Abstract
Hypersensitivity pneumonitis is a complex interstitial lung syndrome and is associated with significant morbimortality, particularly for fibrotic disease. This condition is characterized by sensitization to a specific antigen, whose early identification is associated with improved outcomes. Biomarkers measure objectively biologic processes and may support clinical decisions. These tools evolved to play a crucial role in the diagnosis and management of a wide range of human diseases. This is not the case, however, with hypersensitivity pneumonitis, where there is still great room for research in the path to find consensual diagnostic biomarkers. Gaps in the current evidence include lack of validation, validation against healthy controls alone, small sampling and heterogeneity in diagnostic and classification criteria. Furthermore, discriminatory accuracy is currently limited by overlapping mechanisms of inflammation, damage and fibrogenesis between ILDs. Still, biomarkers such as BAL lymphocyte counts and specific serum IgGs made their way into clinical guidelines, while others including KL-6, SP-D, YKL-40 and apolipoproteins have shown promising results in leading centers and have potential to translate into daily practice. As research proceeds, it is expected that the emergence of novel categories of biomarkers will offer new and thriving tools that could complement those currently available.
1. Introduction
Hypersensitivity pneumonitis (HP) is a complex interstitial lung syndrome characterized by an immunological reaction to a wide variety of organic or low-molecular-weight chemical particles [1]. This immunological reaction arises with repeated and prolonged inhalation of an environmental antigen, to which the individual is sensitized [2].
Early findings suggest the disease pathogenesis is mediated by circulating specific IgG antibodies (precipitins) through the formation of antigen–antibody complexes, in a type III hypersensitivity reaction [3,4,5]. This hypothesis was challenged by the absence of precipitins in some patients, the absence of vasculitis and neutrophilic infiltrates in tissue samples and the lack of histological features of HP in animal models injected with precipitins and exposed to inciting antigens [6,7]. The important role of innate immune response was later described and both mechanisms of type III and type IV hypersensitivity reactions are currently thought to occur [8]. Polymorphisms of genes encoding molecules involved in antigen processing and presentation, remarkably those located on the major histocompatibility complex II region, appear to increase the susceptibility to a pathological sensitization process. Furthermore, the T-cell effector subset response appears to influence histological patterns, with skewing toward Th2 activity correlating with sustained inflammation and fibrotic response [8,9,10,11]. In addition to the inciting antigens, exposure to other external agents such as bacteria, fungi, mites and other inert particles and chemicals is thought to contribute to HP pathogenesis [8].
Despite efforts by international groups and scientific societies, a wide consensus on disease definition is still missing and international clinical practice guidelines on the diagnosis were only recently published for the first time [12,13]. HP was classically categorized as acute, subacute or chronic, depending on the pattern of exposure and duration of illness [14], but patients followed heterogeneous clinical courses irrespective of this category [15]. The need to add prognostic value led to a recent classification of the disease into fibrotic and non-fibrotic, according to clinical, pathological and/or imaging characteristics [12,13,16]. However, heterogeneity in clinical presentation, type and duration of exposure, cultural practices, geographical conditions and host risk factors still contribute for the under-recognition and underdiagnosis of HP. Furthermore, significant overlap of features with other clinical entities helps explain the disappointing agreement levels between multidisciplinary teams on the diagnosis (kappa value 0.29 [0.24–0.40]) [17] and common misdiagnosis with idiopathic pulmonary fibrosis (IPF) and other idiopathic interstitial pneumonias [18,19]. The variety of antigens associated with the disease, currently estimated to be more than 200 [20] and the lack of standardized techniques for testing for hypersensitivity pose as additional obstacles, as the inability to identify the relevant exposure may hamper the diagnosis and is associated with worse outcomes [21].
Altogether, these factors may partially explain discrepancies in HP frequency among different world regions and even within the same country [22,23,24,25]. Even though overall HP frequency appears to be low [26,27,28,29,30], a different perspective might be gained from cohorts and registries of interstitial lung disease (ILD), which report that HP may account for up to 47.3% of incident ILD cases [23,31]. Limited evidence details the epidemiology of acute vs. chronic HP, but approximately 40–50% of the latter corresponds to fibrotic disease [32], whose poor prognosis should be noted. With 7-year survival rates recently estimated in 40.8% [33], fibrotic HP presents poorer outcomes than many cancers [34]. The HP-associated mortality rate appears to be increasing [35], further highlighting the urge for improving accuracy and time to diagnosis and exposure identification, as these are crucial for improving the outcomes of the disease.
2. Biomarkers in Fibrotic HP
Biomarkers can be defined as “characteristics that are objectively measured and evaluated as an indicator of normal biologic processes, pathogenic processes or pharmacologic responses to a therapeutic intervention” [36]. They can be classified according to their putative application as (a) diagnostic, (b) monitoring, (c) pharmacodynamic/response, (d) predictive, (e) prognostic, (f) safety and (g) susceptibility/risk. Even though different subtypes may overlap, it is essential to pursue evidence and validation for each particular use [37]. This is the step where the majority of biomarkers fail before reaching clinical practice [38].
Even though the application of biomarkers has become routine in research and has potential to become central in clinical practice, their adoption strongly depends on their accuracy for each putative use and population, reproducibility, affordability, availability and invasiveness [39].
The development of a biomarker can be made using system biology methods to screen a large quantity of potential biomarkers for their association with the disease (unbiased approach) or selecting individual candidate biomarkers according to pre-existing knowledge on the mechanisms of disease. [39].
