
Liver Transcriptome Analysis of the Black Porgy (Acanthopagrus schlegelii) under Acute Low-Temperature Stress

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Design and Sample Preparation
2.2. Preparation for RNA-Seq Libraries and Sequencing
2.3. Assembly and Annotation
2.4. Sample Relation Analysis
2.5. Differentially Expressed Genes and Enrichment Analysis
2.6. Validation Using Real-Time Quantitative PCR
3. Results
3.1. Transcriptome Sequencing and Assembly
3.2. Sample Relationship
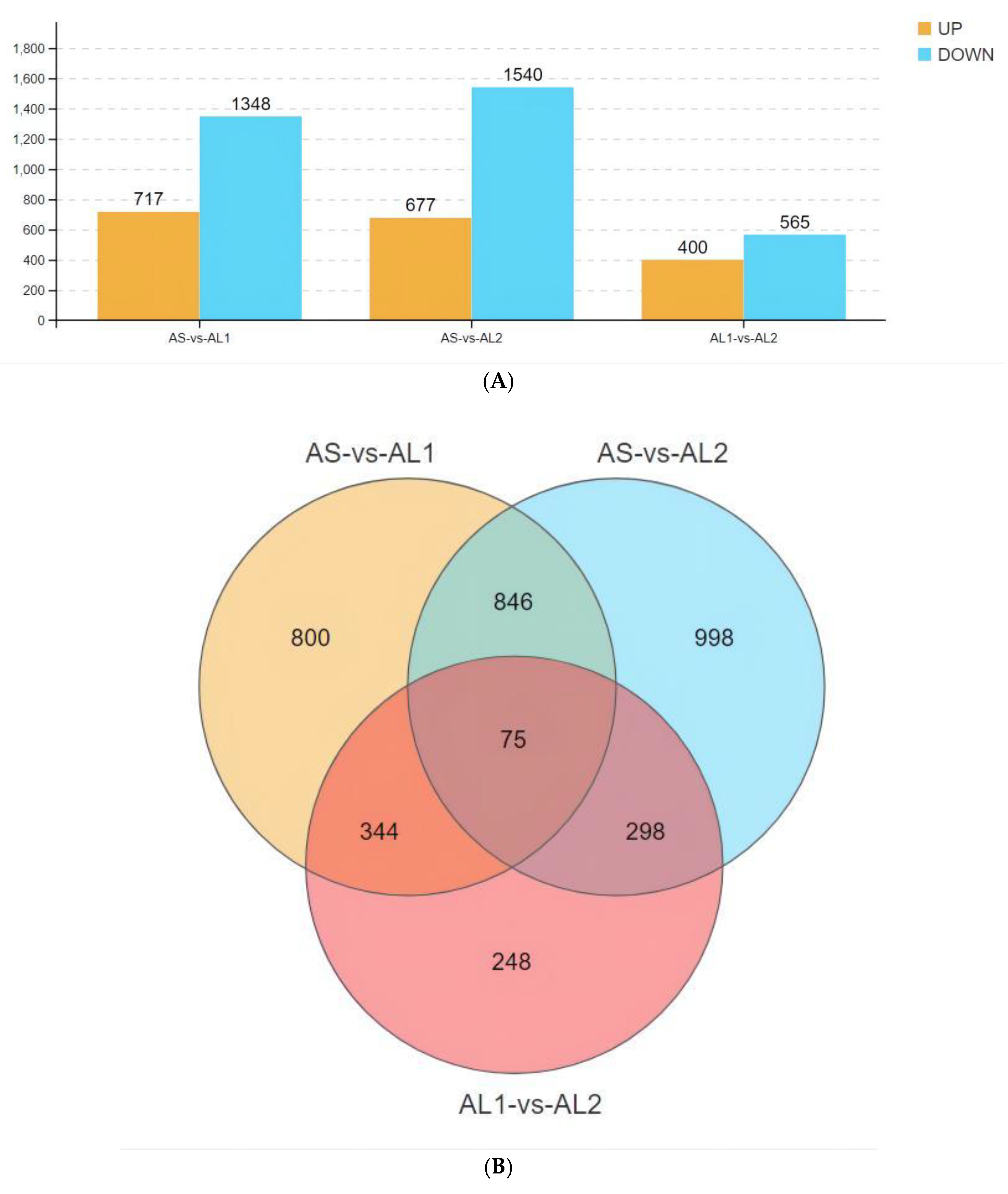
3.3. Basic Characteristics of Identified Differentially Expressed Genes (DEGs)
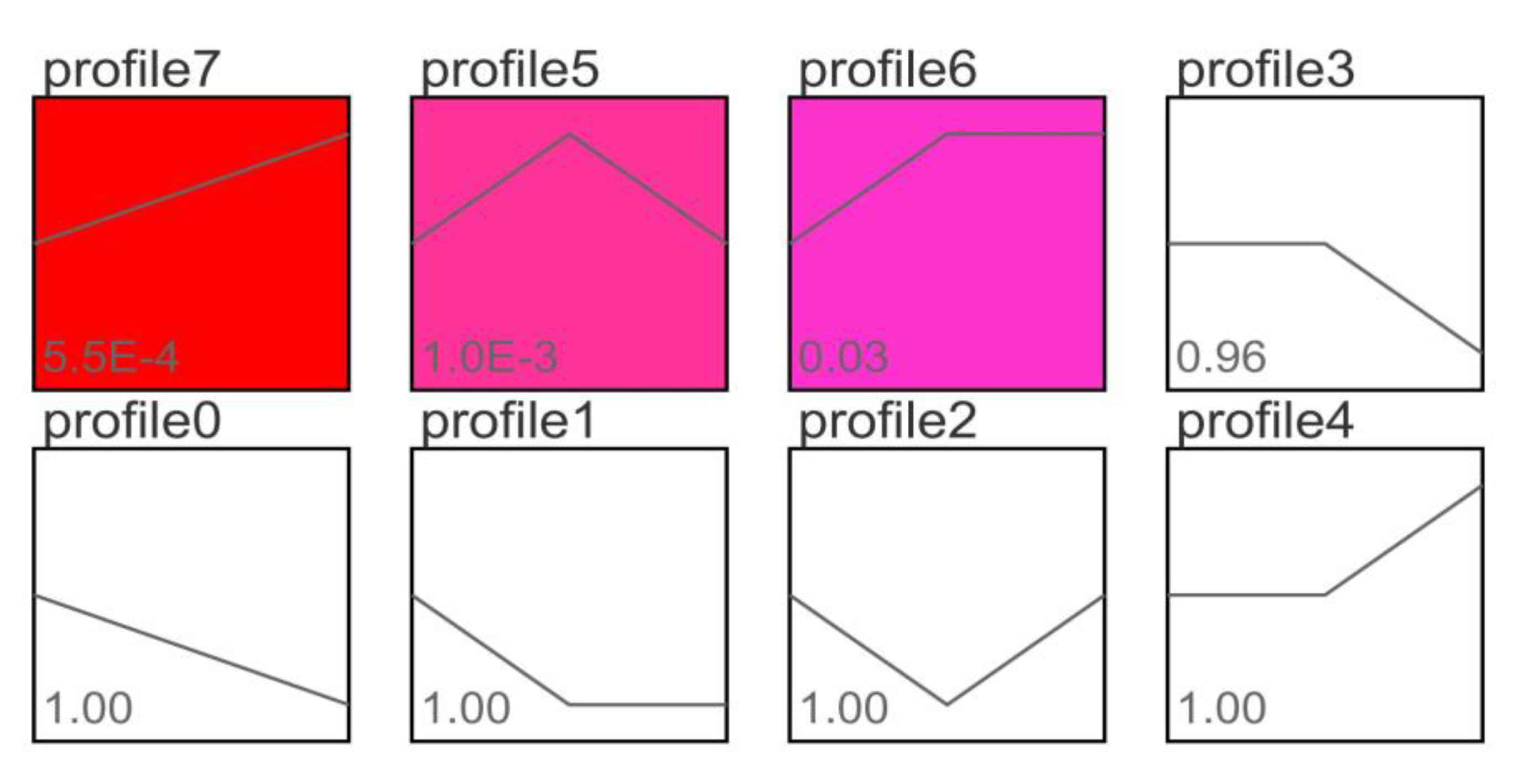
3.4. Trend Analysis of Differentially Expressed Genes
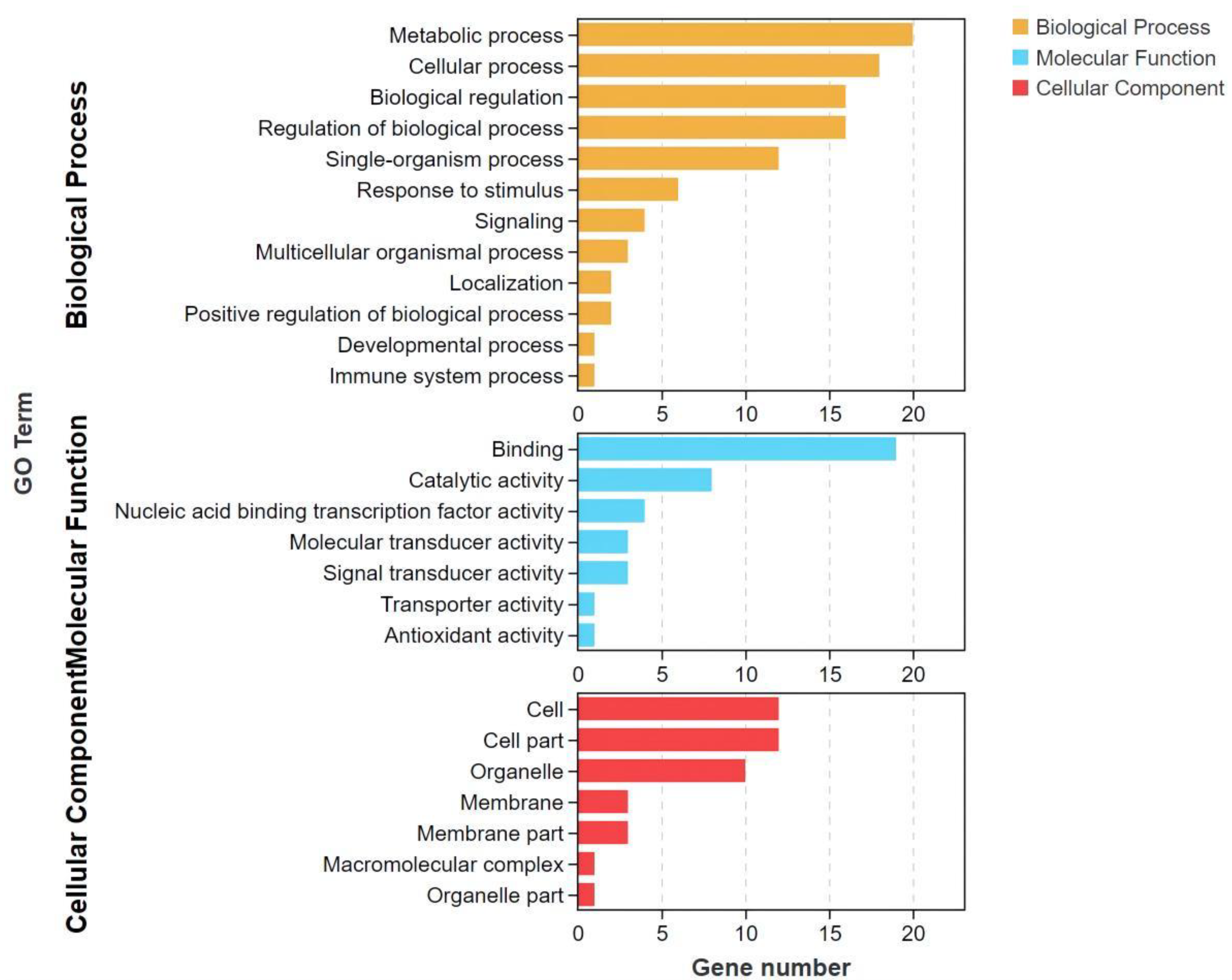
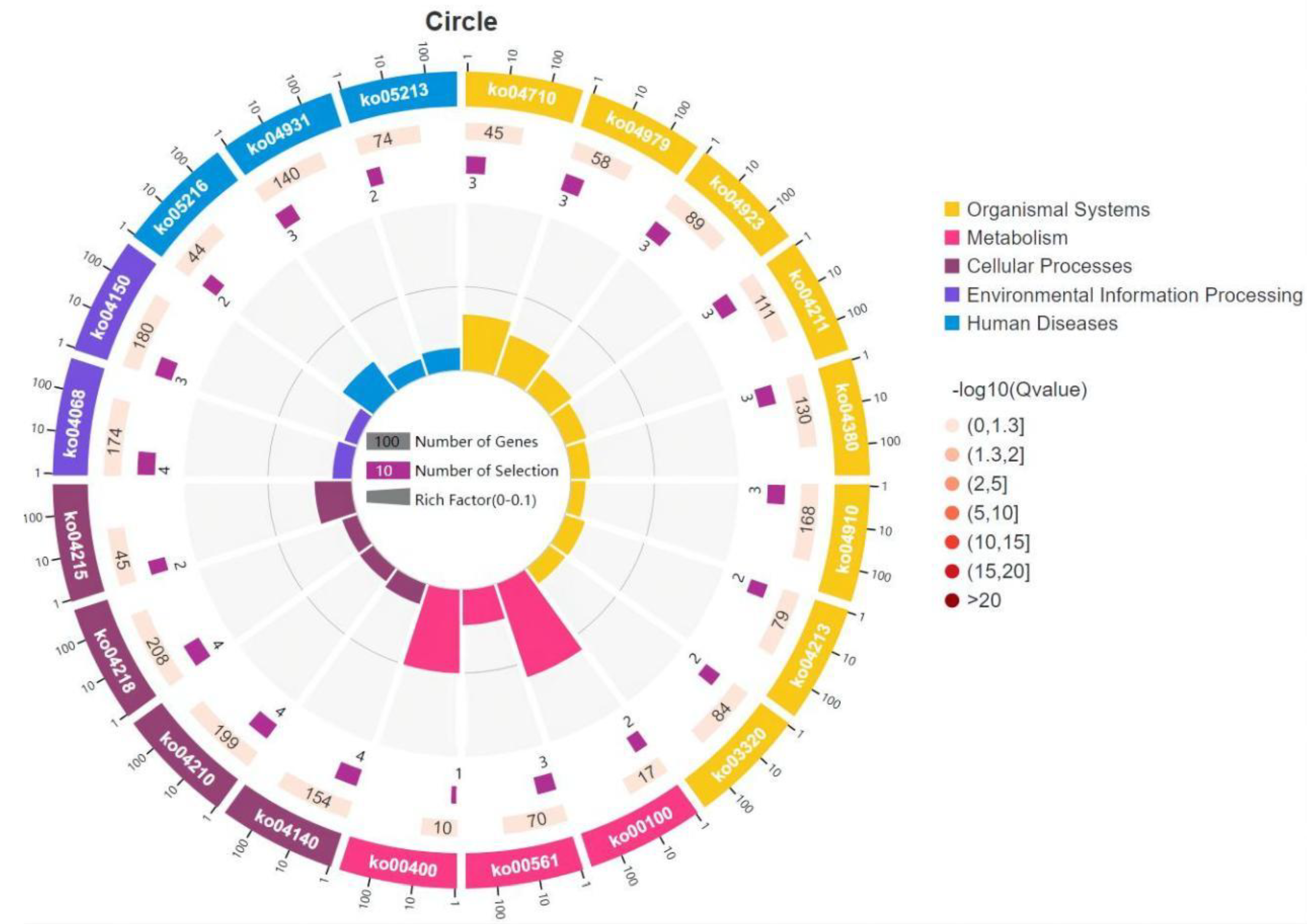
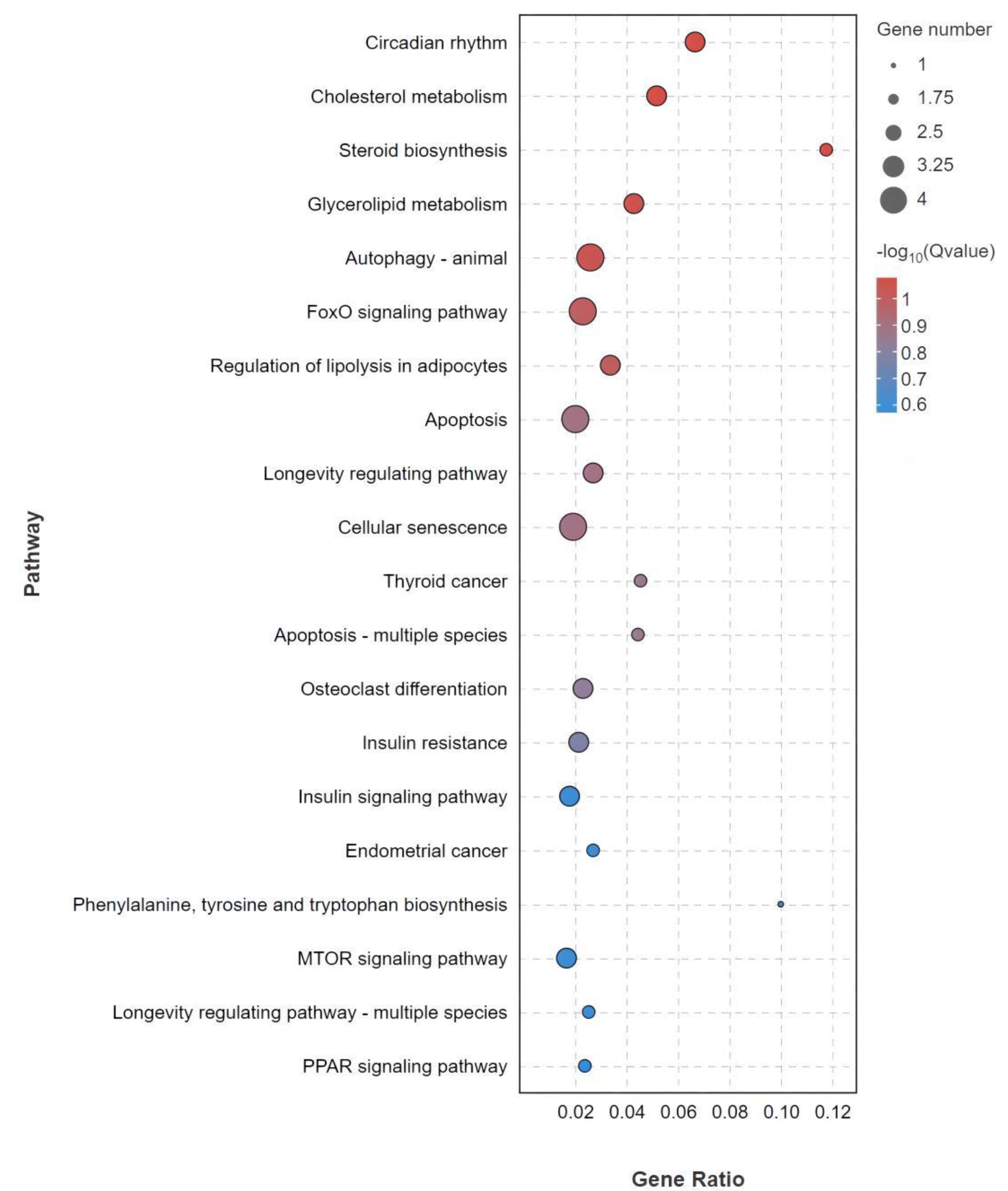
3.5. GO and KEGG Enrichment Analysis
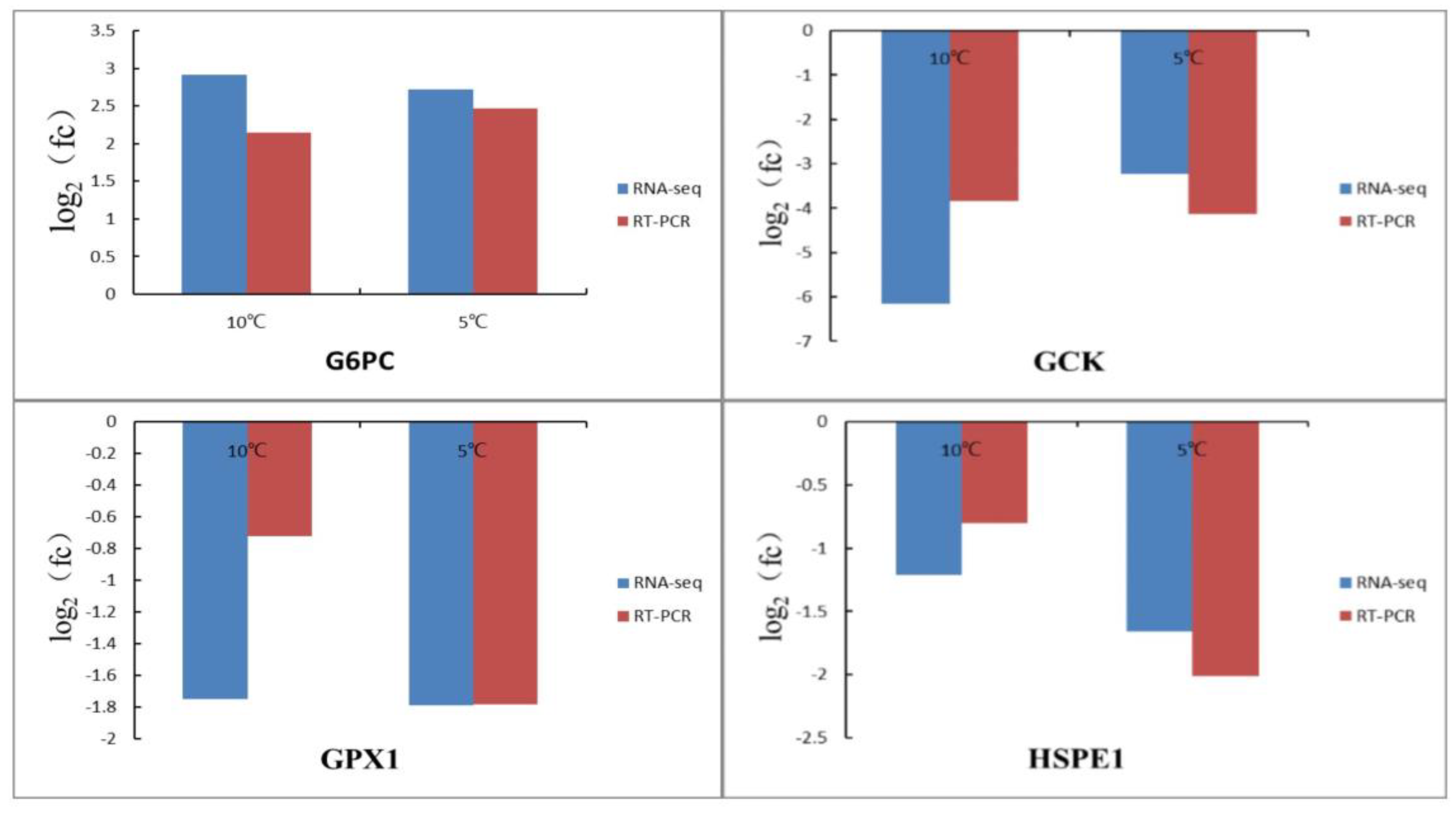
3.6. Validation of RNA-Seq Data by qRT-PCR
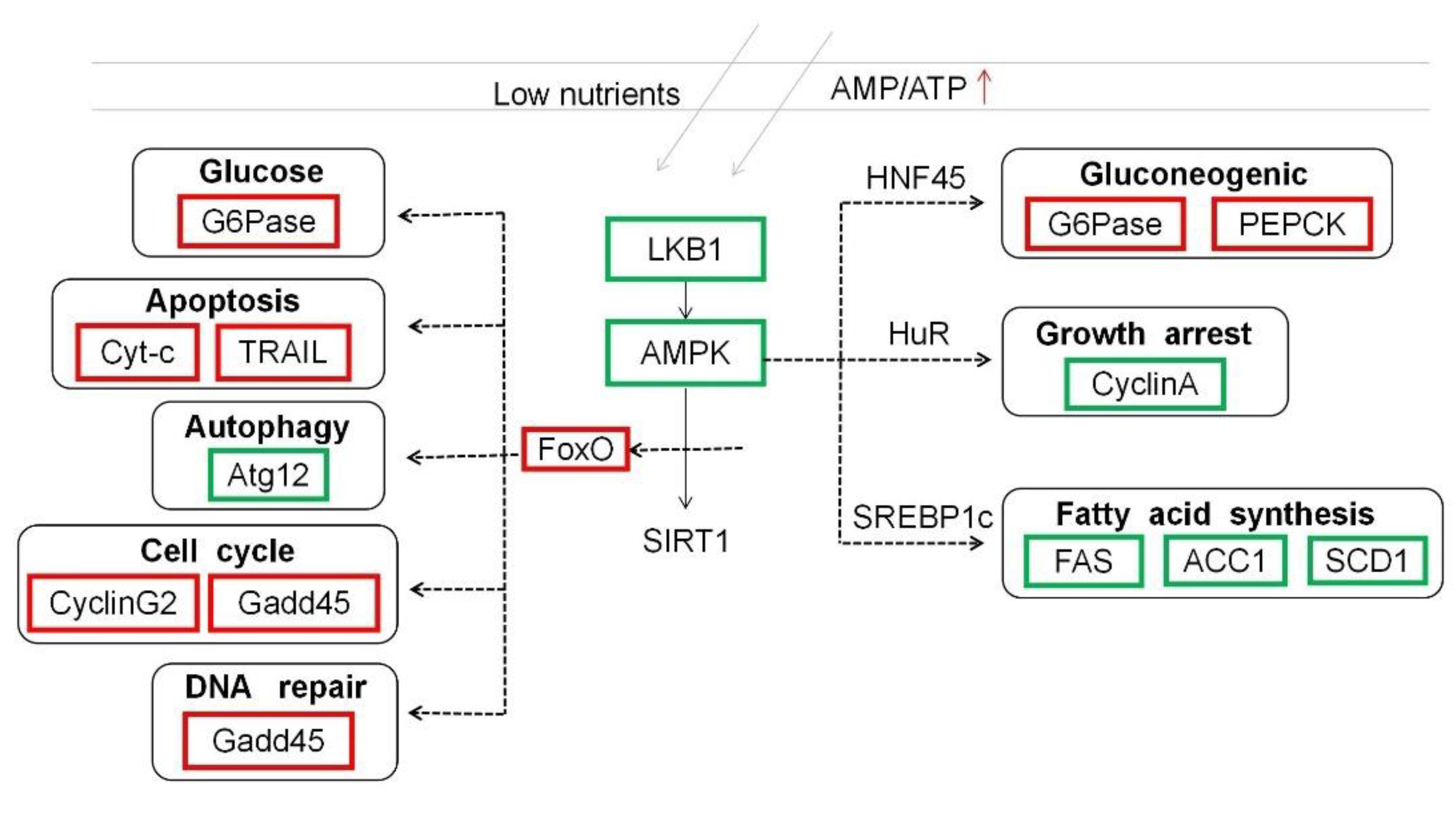
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Abram, Q.H.; Dixon, B.; Katzenback, B.A. Impacts of Low Temperature on the Teleost Immune System. Biology 2017, 6, 39. [Google Scholar] [CrossRef] [PubMed]
- Mininni, A.N.; Milan, M.; Ferraresso, S.; Petochi, T.; Di Marco, P.; Marino, G.; Livi, S.; Romualdi, C.; Bargelloni, L.; Patarnello, T. Liver transcriptome analysis in gilthead sea bream upon exposure to low temperature. BMC Genom. 2014, 15, 11. [Google Scholar] [CrossRef] [PubMed]
