef]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef]
- Wu, H.; Xue, D.; Chen, G.; Han, Z.; Huang, L.; Zhu, C.; Wang, X.; Jin, H.; Wang, J.; Zhu, Y.; et al. The BCL2L1 and PGAM5 axis defines hypoxia-induced receptor-mediated mitophagy. Autophagy 2014, 10, 1712–1725. [Google Scholar] [CrossRef]
- Li, W.; Li, Y.; Siraj, S.; Jin, H.; Fan, Y.; Yang, X.; Huang, X.; Wang, X.; Wang, J.; Liu, L.; et al. FUN14 Domain-Containing 1-Mediated Mitophagy Suppresses Hepatocarcinogenesis by Inhibition of Inflammasome Activation in Mice. Hepatology 2019, 69, 604–621. [Google Scholar] [CrossRef]
- Ji, H.; Wang, J.; Muid, D.; Song, W.; Jiang, Y.; Zhou, H. FUNDC1 activates the mitochondrial unfolded protein response to preserve mitochondrial quality control in cardiac ischemia/reperfusion injury. Cell Signal 2022, 92, 110249. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.T.; Lim, Y.; McCall, M.N.; Huang, K.T.; Haynes, C.M.; Nehrke, K.; Brookes, P.S. Cardioprotection by the mitochondrial unfolded protein response requires ATF5. Am. J. Physiol.-Heart C 2019, 317, H472–H478. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xue, Y.; Xu, X.; Wang, G.; Liu, Y.; Wu, H.; Li, W.; Wang, Y.; Chen, Z.; Zhang, W.; et al. A mitochondrial FUNDC1/HSC70 interaction organizes the proteostatic stress response at the risk of cell morbidity. EMBO J. 2019, 38, e98786. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Agarwal, E.; Bertolini, I.; Seo, J.H.; Caino, M.C.; Ghosh, J.C.; Kossenkov, A.V.; Liu, Q.; Tang, H.Y.; Goldman, A.R.; et al. The mitophagy effector FUNDC1 controls mitochondrial reprogramming and cellular plasticity in cancer cells. Sci. Signal 2020, 13, eaaz8240. [Google Scholar] [CrossRef]
- Aras, S.; Bai, M.; Lee, I.; Springett, R.; Hüttemann, M.; Grossman, L.I. MNRR1 (formerly CHCHD2) is a bi-organellar regulator of mitochondrial metabolism. Mitochondrion 2015, 20, 43–51. [Google Scholar] [CrossRef]
- Aras, S.; Purandare, N.; Gladyck, S.; Somayajulu-Nitu, M.; Zhang, K.; Wallace, D.C.; Grossman, L.I. Mitochondrial Nuclear Retrograde Regulator 1 (MNRR1) rescues the cellular phenotype of MELAS by inducing homeostatic mechanisms. Proc. Natl. Acad. Sci. USA 2020, 117, 32056–32065. [Google Scholar] [CrossRef]
- Mouchiroud, L.; Houtkooper, R.H.; Moullan, N.; Katsyuba, E.; Ryu, D.; Cantó, C.; Mottis, A.; Jo, Y.S.; Viswanathan, M.; Schoonjans, K.; et al. The NAD(+)/Sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell 2013, 154, 430–441. [Google Scholar] [CrossRef]
- Zhang, B.; Tan, Y.; Zhang, Z.; Feng, P.; Ding, W.; Wang, Q.; Liang, H.; Duan, W.; Wang, X.; Yu, S.; et al. Novel PGC-1α/ATF5 axis partly activates UPR(mt) and mediates cardioprotective role of tetrahydrocurcumin in pathological cardiac hypertrophy. Oxid. Med. Cell Longev. 2020, 2020, 9187065. [Google Scholar] [CrossRef]
- Song, S.B.; Jang, S.Y.; Kang, H.T.; Wei, B.; Jeoun, U.W.; Yoon, G.S.; Hwang, E.S. Modulation of mitochondrial membrane potential and ROS generation by nicotinamide in a manner independent of SIRT1 and mitophagy. Mol. Cells 2017, 40, 503–514. [Google Scholar] [CrossRef]
- Kume, S.; Uzu, T.; Horiike, K.; Chin-Kanasaki, M.; Isshiki, K.; Araki, S.; Sugimoto, T.; Haneda, M.; Kashiwagi, A.; Koya, D. Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J. Clin. Invest. 2010, 120, 1043–1055. [Google Scholar] [CrossRef]