Sampling the alveolar milieu with bronchoalveolar lavage (BAL) may be indicated in ILDs, not only to study microbiological agents and cell populations [40], but also cell markers, soluble mediators (e.g., proteins secreted by the epithelium). On the other hand, significant deviations in the serum levels of some molecules may occur due to local overproduction and increased permeability and/or destruction of the air-blood barrier in the diseased lung [41,42,43]. This led to their potential use as serum biomarkers with diverse purposes [44].
This article aims to review the literature on biomarkers that can be quantified in the serum and/or BAL with the potential to contribute to the diagnosis of HP by discriminating patients with fibrotic HP from individuals without documented ILD or those with other ILDs. For brevity, only molecular (excluding genomic) and differential cell biomarkers are reviewed and classified according to their proposed pathobiological mechanisms of origin [45,46,47,48].
We searched MEDLINE using the query “hypersensitivity pneumonitis AND biomarker”. Clinical and preclinical original articles, reviews and statement papers potentially addressing the topic were selected for full-text analysis and included in our review after deemed relevant. This was complemented by a secondary analysis of the references of the manuscripts whose full texts were examined and by a MEDLINE search on specific biomarkers using the query “hypersensitivity pneumonitis AND (specific biomarker)”.
2.1. Immune Dysregulation
As mentioned, the pathogenesis of HP involves a complex, dysregulated cascade of immunological events in predisposed individuals. In the airway, inciting molecules are firstly recognized by pattern-recognition receptors, phagocyted, processed and expressed at the surface of innate immune cells, starting the sensitization process. This is followed by activation of adaptive immune cells and the production of several proteins, including antibodies, cytokines and chemokines [8]. The resulting inflammation leads to the apoptosis of alveolar epithelial cells, which may itself contribute to further recruitment of immune cells [49]. The differential expression of these molecules and cell subtypes has been explored as a potential diagnostic tool in HP and is summarized in Table 1.
2.1.1. BAL Lymphocyte Count
BAL lymphocytosis reflects the degree of alveolitis in chronic HP, a major characteristic of this disease [50]. These counts appear to be less pronounced in the presence of fibrosis [51,52,53,54], but their relationship with antigen exposure, host factors and disease progression is incompletely understood.
In fact, though BAL lymphocyte counts have been proposed as a diagnostic criterion for HP [55,56,57], robust evidence clarifying their role in diagnosing chronic HP [15] and their additive discriminative value in differentiating it from other fibrotic ILDs remains altogether little determined [58].
Tzilas et al. reported an added diagnostic yield for BAL lymphocytosis (using a cutoff of >20%) in a retrospective cohort of ILDs with an indeterminate for UIP pattern on high-resolution computerized tomography. In their study, BAL lymphocytosis led to a change in the diagnosis to HP in 14.7% of the patients [59].
A meta-analysis including 49 case series found significantly higher BAL lymphocyte percentages in HP and in the subgroup of fibrotic HP when compared with IPF and with sarcoidosis. However, threshold analysis for fibrotic HP vs. IPF and fibrotic HP vs. sarcoidosis yielded disappointing discriminative ability (area under curve (AUC) 0.54 (95% CI 0.51–0.58) and 0.44 (95% CI 0.41–0.47), respectively) [60].
Another systematic review (53 studies) and meta-analysis (42 studies) by Adderley et al. found a pooled estimate for the BAL lymphocyte percentage in chronic HP (cHP) of 42.8%, significantly higher than in IPF and non-IPF idiopathic interstitial pneumonias. However, the authors did not find significant differences in the BAL lymphocyte count from CTD-ILDs and sarcoidosis [61].
Inference from the existing evidence may be significantly limited by aggregation of studies with heterogeneous diagnostic criteria and incorporation bias. Still, the role of BAL lymphocyte counts in distinguishing fibrotic HP from IPF and sarcoidosis is currently recognized [12]. However, due to severe heterogeneity and limited number of studies, ideal cutoff values cannot be established, nor their role in differentiating chronic HP from other ILDs.
To conclude, a high BAL lymphocyte count is likely to be more useful for establishing diagnostic likelihood of HP in the setting of multidisciplinary discussion, in conjunction with other diagnostic elements, rather than relied upon its own [12,13].
2.1.2. BAL CD4/CD8 Ratio
CD4 lymphocyte predominance and increased CD4/CD8 ratios on the BAL have been documented in chronic HP, when compared with acute and subacute forms but the use of these markers to diagnose or exclude the disease in the clinical setting is limited by inconsistent findings and significant variation with external factors [9,62,63,64,65,66,67,68,69,70]. Moreover, CD4/CD8 ratio analysis does not appear to improve diagnostic accuracy in ILD when added to cell count analysis [71].
Still, in spite of significant between-study variability, pooled estimates point to lower CD4/CD8 ratios in HP when compared with sarcoidosis [61] and a high ratio (>3.5–4) is currently considered highly specific and useful to support the latter [72,73]. The authors, thus, consider that this parameter may be valuable in specific settings where the differential diagnosis of these two conditions may be challenging.