- Wen, B.; Jin, S.-R.; Chen, Z.-Z.; Gao, J.-Z.; Wang, L.; Liu, Y.; Liu, H.-P. Plasticity of energy reserves and metabolic performance of discus fish (Symphysodon aequifasciatus) exposed to low-temperature stress. Aquaculture 2017, 481, 169–176. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Z.; Yu, Z.; Li, J.; Chen, S.; Sun, R.; Jia, C.; Zhu, F.; Meng, Q.; Xu, S. Molecular cloning and expression pattern of IGFBP-2a in black porgy (Acanthopagrus schlegelii) and evolutionary analysis of IGFBP-2s in the species of Perciformes. Fish Physiol. Biochem. 2019, 45, 1731–1745. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.-C.; Dufour, S.; Chang, C.-F. Molecular and cellular regulation on sex change in hermaphroditic fish, with a special focus on protandrous black porgy, Acanthopagrus schlegelii. Mol. Cell. Endocrinol. 2021, 520, 111069. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Zhang, Z.; Solberg, M.F.; Chen, Z.; Wei, M.; Zhu, F.; Jia, C.; Meng, Q.; Zhang, Z. Comparative transcriptome analysis of mixed tissues of black porgy (Acanthopagrus schlegelii) with differing growth rates. Aquac. Res. 2021, 52, 5800–5813. [Google Scholar] [CrossRef]
- Zhang, Y.; Øverland, M.; Xie, S.; Dong, Z.; Lv, Z.; Xu, J.; Storebakken, T. Mixtures of lupin and pea protein concentrates can efficiently replace high-quality fish meal in extruded diets for juvenile black sea bream (Acanthopagrus schlegeli). Aquaculture 2012, 354–355, 68–74. [Google Scholar] [CrossRef]
- Wen, X.; Hu, Y.; Zhang, X.; Wei, X.; Wang, T.; Yin, S. Integrated application of multi-omics provides insights into cold stress responses in pufferfish Takifugu fasciatus. BMC Genom. 2019, 20, 563. [Google Scholar] [CrossRef]