- Zhang, J.; Xiang, H.; Liu, J.; Chen, Y.; He, R.R.; Liu, B. Mitochondrial Sirtuin 3: New emerging biological function and therapeutic target. Theranostics 2020, 10, 8315–8342. [Google Scholar] [CrossRef]
- Diao, Z.; Ji, Q.; Wu, Z.; Zhang, W.; Cai, Y.; Wang, Z.; Hu, J.; Liu, Z.; Wang, Q.; Bi, S.; et al. SIRT3 consolidates heterochromatin and counteracts senescence. Nucleic Acids Res. 2021, 49, 4203–4219. [Google Scholar] [CrossRef]
- Gariani, K.; Menzies, K.J.; Ryu, D.; Wegner, C.J.; Wang, X.; Ropelle, E.R.; Moullan, N.; Zhang, H.; Perino, A.; Lemos, V.; et al. Eliciting the mitochondrial unfolded protein response by nicotinamide adenine dinucleotide repletion reverses fatty liver disease in mice. Hepatology 2016, 63, 1190–1204. [Google Scholar] [CrossRef]
- Lu, Z.; Chen, Y.; Aponte, A.M.; Battaglia, V.; Gucek, M.; Sack, M.N. Prolonged fasting identifies heat shock protein 10 as a Sirtuin 3 substrate: Elucidating a new mechanism linking mitochondrial protein acetylation to fatty acid oxidation enzyme folding and function. J. Biol. Chem. 2015, 290, 2466–2476. [Google Scholar] [CrossRef]
- Yu, W.; Gao, B.; Li, N.; Wang, J.; Qiu, C.; Zhang, G.; Liu, M.; Zhang, R.; Li, C.; Ji, G.; et al. Sirt3 deficiency exacerbates diabetic cardiac dysfunction: Role of Foxo3A-Parkin-mediated mitophagy. BBA-Mol. Basis Dis. 2017, 1863, 1973–1983. [Google Scholar] [CrossRef]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef]
- Li, Y.; Ruan, D.Y.; Jia, C.C.; Zheng, J.; Wang, G.Y.; Zhao, H.; Yang, Q.; Liu, W.; Yi, S.H.; Li, H.; et al. Aging aggravates hepatic ischemia-reperfusion injury in mice by impairing mitophagy with the involvement of the EIF2α-parkin pathway. Aging 2018, 10, 1902–1920. [Google Scholar] [CrossRef]
- Shen, Z.; Liu, P.; Sun, Q.; Li, Y.; Acharya, R.; Li, X.; Sun, C. FTO inhibits UPR(mt)-induced apoptosis by activating JAK2/STAT3 pathway and reducing m6A level in adipocytes. Apoptosis 2021, 26, 474–487. [Google Scholar] [CrossRef]
- McCarty, M.F.; Lerner, A. Perspective: Low Risk of Parkinson’s Disease in Quasi-Vegan Cultures May Reflect GCN2-Mediated Upregulation of Parkin. Adv. Nutr. 2021, 12, 355–362. [Google Scholar] [CrossRef]
- Reynolds, J.C.; Lai, R.W.; Woodhead, J.S.T.; Joly, J.H.; Mitchell, C.J.; Cameron-Smith, D.; Lu, R.; Cohen, P.; Graham, N.A.; Benayoun, B.A.; et al. MOTS-c is an exercise-induced mitochondrial-encoded regulator of age-dependent physical decline and muscle homeostasis. Nat. Commun. 2021, 12, 470. [Google Scholar] [CrossRef]
- Li, Q.; Lu, H.; Hu, G.; Ye, Z.; Zhai, D.; Yan, Z.; Wang, L.; Xiang, A.; Lu, Z. Earlier changes in mice after D-galactose treatment were improved by mitochondria derived small peptide MOTS-c. Biochem. Biophys. Res. Commun. 2019, 513, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Flippo, K.H.; Potthoff, M.J. Metabolic Messengers: FGF21. Nat. Metab. 2021, 3, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Xu, Z.; Zhou, D.; Fu, T.; Wang, W.; Sun, W.; Xiao, L.; Liu, L.; Ding, C.; Yin, Y.; et al. Mitochondrial proteostasis stress in muscle drives a long-range protective response to alleviate dietary obesity independently of ATF4. Sci. Adv. 2022, 8, eabo0340. [Google Scholar] [CrossRef] [PubMed]