2.1.3. Cytokines and Cytokine Receptors
Upon exposure to the inciting antigen, antigen presenting cells (APCs) release a panoply of cytokines including IFN-γ, IL-1, IL-6, IL-12, IL-18 and TNF-α, and there appears to be no difference in the BAL concentrations of most of these molecules between patients with acute and chronic HP [74]. Meanwhile, upon being presented with processed antigens, T-cell response can evolve to a variety of subsets. While Th1 milieu (e.g., IFN-γ, TNF-α, IL-2, IL-12) appears to predominate and takes part in granuloma formation [75,76], the current evidence suggests that a skew to Th2 response (IL-4, IL-13) plays a significant role in the maintenance of inflammation and development of a fibrotic response in later stages [8,9,10,11,77]. Th17 response (IL-17, IL-22) arises alongside Th1 response and its absence appears to lessen inflammation and fibrosis [78,79,80].
Barrera et al. collected BAL fluid from PH patients, primed it with pigeon serum and assessed a number of cytokines. They found that patients with subacute disease showed higher percentages of CD4+ and CD8+ lymphocytes expressing IFN-γ when compared with those with the chronic form, whose CD8+ lymphocytes expressed higher IL-4 levels [9]. Similarly, culture supernatants obtained from the BAL of chronic HP patients presented higher IL-4 and lower IFN-γ(9). Studying the IL-4/IL-4R axis may also be useful to discriminate HP from sarcoidosis, as IL-4R BAL levels appear to differ, but this requires further validation [81,82]. Indeed, several studies comparing cytokine levels in the BAL of HP patients with controls and/or other ILDs have been published.
TGF-β has been shown to have a critical role in the development of lung fibrosis and elevated levels have been demonstrated in animal models after exposure to bacteria [80,83]. Increased levels in the serum of patients with idiopathic interstitial pneumonias (IIP), including IPF, vs. healthy controls have been reported [84] and the BAL levels in HP appear to be lower when compared with IPF [85], but a complete threshold analysis has not been performed. Similar levels have been observed in patients with HP and sarcoidosis [81].
TNF-α in the BAL of patients with HP appears to be lower than on fibrotic IIP and connective tissue disease-associated ILD (CTD-ILD) [86] but not from those with sarcoidosis [81].
IL-8, a major neutrophil chemoattractant, is overexpressed in the BAL in several ILDs, including HP, sarcoidosis and IPF [85,87,88], and it demonstrated significantly different levels between the former two, even though only cases of acute HP were included [88].
Measuring the change in the BAL cytokine profile with inhalation provocation tests may also allow to distinguish HP from other ILD [89], even though the true discriminatory ability still needs to be assessed.
To conclude, there is a distinctive contribution of cytokines to the inflammatory and fibrogenic processes associated with the disease (Figure 1). Differences in these profiles between HP and other ILDs and/or healthy controls have been documented, especially in the BAL, but validation for diagnostic testing is still lacking.
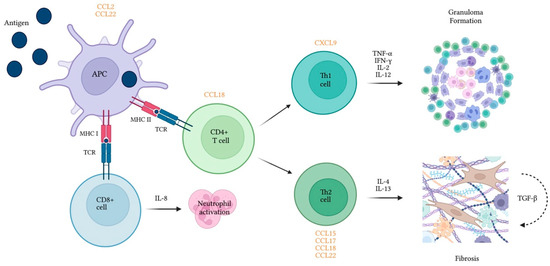
Figure 1.
Cytokines and chemokines released by immune cells play a role in inflammatory and fibrotic processes associated with hypersensitivity pneumonitis. Chemokines are presented in orange in proximity to the cell type they recruit. APC—Antigen presenting cell; CCL—Chemokine ligand; CD—Cluster of differentiation; CXCL—Keratinocyte-derived cytokine ligand; IFN-γ—Interferon gamma; IL—interleukin; MHC—Major histocompatibility complex; TCR—T-cell receptor; TGF-β—Tumor growth factor beta; TNF-α—Tumor necrosis factor alpha.
2.1.4. Chemokines and Receptors
Chemokines are small proteins synthesized by several cell types and are recognized for their ability to stimulate cell migration. Similar to interleukins, a differential expression of chemokines has been shown in chronic HP (Figure 1) and proved useful mostly for predicting prognosis. Some studies, however, reported different levels vs. healthy controls or other ILD groups.
Chemokines such as MIP-1a/CCL3 and CCL5 are released when an APC interacts with the inciting antigen. Additionally, TH2-related chemokines (e.g., TARC/CCL17) were found in the serum of patients with HP, particularly those with an acute exacerbation [90]. Higher serum and BAL levels were also found in patients with UIP-like pattern, when compared with patients with a Nonspecific Interstitial Pneumonia (NSIP)/Organizing Pneumonia (OP) pattern [11].
CCL15 serum levels were higher in chronic HP when compared with IPF and controls [91], but discriminatory analysis remains to be performed. A study by Nukui et al. reported [92] higher CXCL9 (Th1) and CCL17 (Th2) serum levels in chronic bird-related HP compared to healthy controls [93].
The levels of CCL18 (also known as PARC and MIP-4) were initially reported to be higher in tissue biopsies of patients with HP, when compared with IPF and controls [94]. Tissue levels were inversely correlated with the extension of fibrosis and directly correlated with BAL lymphocytes, in line with the results obtained by Cai et al., who reported a correlation between BAL CCL18 and lymphocytes as well as a trend for lower CCL18 in chronic vs. acute/subacute HP. The same group found higher serum and BAL CCL18 levels in HP compared to IPF, COP and sarcoidosis [95].