- Liu, M.; Zhou, Y.-L.; Guo, X.-F.; Wei, W.-Y.; Li, Z.; Zhou, L.; Wang, Z.-W.; Gui, J.-F. Comparative transcriptomes and metabolomes reveal different tolerance mechanisms to cold stress in two different catfish species. Aquaculture 2022, 560, 738543. [Google Scholar] [CrossRef]
- Huang, D.; Liang, H.; Zhu, J.; Ren, M.; Ge, X. Transcriptome reveals insights into hepatic nutritional metabolism and gill immune responses adapted to cold stress in genetically improved farmed tilapia (GIFT: Oreochromis niloticus). Aquac. Rep. 2022, 26, 101297. [Google Scholar] [CrossRef]
- Zhang, M.; Hu, J.; Zhu, J.; Wang, Y.; Zhang, Y.; Li, Y.; Xu, S.; Yan, X.; Zhang, D. Transcriptome, antioxidant enzymes and histological analysis reveal molecular mechanisms responsive to long-term cold stress in silver pomfret (Pampus argenteus). Fish Shellfish. Immunol. 2022, 121, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Ren, M.; Liang, H.; Ge, X.; Xu, H.; Wu, L. Transcriptome analysis of the effect of high-temperature on nutrient metabolism in juvenile grass carp (Ctenopharyngodon idellus). Gene 2021, 809, 146035. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, K.; Chen, S.; Zhang, Z.; Zhang, J.; You, X.; Bian, C.; Xu, J.; Jia, C.; Qiang, J.; et al. Draft genome of the protandrous Chinese black porgy, Acanthopagrus schlegelii. Gigascience 2018, 7, giy012. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; You, Q.; Chi, C.; Luo, S.; Song, H.; Lou, B.; Takeuchi, Y. Transcriptional response to low temperature in the yellow drum (Nibea albiflora) and identification of genes related to cold stress. Comp. Biochem. Physiol. Part D Genom. Proteom. 2018, 28, 80–89. [Google Scholar] [CrossRef]