- Fu, T.; Xu, Z.; Liu, L.; Guo, Q.; Wu, H.; Liang, X.; Zhou, D.; Xiao, L.; Liu, L.; Liu, Y.; et al. Mitophagy Directs Muscle-Adipose Crosstalk to Alleviate Dietary Obesity. Cell Rep. 2018, 23, 1357–1372. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.M.; Albarado, D.C.; Coco, L.G.; Spann, R.A.; Khan, M.S.; Qualls-Creekmore, E.; Burk, D.H.; Burke, S.J.; Collier, J.J.; Yu, S.; et al. FGF21 is required for protein restriction to extend lifespan and improve metabolic health in male mice. Nat. Commun. 2022, 13, 1897. [Google Scholar] [CrossRef]
- Oost, L.J.; Kustermann, M.; Armani, A.; Blaauw, B.; Romanello, V. Fibroblast growth factor 21 controls mitophagy and muscle mass. J. Cachexia Sarcopenia Muscle 2019, 10, 630–642. [Google Scholar] [CrossRef]
- Wang, X.M.; Xiao, H.; Liu, L.L.; Cheng, D.; Li, X.J.; Si, L.Y. FGF21 represses cerebrovascular aging via improving mitochondrial biogenesis and inhibiting p53 signaling pathway in an AMPK-dependent manner. Exp. Cell Res. 2016, 346, 147–156. [Google Scholar] [CrossRef]
- Ljubicic, V.; Joseph, A.M.; Adhihetty, P.J.; Huang, J.H.; Saleem, A.; Uguccioni, G.; Hood, D.A. Molecular basis for an attenuated mitochondrial adaptive plasticity in aged skeletal muscle. Aging 2009, 1, 818–830. [Google Scholar] [CrossRef]
- Chabi, B.; Ljubicic, V.; Menzies, K.J.; Huang, J.H.; Saleem, A.; Hood, D.A. Mitochondrial function and apoptotic susceptibility in aging skeletal muscle. Aging Cell 2008, 7, 2–12. [Google Scholar] [CrossRef]
- Sebastián, D.; Sorianello, E.; Segalés, J.; Irazoki, A.; Ruiz-Bonilla, V.; Sala, D.; Planet, E.; Berenguer-Llergo, A.; Muñoz, J.P.; Sánchez-Feutrie, M.; et al. Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J. 2016, 35, 1677–1693. [Google Scholar] [CrossRef]
- Leduc-Gaudet, J.P.; Picard, M.; St-Jean Pelletier, F.; Sgarioto, N.; Auger, M.J.; Vallée, J.; Robitaille, R.; St-Pierre, D.H.; Gouspillou, G. Mitochondrial morphology is altered in atrophied skeletal muscle of aged mice. Oncotarget 2015, 6, 17923–17937. [Google Scholar] [CrossRef]
- Cordeiro, A.V.; Brícola, R.S.; Braga, R.R.; Lenhare, L.; Silva, V.R.R.; Anaruma, C.P.; Katashima, C.K.; Crisol, B.M.; Simabuco, F.M.; Silva, A.S.R.; et al. Aerobic exercise training induces the mitonuclear imbalance and UPRmt in the skeletal muscle of aged mice. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 2258–2261. [Google Scholar] [CrossRef]
- Migliavacca, E.; Tay, S.K.H.; Patel, H.P.; Sonntag, T.; Civiletto, G.; McFarlane, C.; Forrester, T.; Barton, S.J.; Leow, M.K.; Antoun, E.; et al. Mitochondrial oxidative capacity and NAD(+) biosynthesis are reduced in human sarcopenia across ethnicities. Nat. Commun. 2019, 10, 5808. [Google Scholar] [CrossRef]
- Zhang, H.; Ryu, D.; Wu, Y.; Gariani, K.; Wang, X.; Luan, P.; D’Amico, D.; Ropelle, E.R.; Lutolf, M.P.; Aebersold, R.; et al. NAD⁺ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 2016, 352, 1436–1443. [Google Scholar] [CrossRef]
- Tian, Y.; Garcia, G.; Bian, Q.; Steffen, K.K.; Joe, L.; Wolff, S.; Meyer, B.J.; Dillin, A. Mitochondrial stress induces chromatin reorganization to promote longevity and UPR(mt). Cell 2016, 165, 1197–1208. [Google Scholar] [CrossRef]
- Shao, L.W.; Peng, Q.; Dong, M.; Gao, K.; Li, Y.; Li, Y.; Li, C.Y.; Liu, Y. Histone deacetylase HDA-1 modulates mitochondrial stress response and longevity. Nat. Commun. 2020, 11, 4639. [Google Scholar] [CrossRef]