Abnormal expression of CCL2 (MCP-1), an important monocyte/macrophage-mediated chemoattractant, has been documented in the lungs of patients with IPF and other ILDs [96]. Lymphocytes from patients with HP overexpress this protein in the presence of fibrocytes [97] and may display elevated BAL levels when compared with healthy controls [88,98]. Interestingly, even though serum and BAL elevations are also found in IPF, sarcoidosis and CTD-ILD [98], serum levels appear to be lower in fibrotic HP compared to IPF, with acceptable discriminative ability (AUC 74%) [99].
CCL22 (MDC) participates in macrophage recruitment and plays a pivotal role in several Th2 conditions such as asthma and eosinophilic pneumonia [100,101]. High levels in the BAL of patients with IPF and HP have been reported [85].
2.1.5. Specific IgG Antibodies
Even though HP is believed to be mostly a consequence of T-cell-mediated immunity, B lymphocytes are also presented with processed antigens by innate immune cells, triggering the production of specific IgGs [8]. Resulting immune complexes appear to mediate lung injury [49,102].
Even though lists of proven and suspicious exposures and antigens are growing [8], testing for specific IgG antibodies against potential antigens plays an important role in the diagnostic approach. An elevation of these biomarkers, however, reflects previous exposure and sensitization rather than the disease itself [103]. This is corroborated by (a) the evidence of high serum levels among those regularly exposed but not diseased (b) the absence of vasculitis and neutrophilic infiltration in diseased lung tissue and (c) the absence of histologic features of HP in animal models exposed to antigens after being injected with precipitins [7,103].
There are several methods for assessing serum IgG antibodies, both qualitive and quantitative. The major limitations for their use for diagnosis are the lack of technical standardization for immunoassays and antigen preparations, cross-reactivity among antigens, variable cutoffs for quantitative assays and limited test-performance validation in specific populations [13]. Nonetheless, current clinical guidelines endorse the role of specific IgG antibodies as ancillary methods to document exposure to a culprit antigen and increase the likelihood of HP [12,13].
Jenkins et al. systematically reviewed and pooled studies on serum IgG testing using ELISA assays or precipitins and concluded that this parameter was able to discriminate HP from other ILDs with a sensitivity of 83% (95% CI, 28–98%) and a specificity of 68% (95% CI, 38–90%) [104], an suboptimal performance for it to be used as an individual diagnostic test in most clinical settings. Results from the same study revealed a capacity to distinguish patients with HP from healthy exposed subjects with a sensitivity and specificity of 90% (95% CI, 62–98%) and 91% (95% CI, 69–98%), and from healthy unexposed individuals with 93% (95% CI, 59–100%) and 100% (95% CI, 99–100%) sensitivity and specificity, respectively [104].
In an interesting prospective case-cohort study, Morell et al. found a positivity rate of 63% (n = 29) for specific IgG tests in a population of patients fulfilling the 2011 ATS/JRS/ALAT Guidelines IPF criteria, 18 of whom were confirmed to be fibrotic HP in the follow-up period [19].
Summing up, in the appropriate clinical setting and when added to other relevant sources of information, IgG testing may be useful to support the diagnosis of HP or even suggest a previously unnoticed inciting source [15]. This is particularly relevant in the case of fibrotic HP, where failure to identify an occult exposure may lead to a misdiagnosis (namely of IPF) and deprive the patient of adequate counseling for antigen avoidance and immunomodulatory therapy [105].
2.1.6. Markers of Macrophage Activation
YKL-40
YKL-40 (also chitinase 3-like 1) is a chitinase-like protein expressed by a variety of cells, including neutrophils, epithelial cells and APCs, in response to specific stimuli [106,107,108,109]. It plays an important role in several biological cascades in the respiratory system, inhibiting oxidant-induced injury, stimulating adaptive Th2 immunity, regulating cell apoptosis and alternative macrophage activation, ultimately contributing to fibrosis and wound healing [106]. It is not surprising, thus, that it has been described as a biomarker in diseases characterized by inflammation, fibrosis and remodeling, IPF and sarcoidosis among them [109,110,111,112]. In fact, the usefulness of YKL-40 in the diagnosis of ILD (versus healthy controls) was summarized in a systematic review and meta-analysis, which revealed overall highest serum levels in sarcoidosis when compared with other ILDs [113]. Unfortunately, studies on HP were not included. Patients with HP appear to have higher serum levels than healthy controls, with the most favorable cutoffs showing, however, suboptimal discriminatory ability [114,115,116]. On the other hand, serum YKL-40 levels seem to be lower than in other ILDs, such as idiopathic NSIP, OP and IPF [114,115]. In fact, this biomarker’s acceptable ability to discriminate between HP and IPF [114,115] is worth noting and further exploring in the future, given the clinical relevance of this issue.
2.1.7. Acute Phase Reactants
Serum Amyloid A (SSA)
SSA is an apo-lipoprotein mainly produced in the liver by activated monocyte cells in response to proinflammatory cytokines but is also expressed by other extrahepatic cell types [117,118]. It has shown several functions, including the ability to induce the synthesis of cytokines and to attract neutrophils and mast cells.