- Makrinos, D.L.; Bowden, T.J. Natural environmental impacts on teleost immune function. Fish Shellfish Immunol. 2016, 53, 50–57. [Google Scholar] [CrossRef]
- Jiao, L.; Dai, T.; Lu, J.; Tao, X.; Jin, M.; Sun, P.; Zhou, Q. Effects of dietary manganese supplementation on growth performance, antioxidant capacity, immune function and intestinal microbiota in Pacific white shrimp Litopenaeus vannamei. Aquac. Nutr. 2021, 27, 1972–1982. [Google Scholar] [CrossRef]
- Sun, M.; Li, S.; Zhang, X.; Xiang, J.; Li, F. Isolation and transcriptome analysis of three subpopulations of shrimp hemocytes reveals the underlying mechanism of their immune functions. Dev. Comp. Immunol. 2020, 108, 103689. [Google Scholar] [CrossRef]
- Song, Y.; Wu, M.; Pang, Y.; Song, X.; Shi, A.; Shi, X.; Niu, C.; Cheng, Y.; Yang, X. Effects of melatonin feed on the changes of hemolymph immune parameters, antioxidant capacity, and mitochondrial functions in Chinese mitten crab (Eriocheir sinensis) caused by acute hypoxia. Aquaculture 2021, 535, 736374. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, Y.; Gul, Y.; Li, Q.; Song, J.; Hu, M. Effects of copper supplement on the immune function and blood-chemistry in adult Chinese horseshoe crab Tachypleus tridentatus. Aquaculture 2020, 515, 734576. [Google Scholar] [CrossRef]
- Kapila, R.; Pant, R.; Gaur, A.K.; Mahanta, P.C. Effect of low temperature on metabolic enzymes and HSP-70 expression of coldwater fish Barilius bendelisis. Asian Fish. Sci. 2009, 22, 125–136. [Google Scholar] [CrossRef]
- Ji, L.; Jiang, K.; Liu, M.; Wang, B.; Han, L.; Zhang, M.; Wang, L. Low temperature stress on the hematological parameters and HSP gene expression in the turbot Scophthalmus maximus. Chin. J. Oceanol. Limnol. 2015, 34, 430–440. [Google Scholar] [CrossRef]