- Dillin, A.; Hsu, A.L.; Arantes-Oliveira, N.; Lehrer-Graiwer, J.; Hsin, H.; Fraser, A.G.; Kamath, R.S.; Ahringer, J.; Kenyon, C. Rates of behavior and aging specified by mitochondrial function during development. Science 2002, 298, 2398–2401. [Google Scholar] [CrossRef]
- Angeli, S.; Foulger, A.; Chamoli, M.; Peiris, T.H.; Gerencser, A.; Shahmirzadi, A.A.; Andersen, J.; Lithgow, G. The mitochondrial permeability transition pore activates the mitochondrial unfolded protein response and promotes aging. Elife 2021, 10, e63453. [Google Scholar] [CrossRef]
- Rea, S.L.; Ventura, N.; Johnson, T.E. Relationship between mitochondrial electron transport chain dysfunction, development, and life extension in Caenorhabditis elegans. PLoS Biol. 2007, 5, e259. [Google Scholar] [CrossRef]
- Lee, S.S.; Lee, R.Y.; Fraser, A.G.; Kamath, R.S.; Ahringer, J.; Ruvkun, G. A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat. Genet. 2003, 33, 40–48. [Google Scholar] [CrossRef]
- Tian, Y.; Merkwirth, C.; Dillin, A. Mitochondrial UPR: A Double-Edged Sword. Trends Cell Biol. 2016, 26, 563–565. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.F.; Vander Wende, H.; Simko, M.; Klum, S.; Barfield, S.; Choi, H.; Pineda, V.V.; Kaeberlein, M. Activation of the mitochondrial unfolded protein response does not predict longevity in Caenorhabditis elegans. Nat. Commun. 2014, 5, 3483. [Google Scholar] [CrossRef] [PubMed]
- Kenny, T.C.; Craig, A.J.; Villanueva, A.; Germain, D. Mitohormesis Primes Tumor Invasion and Metastasis. Cell Rep. 2019, 27, 2292–2303.e2296. [Google Scholar] [CrossRef] [PubMed]
- Brearley-Sholto, M.C.; Loczenski-Brown, D.M.; Jones, S.; Daniel, Z.; Ebling, F.J.P.; Parr, T.; Brameld, J.M. Effect of AAV-mediated overexpression of ATF5 and downstream targets of an integrated stress response in murine skeletal muscle. Sci. Rep. 2021, 11, 19796. [Google Scholar] [CrossRef]
- Chen, C.C.W.; Erlich, A.T.; Crilly, M.J.; Hood, D.A. Parkin is required for exercise-induced mitophagy in muscle: Impact of aging. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E404–E415. [Google Scholar] [CrossRef]
- Zhou, J.; Chong, S.Y.; Lim, A.; Singh, B.K.; Sinha, R.A.; Salmon, A.B.; Yen, P.M. Changes in macroautophagy, chaperone-mediated autophagy, and mitochondrial metabolism in murine skeletal and cardiac muscle during aging. Aging 2017, 9, 583–599. [Google Scholar] [CrossRef]
- Yeo, D.; Kang, C.; Gomez-Cabrera, M.C.; Vina, J.; Ji, L.L. Intensified mitophagy in skeletal muscle with aging is downregulated by PGC-1alpha overexpression in vivo. Free Radic. Biol. Med. 2019, 130, 361–368. [Google Scholar] [CrossRef]
- Gong, Y.; Luo, Y.; Liu, S.; Ma, J.; Liu, F.; Fang, Y.; Cao, F.; Wang, L.; Pei, Z.; Ren, J. Pentacyclic triterpene oleanolic acid protects against cardiac aging through regulation of mitophagy and mitochondrial integrity. BBA-Mol. Basis Dis. 2022, 1868, 166402. [Google Scholar] [CrossRef]
- Gouspillou, G.; Godin, R.; Piquereau, J.; Picard, M.; Mofarrahi, M.; Mathew, J.; Purves-Smith, F.M.; Sgarioto, N.; Hepple, R.T.; Burelle, Y.; et al. Protective role of Parkin in skeletal muscle contractile and mitochondrial function. J. Physiol. 2018, 596, 2565–2579. [Google Scholar] [CrossRef]
- Leduc-Gaudet, J.P.; Mayaki, D.; Reynaud, O.; Broering, F.E.; Chaffer, T.J.; Hussain, S.N.A.; Gouspillou, G. Parkin Overexpression Attenuates Sepsis-Induced Muscle Wasting. Cells 2020, 9, 1454. [Google Scholar] [CrossRef]