The elevation of SSA levels has long been described in sarcoidosis [119,120,121,122], a finding substantiated by the description of the role of this molecule as an innate regulator of granulomatous inflammation in this disease [118]. Chen et al. reported the overexpression of SSA in granulomatous tissue on lung and lymph nodes of sarcoidosis patients, with a greater extent when compared to other granulomatous diseases including HP [118]. This study also showed a correlation with collagen deposition and tissue fibrosis. These findings are in line with those of higher SSA levels in patients with fibrotic vs. non-fibrotic sarcoidosis [123] and with the proposed role for SSA in ECM remodeling and fibrogenesis, favoring a profibrotic cytokine environment and metalloprotease expression by fibroblasts [124,125]. In fact, the increase in SSA levels in other fibrotic diseases has also been widely demonstrated [123,124,126,127,128]. For HP, particular emphasis should be given to the different serum levels of patients with HP vs. IPF and controls, translating into an acceptable-to-excellent discriminatory ability [126,128]. Later conflicting evidence [123], however, highlighted the need to further explore the abilities of this biomarker for the diagnosis of fibrotic HP (fHP).

Table 1.
Studies exploring biomarkers of immune dysregulation in individuals with hypersensitivity pneumonitis.
Table 1.
Studies exploring biomarkers of immune dysregulation in individuals with hypersensitivity pneumonitis.
| Biomarker | Investigator | Type of Study | Population/Methods | Threshold Analysis (Cutoffs)/Other Results | Conclusion(s) of Interest | Main Limitations |
|---|---|---|---|---|---|---|
| BAL Lymphocytes | Patolia et al. [60] | Systematic review and meta-analysis | Studies on BAL cell counts in ILDs; comparison of BAL% lymphocytes in HP vs. IPF or sarcoidosis | No 20% BAL lymphocytes: fHP vs. IPF: Sen 69% Sp 61% fHP vs. sarcoidosis: Sen 69% Sp 26% | BAL lymphocytes: Higher in HP vs. IPF Higher in HP vs. sarcoidosis | No RCTs Possible incorporation bias of the studies reviewed cHP used as surrogate for fibrotic HP |
| Adderley et al. [61] | Systematic review and meta-analysis | Studies on BAL cell analysis in ILDs; estimation of pooled BAL% lymphocytes for cHP vs. non-cHP ILDs | Yes 21.3% BAL lymphocytes: CHP vs. non-CHP ILD Sen 66.5% Sp 65.9% 21% BAL lymphocytes CHP vs. IPF/non-IPF IIP Sen 70.7% Sp 67.6% | BAL lymphocytes: Higher in cHP vs. non-cHP ILD | No RCTs Possible incorporation bias affecting the included studies Heterogeneity of cHP diagnostic criteria among studies | |
| BAL CD4/CD8 cell ratio | Adderley et al. [61] | Systematic review and meta-analysis | Studies on BAL cell analysis in ILDs; estimation of pooled BAL ratios for CHP vs. non-cHP ILDs | No. CD4/CD8 Ratio pooled estimates: cHP 1.6 IPF 1.6 Sarc 4.6 | CD4/CD8 Ratio: CHP vs. IPF not different cHP ≠ sarcoidosis | Small sample size of non-CHP groups Possible incorporation bias affecting the included studies High between-study heterogeneity |
| Barrera et al. [9] | Prospective cohort | Patients with aHP, saHP, cHP and healthy controls; comparison of BAL CD4/CD8 and cytokines in BAL cell culture supernatant | No. Median CD4/CD8 Ratio: cHP 3.05 SaHP 1.3 Controls 1.3 IL-4 (pg/mL): cHP 80 saHP 25 IFN-γ (pg/mL): cHP 3.82 saHP 100 TGF-β1: No difference | CD4/CD8 Ratio: Higher in CHP vs. subacute HP and healthy controls | Former diagnostic criteria and classification of HP Only Pidgeon Breeder’s Disease included Single center study: small sample | |
| Cytokines, Chemokines and receptors | Ye et al. [74] | Cross-sectional study | Patients with aHP, cHP and healthy controls; comparison of cytokine concentration in alveolar macrophage culture supernatant | No. IL-12 (pg/mL): cHP 15.4 Controls 1.1 IL-18 (pg/mL): aHP 201 cHP 224 Controls 56 TNF-α (pg/mL): aHP 3219 cHP 1522 controls 249 | No difference between aHP and cHP | Own diagnostic and classification criteria Single center study; Small sample size |
| Matěj et al. [81] | Cross-sectional study (case-control analysis) | Patients with HP and sarcoidosis; comparison of BAL cytokine concentration at diagnosis | No IL-4R (pg/mL): HP 1182 Sarc 303 PAR-2 (pg/mL): HP 2009 Sarc 330 TGF- β and TNF-α: No difference | PAR-2 as a potential tool in the differential diagnosis | Single center study; small sample HP diagnostic criteria and inciting antigens not detailed | |
| Sterclova et al. [82] | Cross-sectional study (case-control analysis) | Patients with cHP and sarcoidosis; determination of IL-4/IL-4R axis role | No. IL-4Ra: cHP 1190 Sarc 303 IL-4: No difference | BAL IL-4Ra significantly higher in cHP vs. sarcoidosis | Own HP diagnostic criteria Single center study; Small sample size | |