- Somero, G.N. The cellular stress response and temperature: Function, regulation, and evolution. J. Exp. Zool. Part A Ecol. Integr. Physiol. 2020, 333, 379–397. [Google Scholar] [CrossRef] [PubMed]
- Rocha-Santos, C.; Bastos, F.; Dantas, R.; Hauser-Davis, R.; Rodrigues, L.; Bastos, V.C.; Bastos, J.C. Glutathione peroxidase and glutathione S-transferase in blood and liver from a hypoxia-tolerant fish under oxygen deprivation. Ecotoxicol. Environ. Saf. 2018, 163, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Gao, J.; Lu, L.; Bold, T.; Li, X.; Wang, S.; Chang, Z.; Chen, J.; Kong, X.; Zheng, Y.; et al. Intracellular GSH/GST antioxidants system change as an earlier biomarker for toxicity evaluation of iron oxide nanoparticles. Nanoimpact 2021, 23, 100338. [Google Scholar] [CrossRef]
- Sun, Z.; Tan, X.; Xu, M.; Liu, Q.; Ye, H.; Zou, C.; Ye, C. Liver transcriptome analysis and de novo annotation of the orange-spotted groupers (Epinephelus coioides) under cold stress. Comp. Biochem. Physiol. Part D Genom. Proteom. 2019, 29, 264–273. [Google Scholar] [CrossRef]
- Song, H.; Xu, D.; Tian, L.; Chen, R.; Wang, L.; Tan, P.; You, Q. Overwinter mortality in yellow drum (Nibea albiflora): Insights from growth and immune responses to cold and starvation stress. Fish Shellfish Immunol. 2019, 92, 341–347. [Google Scholar] [CrossRef]
- Bondar, A.-N. Introduction: Biomembrane Structure, Dynamics, and Reactions. Chem. Rev. 2019, 119, 5535–5536. [Google Scholar] [CrossRef]