- Yu, Y.; Zhao, Y.; Teng, F.; Li, J.; Guan, Y.; Xu, J.; Lv, X.; Guan, F.; Zhang, M.; Chen, L. Berberine Improves Cognitive Deficiency and Muscular Dysfunction via Activation of the AMPK/SIRT1/PGC-1a Pathway in Skeletal Muscle from Naturally Aging Rats. J. Nutr. Health Aging 2018, 22, 710–717. [Google Scholar] [CrossRef]
- Koltai, E.; Szabo, Z.; Atalay, M.; Boldogh, I.; Naito, H.; Goto, S.; Nyakas, C.; Radak, Z. Exercise alters SIRT1, SIRT6, NAD and NAMPT levels in skeletal muscle of aged rats. Mech. Ageing Dev. 2010, 131, 21–28. [Google Scholar] [CrossRef]
- Hao, J.; Xi, Y.; Jiao, L.; Wen, X.; Wu, R.; Chang, G.; Sun, F.; Wei, C.; Li, H. Exogenous hydrogen sulfide inhibits the senescence of cardiomyocytes through modulating mitophagy in rats. Cell Signal 2022, 100, 110465. [Google Scholar] [CrossRef]
- Li, Y.; Ma, Y.; Song, L.; Yu, L.; Zhang, L.; Zhang, Y.; Xing, Y.; Yin, Y.; Ma, H. SIRT3 deficiency exacerbates p53/Parkin-mediated mitophagy inhibition and promotes mitochondrial dysfunction: Implication for aged hearts. Int. J. Mol. Med. 2018, 41, 3517–3526. [Google Scholar] [CrossRef]
- Chalil, S.; Pierre, N.; Bakker, A.D.; Manders, R.J.; Pletsers, A.; Francaux, M.; Klein-Nulend, J.; Jaspers, R.T.; Deldicque, L. Aging related ER stress is not responsible for anabolic resistance in mouse skeletal muscle. Biochem. Biophys. Res. Commun. 2015, 468, 702–707. [Google Scholar] [CrossRef]
- D’Souza, R.F.; Woodhead, J.S.T.; Hedges, C.P.; Zeng, N.; Wan, J.; Kumagai, H.; Lee, C.; Cohen, P.; Cameron-Smith, D.; Mitchell, C.J.; et al. Increased expression of the mitochondrial derived peptide, MOTS-c, in skeletal muscle of healthy aging men is associated with myofiber composition. Aging 2020, 12, 5244–5258. [Google Scholar] [CrossRef]
- Lee, C.; Zeng, J.; Drew, B.G.; Sallam, T.; Martin-Montalvo, A.; Wan, J.; Kim, S.J.; Mehta, H.; Hevener, A.L.; de Cabo, R.; et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab. 2015, 21, 443–454. [Google Scholar] [CrossRef]
- Villarroya, J.; Gallego-Escuredo, J.M.; Delgado-Anglés, A.; Cairó, M.; Moure, R.; Gracia Mateo, M.; Domingo, J.C.; Domingo, P.; Giralt, M.; Villarroya, F. Aging is associated with increased FGF21 levels but unaltered FGF21 responsiveness in adipose tissue. Aging Cell 2018, 17, e12822. [Google Scholar] [CrossRef]
- Yoo, S.Z.; No, M.H.; Heo, J.W.; Park, D.H.; Kang, J.H.; Kim, S.H.; Kwak, H.B. Role of exercise in age-related sarcopenia. J. Exerc. Rehabil. 2018, 14, 551–558. [Google Scholar] [CrossRef]
- Memme, J.M.; Oliveira, A.N.; Hood, D.A. Chronology of UPR activation in skeletal muscle adaptations to chronic contractile activity. Am. J. Physiol. Cell Physiol. 2016, 310, C1024–C1036. [Google Scholar] [CrossRef]
- Mesbah Moosavi, Z.S.; Hood, D.A. The unfolded protein response in relation to mitochondrial biogenesis in skeletal muscle cells. Am. J. Physiol. Cell Physiol. 2017, 312, C583–C594. [Google Scholar] [CrossRef] [PubMed]
- Slavin, M.B.; Kumari, R.; Hood, D.A. ATF5 is a regulator of exercise-induced mitochondrial quality control in skeletal muscle. Mol. Metab. 2022, 66, 101623. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Bo, H.; Song, Y.; Li, C.; Zhang, Y. Mitochondrial ROS produced by skeletal muscle mitochondria promote the decisive signal for UPRmt activation. Biomed. Res. Int. 2022, 2022, 7436577. [Google Scholar] [CrossRef] [PubMed]