| Stijn et al. [85] | Cross-sectional study (case-control analysis) | Patients with cHP, IPF and controls without pulmonary disease. BAL protein comparison (multiplex) at diagnosis | No. TGF-β1 lower in cHP vs. IPF and controls IL-8 lower in cHP vs. controls MMP-8, MMP-9, MCP-1, MDC, MPO and Protein-C higher in cHP vs. controls IL-17a and IL-23, RAGE, SP-C, TIMP-1, fibronectin, eotaxin, IL-17A, IL-23, PARC, RANTES, TSLP, PlGF, FGFb, and tissue factor: no difference | No considerations on use as tools in differential diagnosis. | Multidisciplinary meeting diagnoses—criteria and inciting antigens not detailed Single center study; Small sample size | |
| Bruzova et al. [86] | Cross-sectional study (case-control analysis) | Patients with IPF, fIIP, CTD-ILD and cHP Comparison of BAL supernatant protein levels | No TNF-α HP 3.41 fIIP 1.52 CTD-ILD 2.33 IL4R-α and PAR2: no differences MMP-7: no differences | TNF-α higher in cHP vs. fIIP and CTD-ILD No differences in IL-4Ra, PAR-2 and MMP-7 TNF-α as a potential diagnostic tool in IIP | Inciting antigens not detailed Predominance of non-fibrotic cases in HP Single center study; relatively small sample | |
| Sterclova et al. [87] | Cross-sectional study (case-control analysis) | Patients with IPF and cHP. Comparison of BAL supernatant protein levels | No. I-TAC/CXCL11, IP-10/CXCL10, IL-8/ CXCL8 and ENA-78/CXCL5: no differences | No difference in the studied proteins | Former diagnostic criteria of IPF; own criteria of HP and little detailed Inciting antigens not detailed Single center study; Small sample size | |
| Sugiyama et al. [88] | Cross-sectional study (case-control analysis) | Patients with summer-type aHP, pulmonary sarcoidosis and control. Comparison of BAL supernatant protein levels | No. IL-8: aHP 30.8 Sarc 11.7 Controls 7.4 MCP-1: aHP 34.8 Sarc 45 Controls 10.6 | IL-8 higher in HP than sarcoidosis MCP-1 higher in aHP vs. controls, not sarcoidosis | Only summer-type HP included Single center study; Small sample size | |
| Inoue et al. [89] | Cross-sectional study (case-control analysis) | Patients with HP and other ILDs; comparison of BAL and serum potential biomarkers after IPT with pigeon extract in avian HP and other ILDs (including non-avian HP) | No BAL G-CSF, IL-6, and IL-17 higher after IPT only in avian HP Serum leukocytes and neutrophils elevated after IPT only in avian HP | Neutrophils and the cytokine/chemokine-associated millieu increase with IPT in avian HP but changes in cytokines/chemokines should be carefully interpreted (risks of multiple comparisons) | Former diagnostic/classification criteria Single center study; small sample Only avian IPT performed | |
| Watanabe et al. [91] | Cross-sectional study (case-control analysis) | Patients with cHP, IPF and healthy controls; comparison of serum and BAL CC15 | No Serum (μg/mL): cHP 29.1 IPF 19.7 controls19.5 BAL (μg/mL): cHP 0.76 IPF 0.54 | Serum CCL15 higher in cHP vs. IPF; | Former diagnostic/classification criteria Single center study; Small sample size The assessment of inciting antigens was performed using precipitins only | |
| Nukui et al. [93] | Retrospective cohort (case-control analysis) | Patients with cHP and healthy volunteers; comparison of serum CXCL9, CCL17 and KL-6 | No KL-6 (U/mL): cHP 1182 controls 184 CXCL9 (pg/mL) cHP 19.3 controls 10.5 CCL17 (pg/mL) cHP 543 controls 274 | KL-6, CXCL9 and CCL17 higher in avian cHP vs. healthy controls | Former diagnostic criteria Only avian cHP defined only by IPT | |
| Cai et al. [95] | Cross-sectional study (case-control analysis) | Patients with HP and other ILDs; comparison of serum and BAL CCL18 levels | No Serum (ng/mL): HP 190 IPF 149 COP 146 Sarc 108 BAL (ng/mL) HP 13 IPF 6 RB-IL/DIP 54 COP 5 Sarc 7 iNSIP 9 | CL18 level highest in HP among the investigated ILDs | Own diagnostic criteria for HP and former for IIP; saHP and cHP considered in the same group Single center study; Small sample size | |
| Garcia de Alba [97] | Cross-sectional study (case-control analysis) | Patients with cHP and healthy controls; comparison of serum and BAL CXCL12 levels | No Serum (pg/mL): cHP 2302 controls 813 BAL (pg/mL) cHP 493 controls undetectable | CXCL12 levels higher in cHP vs. controls. No considerations on use as diagnostic tool | Own/former HP criteria Only bird-related HP Single center study; Small sample size | |