- Hasegawa, S.; Yamasaki, M.; Fukui, T. Degradation of acetoacetyl-CoA synthetase, a ketone body-utilizing enzyme, by legumain in the mouse kidney. Biochem. Biophys. Res. Commun. 2014, 453, 631–635. [Google Scholar] [CrossRef]
- Yan, Y.; Mukherjee, S.; Harikumar, K.G.; Strutzenberg, T.S.; Zhou, X.E.; Suino-Powell, K.; Xu, T.-H.; Sheldon, R.D.; Lamp, J.; Brunzelle, J.S.; et al. Structure of an AMPK complex in an inactive, ATP-bound state. Science 2021, 373, 413–419. [Google Scholar] [CrossRef]
- Paiva, P.; Medina, F.E.; Viegas, M.; Ferreira, P.; Neves, R.P.P.; Sousa, J.P.M.; Ramos, M.J.; Fernandes, P.A. Animal Fatty Acid Synthase: A Chemical Nanofactory. Chem. Rev. 2021, 121, 9502–9553. [Google Scholar] [CrossRef] [PubMed]
- Barber, M.C.; Price, N.T.; Travers, M.T. Structure and regulation of acetyl-CoA carboxylase genes of metazoa. Biochim. Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2005, 1733, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Perera, E.; Turkmen, S.; Simó-Mirabet, P.; Zamorano, M.J.; Xu, H.; Naya-Català, F.; Izquierdo, M.; Pérez-Sánchez, J. Stearoyl-CoA desaturase (scd1a) is epigenetically regulated by broodstock nutrition in gilthead sea bream (Sparus aurata). Epigenetics 2019, 15, 536–553. [Google Scholar] [CrossRef] [PubMed]
- Bionaz, M.; Loor, J.J. Gene networks driving bovine milk fat synthesis during the lactation cycle. BMC Genom. 2008, 9, 366. [Google Scholar] [CrossRef]
- Gross, D.N.; Heuvel, A.P.J.V.D.; Birnbaum, M.J. The role of FoxO in the regulation of metabolism. Oncogene 2008, 27, 2320–2336. [Google Scholar] [CrossRef]