- Laker, R.C.; Drake, J.C.; Wilson, R.J.; Lira, V.A.; Lewellen, B.M.; Ryall, K.A.; Fisher, C.C.; Zhang, M.; Saucerman, J.J.; Goodyear, L.J.; et al. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat. Commun. 2017, 8, 548. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.C.; Laker, R.C.; Wilson, R.J.; Zhang, M.; Yan, Z. Exercise-induced mitophagy in skeletal muscle occurs in the absence of stabilization of Pink1 on mitochondria. Cell Cycle 2019, 18, 1–6. [Google Scholar] [CrossRef]
- Drake, J.C.; Wilson, R.J.; Laker, R.C.; Guan, Y.; Spaulding, H.R.; Nichenko, A.S.; Shen, W.; Shang, H.; Dorn, M.V.; Huang, K.; et al. Mitochondria-localized AMPK responds to local energetics and contributes to exercise and energetic stress-induced mitophagy. Proc. Natl. Acad. Sci. USA 2021, 118, e2025932118. [Google Scholar] [CrossRef]
- Vainshtein, A.; Desjardins, E.M.; Armani, A.; Sandri, M.; Hood, D.A. PGC-1α modulates denervation-induced mitophagy in skeletal muscle. Skelet. Muscle 2015, 5, 9. [Google Scholar] [CrossRef]
- Brandt, N.; Gunnarsson, T.P.; Bangsbo, J.; Pilegaard, H. Exercise and exercise training-induced increase in autophagy markers in human skeletal muscle. Physiol. Rep. 2018, 6, e13651. [Google Scholar] [CrossRef]
- Lo Verso, F.; Carnio, S.; Vainshtein, A.; Sandri, M. Autophagy is not required to sustain exercise and PRKAA1/AMPK activity but is important to prevent mitochondrial damage during physical activity. Autophagy 2014, 10, 1883–1894. [Google Scholar] [CrossRef]
- He, C.; Bassik, M.C.; Moresi, V.; Sun, K.; Wei, Y.; Zou, Z.; An, Z.; Loh, J.; Fisher, J.; Sun, Q.; et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 2012, 481, 511–515. [Google Scholar] [CrossRef]
- Chen, C.C.W.; Erlich, A.T.; Hood, D.A. Role of Parkin and endurance training on mitochondrial turnover in skeletal muscle. Skelet. Muscle 2018, 8, 10. [Google Scholar] [CrossRef]
- Dethlefsen, M.M.; Halling, J.F.; Møller, H.D.; Plomgaard, P.; Regenberg, B.; Ringholm, S.; Pilegaard, H. Regulation of apoptosis and autophagy in mouse and human skeletal muscle with aging and lifelong exercise training. Exp. Gerontol. 2018, 111, 141–153. [Google Scholar] [CrossRef]
- O’Leary, M.F.; Vainshtein, A.; Iqbal, S.; Ostojic, O.; Hood, D.A. Adaptive plasticity of autophagic proteins to denervation in aging skeletal muscle. Am. J. Physiol. Cell Physiol. 2013, 304, C422–C430. [Google Scholar] [CrossRef]
- Gao, J.; Yu, L.; Wang, Z.; Wang, R.; Liu, X. Induction of mitophagy in C2C12 cells by electrical pulse stimulation involves increasing the level of the mitochondrial receptor FUNDC1 through the AMPK-ULK1 pathway. Am. J. Transl. Res. 2020, 12, 6879–6894. [Google Scholar]
- Jamart, C.; Naslain, D.; Gilson, H.; Francaux, M. Higher activation of autophagy in skeletal muscle of mice during endurance exercise in the fasted state. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E964–E974. [Google Scholar] [CrossRef]
- Morville, T.; Sahl, R.E.; Trammell, S.A.; Svenningsen, J.S.; Gillum, M.P.; Helge, J.W.; Clemmensen, C. Divergent effects of resistance and endurance exercise on plasma bile acids, FGF19, and FGF21 in humans. JCI Insight 2018, 3, e122737. [Google Scholar] [CrossRef] [PubMed]