| Suga et al. [98] | Cohort study (case-control analysis) | Patients with ILD (IPF, AIP, CTD-ILD, OP and HP); Comparison of serum and BAL MCP-1/CCL2 levels | No BAL levels higher in IPF vs. HP Serum levels higher in IPF, Sarc, and CTD-ILD vs. HP and controls | Differences in the pattern of MCP-1 in BALF and serum may help in the differential diagnosis of ILD. | Former diagnostic criteria Single center study; Small sample size | |
| Specific IgG antibodies | Jenkins et al. [104] | Systematic Review and meta-analysis | Studies on serum IgG testing and questionnaires in HP | No. HP vs. other ILDs Sen 83% Sp 68% HP vs. E-controls Sen 90% Sp 91% HP vs. u-controls Sen 93% Sp 100% | IgG testing insufficient to distinguish HP from other ILDs but can be useful in screening for exposures and provide supportive of HP | No RCTs Small studies included Risk of incorporation bias |
| Chitinase-3-like protein 1 (YKL-40) | Long et al. [114] | Retrospective cohort (case-control analysis) | Patients with HP, IPF, iNSIP, COP and healthy controls; Comparison of serum and BAL YKL-40 | Yes Serum (ng/mL) HP 127 IPF 214 iNSIP 184 COP 213 controls 39 a/saHP 179 cHP 117 BAL (ng/mL) HP 21 IPF 9 a/saHP 42 cHP 15 control 3 Optimal cutoff serum: HP vs. controls: 47 AUC 0.90 HP vs. IPF: 134 AUC 0.727 | Serum YKL-40 higher in HP vs. controls but lower vs. other ILDs BAL YKL-40 higher in HP vs. controls and IPF No considerations on use as diagnostic tool | Former diagnostic and classification criteria Single center study; Small sample size |
| Sanchez-Diaz et al. [115] | Cross-sectional study (case-control analysis) | Patients with HP, IPF, sarcoidosis and healthy controls; Comparison of serum YKL-40 and KL-6 | Yes Serum YKL-40 HP 56 ng/mL IPF NM Optimal cutoff for HP vs. IPF: Serum YKL-40 121 ng/mL AUC 0.741 Serum KL-60: 1441 U/mL AUC 0.702 | Serum HP higher in HP vs. IPF | Ancient diagnostic criteria; own classification criteria Single center study; Small sample size | |
| Serum Amyloid A | Vietri et al. [127] | Prospective cohort (case-control analysis) | Patients with IPF and non-IPF ILDs including sarcoidosis, cHP, PLC and healthy controls; comparison of SAA concentrations in the different groups | No SAA: IPF6418 ng/mL; cHP 4494 ng/mL Optimal cutoff for IPF vs. cHP: 5397 ng/mL AUC 0.79 | SAA Higher in IP levels vs. other ILDs; potential to differentiate IPF from cHP | Diagnostic criteria of diseases not detailed; inciting antigens not detailed Single center study; Small sample size |
| Bergantini et al. [128] | Coss-sectional study (case-control analysis) | Patients with IPF, sarcoidosis, cH. Comparison of fibrotic/inflammatory markers | Yes. KL-6 (U/mL) cHP 1146 Sarc 537 IPF 2062 SAA (ng/mL) cHP 4022 Sarc 4370 IPF 7031 Optimal cutoff for cHP vs. IPF: KL-6: 2206 AUC 0.74 SAA: 53,971 AUC 0.85 KL-6 + SAA: AUC 0.79 | KL-5 different in cHP vs. IPF and sarcoidosis SAA different in cHP vs. IPF Combined panel might improve diagnostic accuracy in multidisciplinary setting | Diagnostic and classification criteria not detailed Single center study; Small sample size |
Studies comparing biomarkers of immune dysregulation in human individuals with HP vs. other ILDs and/or healthy controls. Biomarkers of other classes are highlighted. aHP—Acute hypersensitivity pneumonitis; AIP—Acute interstitial pneumonia cHP—Chronic Hypersensitivity Pneumonitis; COP—Cryptogenic Organizing Pneumonia CTD—Connective Tissue Disease; fIIP—Fibrosing Idiopathic Interstitial Pneumonia; ILD—Interstitial Lung Disease; iNSIP—idiopathic nonspecific interstitial pneumonia IPF—Idiopathic Pulmonary Fibrosis; IPT—inhalation provocation testing; PLCH—Pulmonary Langerhans Cell Histiocytosis; RCT—Randomized Controlled Trial; SAA—Serum Amuloid A; saHP—subacute hypersensitivity pneumonitis; Sarc—Sarcoidosis; Sen -Sensitivity; Sp—Specificity.
2.2. Epithelial Cell Dysfunction
Lung epithelial cells have a crucial role in defending the host from external aggressions through different mechanisms, including secretion of mucus, surfactant and several other defense proteins [129]. Epithelial cell injury and apoptosis are well documented in hypersensitivity pneumonitis [130], as well as significant changes in levels of epithelial cell-related biomarkers (Figure 2, Table 2).
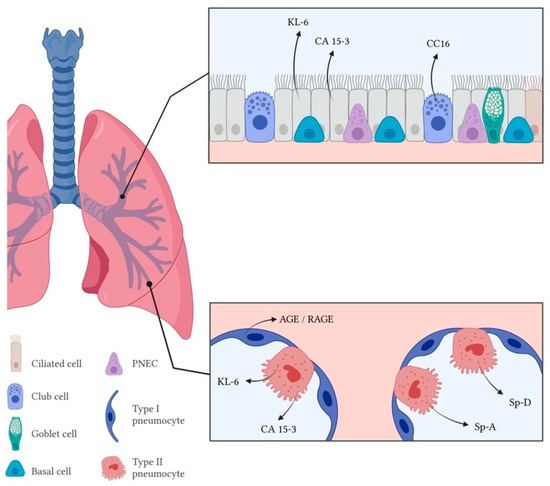
Figure 2.
Markers of epithelial cell dysfunction of respiratory epithelium in the airway (upper rectangle) and alveolar space (lower rectangle) in hypersensitivity pneumonitis. PNEC—Pulmonary neuroendocrine cells; KL-6—Krebs von den Lungen-6; CA 15-3—Carcinoma antigen 15-3; CC16—Club-cell protein 16; RAGE—Receptor for advanced glycation end products; Sp-A—Surfactant protein A; Sp-D—Surfactant protein D.