- Smaldone, G.; Caruso, D.; Sandomenico, A.; Iaccarino, E.; Focà, A.; Ruggiero, A.; Ruvo, M.; Vitagliano, L. Members of the GADD45 Protein Family Show Distinct Propensities to form Toxic Amyloid-Like Aggregates in Physiological Conditions. Int. J. Mol. Sci. 2021, 22, 10700. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Sequence (5′–3′) | Amplification Efficiency/% |
|---|---|---|
| β-actin | F: CGACGGTCAGGTCATCAC | 91.957 |
| R: GCCAGCAGACTCCATTCC | ||
| G6PC | F: TACACCGAGACCAAGAAG | 92.611 |
| R: GACAGCAGGTAGAAGAGG | ||
| GCK | F: GACATCATCTCAAGCACAAG | 94.953 |
| R: GGCAACCACATCCATCTC | ||
| GPX1 | F: ATCTGCCCTGATGACTGAC | 93.368 |
| R: CCTGCTGTAACGCTTGAAG | ||
| HSPE1 | F: AGAAGGCTCAAGGCAAAG | 93.504 |
| R: TCACGGAACAGGAAATAGTC |
| Sample | Rawdata | CleanData (%) | Adapter (%) | LowQuality (%) |
|---|---|---|---|---|
| AS-1 | 41,124,586 | 99.84 | 0.02 | 0.14 |
| AS-2 | 44,151,136 | 99.81 | 0.02 | 0.16 |
| AS-3 | 44,722,244 | 99.84 | 0.02 | 0.14 |
| AL1-1 | 50,589,846 | 99.78 | 0.03 | 0.19 |
| AL1-2 | 44,156,390 | 99.76 | 0.04 | 0.20 |
| AL1-3 | 50,278,754 | 99.82 | 0.02 | 0.15 |
| AL2-1 | 37,965,772 | 99.85 | 0.02 | 0.13 |
| AL2-2 | 38,403,050 | 99.82 | 0.02 | 0.15 |
| AL2-3 | 40,782,160 | 99.82 | 0.03 | 0.15 |
| Sample | Refer Genes | Sequenced Refer Genes | Average Sequenced Refer Genes | Novel Genes | Sequenced Novel Genes | Average Sequenced Novel Genes | Total Genes | Sequenced Total Genes | Average Sequenced Total Genes |
|---|---|---|---|---|---|---|---|---|---|
| all | 19,465 | 16,608 | 1064 | 1064 | 20,529 | 17,672 | |||
| AS-1 | 19,465 | 13,897 | 14,009 | 1064 | 999 | 1005 | 20,529 | 14,896 | 15,614 |
| AS-2 | 19,465 | 14,070 | 1064 | 1011 | 20,529 | 15,081 | |||
| AS-3 | 19,465 | 14,060 | 1064 | 1004 | 20,529 | 15,064 | |||