- Khalafi, M.; Alamdari, K.A.; Symonds, M.E.; Nobari, H.; Carlos-Vivas, J. Impact of acute exercise on immediate and following early post-exercise FGF-21 concentration in adults: Systematic review and meta-analysis. Hormones 2021, 20, 23–33. [Google Scholar] [CrossRef]
- Goncalves, R.L.; Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Brand, M.D. Sites of superoxide and hydrogen peroxide production by muscle mitochondria assessed ex vivo under conditions mimicking rest and exercise. J. Biol. Chem. 2015, 290, 209–227. [Google Scholar] [CrossRef]
- Wang, Y.; Nartiss, Y.; Steipe, B.; McQuibban, G.A.; Kim, P.K. ROS-induced mitochondrial depolarization initiates PARK2/PARKIN-dependent mitochondrial degradation by autophagy. Autophagy 2012, 8, 1462–1476. [Google Scholar] [CrossRef]
- Bouviere, J.; Fortunato, R.S.; Dupuy, C.; Werneck-de-Castro, J.P.; Carvalho, D.P.; Louzada, R.A. Exercise-stimulated ROS sensitive signaling pathways in skeletal muscle. Antioxidants 2021, 10, 537. [Google Scholar] [CrossRef]
- Dong, K.; Wu, M.; Liu, X.; Huang, Y.; Zhang, D.; Wang, Y.; Yan, L.J.; Shi, D. Glutaredoxins concomitant with optimal ROS activate AMPK through S-glutathionylation to improve glucose metabolism in type 2 diabetes. Free Radic. Biol. Med. 2016, 101, 334–347. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Zhao, A.; Yan, X.; Gao, H.; Zhang, C.; Liu, X.; Luo, Q.; Xie, F.; Liu, S.; Shi, D. Hepatic AMPK signaling activation in response to dynamic REDOX balance is a biomarker of exercise to improve blood glucose control. Elife 2022, 11, e79939. [Google Scholar] [CrossRef] [PubMed]
- Møller, A.B.; Vendelbo, M.H.; Christensen, B.; Clasen, B.F.; Bak, A.M.; Jørgensen, J.O.; Møller, N.; Jessen, N. Physical exercise increases autophagic signaling through ULK1 in human skeletal muscle. J. Appl. Physiol. 2015, 118, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.R.; Lee, G.H.; Cho, E.Y.; Chae, S.W.; Ahn, T.; Chae, H.J. Bax inhibitor 1 regulates ER-stress-induced ROS accumulation through the regulation of cytochrome P450 2E1. J. Cell Sci. 2009, 122, 1126–1133. [Google Scholar] [CrossRef] [PubMed]
- Cantó, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef]
- Lan, F.; Cacicedo, J.M.; Ruderman, N.; Ido, Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J. Biol. Chem. 2008, 283, 27628–27635. [Google Scholar] [CrossRef]
- Baker, J.R.; Vuppusetty, C.; Colley, T.; Papaioannou, A.I.; Fenwick, P.; Donnelly, L.; Ito, K.; Barnes, P.J. Oxidative stress dependent microRNA-34a activation via PI3Kα reduces the expression of sirtuin-1 and sirtuin-6 in epithelial cells. Sci. Rep. 2016, 6, 35871. [Google Scholar] [CrossRef]
- Rippo, M.R.; Olivieri, F.; Monsurrò, V.; Prattichizzo, F.; Albertini, M.C.; Procopio, A.D. MitomiRs in human inflamm-aging: A hypothesis involving miR-181a, miR-34a and miR-146a. Exp. Gerontol. 2014, 56, 154–163. [Google Scholar] [CrossRef]
- Guo, Z.; Tuo, H.; Tang, N.; Liu, F.Y.; Ma, S.Q.; An, P.; Yang, D.; Wang, M.Y.; Fan, D.; Yang, Z.; et al. Neuraminidase 1 deficiency attenuates cardiac dysfunction, oxidative stress, fibrosis, inflammatory via AMPK-SIRT3 pathway in diabetic cardiomyopathy mice. Int. J. Biol. Sci. 2022, 18, 826–840. [Google Scholar] [CrossRef]