| AL1-1 | 19,465 | 13,959 | 13,891 | 1064 | 1005 | 1004 | 20,529 | 14,964 | 14,895 |
| AL1-2 | 19,465 | 13,602 | 1064 | 1001 | 20,529 | 14,603 | |||
| AL1-3 | 19,465 | 14,113 | 1064 | 1007 | 20,529 | 15,120 | |||
| AL2-1 | 19,465 | 13,905 | 14,009 | 1064 | 1003 | 1012 | 20,529 | 14,908 | 15,021 |
| AL2-2 | 19,465 | 14,081 | 1064 | 1021 | 20,529 | 15,102 | |||
| AL2-3 | 19,465 | 14,041 | 1064 | 1011 | 20,529 | 15,052 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Chen, Z.; Wei, M.; Lin, Z.; Shen, M.; Zhu, F.; Jia, C.; Meng, Q.; Xu, D.; Du, S.; et al. Liver Transcriptome Analysis of the Black Porgy (Acanthopagrus schlegelii) under Acute Low-Temperature Stress. Life 2023, 13, 721. https://doi.org/10.3390/life13030721
Wang Y, Chen Z, Wei M, Lin Z, Shen M, Zhu F, Jia C, Meng Q, Xu D, Du S, et al. Liver Transcriptome Analysis of the Black Porgy (Acanthopagrus schlegelii) under Acute Low-Temperature Stress. Life. 2023; 13(3):721. https://doi.org/10.3390/life13030721
Chicago/Turabian StyleWang, Yue, Ziqiang Chen, Mingliang Wei, Zhijie Lin, Mingjun Shen, Fei Zhu, Chaofeng Jia, Qian Meng, Dafeng Xu, Shuran Du, and et al. 2023. "Liver Transcriptome Analysis of the Black Porgy (Acanthopagrus schlegelii) under Acute Low-Temperature Stress" Life 13, no. 3: 721. https://doi.org/10.3390/life13030721
APA StyleWang, Y., Chen, Z., Wei, M., Lin, Z., Shen, M., Zhu, F., Jia, C., Meng, Q., Xu, D., Du, S., Liu, Y., Chen, S., Zhang, C., Zhang, Z., & Zhang, Z. (2023). Liver Transcriptome Analysis of the Black Porgy (Acanthopagrus schlegelii) under Acute Low-Temperature Stress. Life, 13(3), 721. https://doi.org/10.3390/life13030721