- Han, L.; Li, J.; Li, J.; Pan, C.; Xiao, Y.; Lan, X.; Wang, M. Activation of AMPK/Sirt3 pathway by phloretin reduces mitochondrial ROS in vascular endothelium by increasing the activity of MnSOD via deacetylation. Food Funct. 2020, 11, 3073–3083. [Google Scholar] [CrossRef]
- Brandauer, J.; Andersen, M.A.; Kellezi, H.; Risis, S.; Frøsig, C.; Vienberg, S.G.; Treebak, J.T. AMP-activated protein kinase controls exercise training- and AICAR-induced increases in SIRT3 and MnSOD. Front. Physiol. 2015, 6, 85. [Google Scholar] [CrossRef]
- Yeo, D.; Kang, C.; Ji, L.L. Aging alters acetylation status in skeletal and cardiac muscles. GeroScience 2020, 42, 963–976. [Google Scholar] [CrossRef]
- Ji, L.L.; Yeo, D. NAD(+) deficit, protein acetylation and muscle aging. Aging 2021, 13, 14546–14548. [Google Scholar] [CrossRef]
- Papa, L.; Germain, D. SirT3 regulates the mitochondrial unfolded protein response. Mol. Cell Biol. 2014, 34, 699–710. [Google Scholar] [CrossRef]
- He, J.; Shangguan, X.; Zhou, W.; Cao, Y.; Zheng, Q.; Tu, J.; Hu, G.; Liang, Z.; Jiang, C.; Deng, L.; et al. Glucose limitation activates AMPK coupled SENP1-Sirt3 signalling in mitochondria for T cell memory development. Nat. Commun. 2021, 12, 4371. [Google Scholar] [CrossRef]
- Li, K.; Chen, L.; Lin, Z.; Zhu, J.; Fang, Y.; Du, J.; Shen, B.; Wu, K.; Liu, Y. Role of the AMPK/ACC Signaling Pathway in TRPP2-Mediated Head and Neck Cancer Cell Proliferation. Biomed. Res. Int. 2020, 2020, 4375075. [Google Scholar] [CrossRef]
- Iqbal, S.; Hood, D.A. Oxidative stress-induced mitochondrial fragmentation and movement in skeletal muscle myoblasts. Am. J. Physiol. Cell Physiol. 2014, 306, C1176–C1183. [Google Scholar] [CrossRef]
- Liu, L.; Wise, D.R.; Diehl, J.A.; Simon, M.C. Hypoxic reactive oxygen species regulate the integrated stress response and cell survival. J. Biol. Chem. 2008, 283, 31153–31162. [Google Scholar] [CrossRef]
- Kang, G.M.; Min, S.H.; Lee, C.H.; Kim, J.Y.; Lim, H.S.; Choi, M.J.; Jung, S.B.; Park, J.W.; Kim, S.; Park, C.B.; et al. Mitohormesis in Hypothalamic POMC Neurons Mediates Regular Exercise-Induced High-Turnover Metabolism. Cell Metab. 2021, 33, 334–349.e336. [Google Scholar] [CrossRef]
- Kim, K.H.; Son, J.M.; Benayoun, B.A.; Lee, C. The Mitochondrial-Encoded Peptide MOTS-c Translocates to the Nucleus to Regulate Nuclear Gene Expression in Response to Metabolic Stress. Cell Metab. 2018, 28, 516–524.e517. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Li, J.; Zhang, Z.; Wang, R.; Bo, H.; Zhang, Y. Exercise Improves the Coordination of the Mitochondrial Unfolded Protein Response and Mitophagy in Aging Skeletal Muscle. Life 2023, 13, 1006. https://doi.org/10.3390/life13041006
Wang Y, Li J, Zhang Z, Wang R, Bo H, Zhang Y. Exercise Improves the Coordination of the Mitochondrial Unfolded Protein Response and Mitophagy in Aging Skeletal Muscle. Life. 2023; 13(4):1006. https://doi.org/10.3390/life13041006
Chicago/Turabian StyleWang, Yan, Jialin Li, Ziyi Zhang, Runzi Wang, Hai Bo, and Yong Zhang. 2023. "Exercise Improves the Coordination of the Mitochondrial Unfolded Protein Response and Mitophagy in Aging Skeletal Muscle" Life 13, no. 4: 1006. https://doi.org/10.3390/life13041006
APA StyleWang, Y., Li, J., Zhang, Z., Wang, R., Bo, H., & Zhang, Y. (2023). Exercise Improves the Coordination of the Mitochondrial Unfolded Protein Response and Mitophagy in Aging Skeletal Muscle. Life, 13(4), 1006. https://doi.org/10.3390/life13041006