2.2.1. Mucin-Associated Antigens
Krebs Von Den Lungen-6 (KL-6)
KL-6, a sub-molecule of Mucin 1, is a high molecular weight glycoprotein expressed by type 2 pneumocytes and bronchiolar epithelial cells and has been one of the first biomarkers of lung damage to be reported, both in the serum and the BAL [41,131,132]. Correlation with bronchiolar epithelial cell damage and regeneration and with progressive interstitial thickening boosted it as a promising biomarker for detecting, assessing activity and predicting prognosis of several ILDs [133,134].
Evidence has consolidated the utility of serum levels in differentiating patients with ILD, including HP, from subjects without ILD [41,93,135,136,137,138], but low diagnostic accuracy limits the utility in ILD differential diagnosis [133,139,140].
In an early study, Takahashi et al. described significantly higher serum KL-6 concentrations in patients with farmer’s lung disease when compared with farmers without the disease (including the same subjects prior to developing the disease) [137]. Interestingly, lower diffusing capacity for carbon monoxide (DLCO) and KCO levels were also noted in farmers with high serum KL-6 levels, regardless of fulfillment of the criteria for HP [137]. Ji et al. later found higher serum KL-6 levels in bird fanciers vs. controls, and in acutely symptomatic fanciers vs. asymptomatic ones [141]. In this study, KL-6 was also associated with specific IgG antibodies and this correlation was stronger in symptomatic subjects.
Okamoto et al. reported an increased serum value of KL-6 in chronic HP when compared with IPF, CTD-ILD and sarcoidosis and were able to determine an optimal cutoff value of 1115 U/mL for the differential diagnosis with IPF, noting low specificity as a major limitation [142]. Onishi et al. showed significantly higher serum levels of KL-6 in cHP patients when compared with IPF [143].
Besides the study from Lanzarone et al., who found a correlation between KL-6 levels and ground glass opacities score [140], we found no other studies on KL-6 discriminating fHP, but there is some evidence on the correlation between KL-6 levels and fibrotic extension in other entities [144,145]. These data point to a plausible role of KL-6 in identifying individuals with fibrotic HP among healthy subjects and as a surrogate marker for antibody titration in individuals whose exposure cannot be identified.
CA 15-3 and CA125
Like KL-6, Carcinoma Antigen 15-3 (CA 15-3) assays target a Mucin 1 (MUC1)-derived glycoprotein and are believed to reflect fibrotic processes and immunologic disease activity in several ILDs [146,147]. Compared with KL-6, this test has the advantages of wider implementation and lower cost associated with its use in breast cancer. CA 15-3 levels were found to correlate with KL-6 levels in patients with fibrotic ILDs, including HP, and to correlate with fibrosis scores [147,148,149,150]. The diagnostic ability of the test has not, however, been explored, to the best of our knowledge, but it provides a potential field for future research.
2.2.2. Surfactant-Associated Proteins
Surfactant protein A (Sp-A) and surfactant protein D (Sp-D) are members of the epithelial collectins, a group of soluble pattern-recognition receptors produced by type II pneumocytes and involved in the innate immune response in both pulmonary and non-pulmonary tissues [151,152,153]. They are major protein components of lung surfactant, being responsible for its homeostasis [151,152,153]. Alongside KL-6, Sp-D is one of the most widely accepted diagnostic tests for ILD, particularly in Japan, being useful as a marker of disease activity as well [142]. Tanaka et al. first described the elevation of serum Sp-D levels in two patients with mushroom worker’s lung, reporting a decrease with antigen avoidance and steroid therapy [154].
Janssen et al. demonstrated a relevant discriminatory capacity of serum Sp-D in patients with bird fancier’s lung vs. controls (AUC 0.96) [155].
Okamoto et al. analyzed serum Sp-D levels in acute and chronic HP and compared them with three other groups of patients diagnosed with IPF, sarcoidosis and CTD-ILD. The authors report higher Sp-D levels in acute vs. chronic HP, and in both when compared with every other group. Additionally, at an optimal cutoff of 209 ng/mL, this biomarker could reasonably discriminate between chronic HP and IPF (AUC = 0.729) [142]. Similarly, Onishi et al. also demonstrated higher levels of Sp-D in chronic summer-type hypersensitivity pneumonitis when compared with IPF [143].
Guzman et al. first reported the increase in Sp-A+ alveolar macrophages in HP [156]. This was later corroborated by other groups, who reported significant increases in Sp-A in the BAL of patients with HP vs. healthy controls [157,158,159]. Hamm et al. and Phelps et al. reported increased levels in other ILDs, namely sarcoidosis and IPF, but did not find differences from patients with HP [157,159].
This evidence underlines the potential utility of surfactant proteins, particularly Sp-D, in the diagnosis of HP, although both still require proper validation for clinical use.
2.2.3. Club-Cell Protein
Club-Cell Protein 16 (CC16, also known as CC10 and uteroglobin) is secreted primarily by the Club Cells (non-ciliated bronchial epithelial secretory cells). Its functions are not completely understood, but it appears to prevent fibrosis in animal models, possibly by reducing profibrotic cytokine and metalloproteinase activity [160,161,162]. On the other hand, cytokines such as IFN-γ, TNF-α and IL-10 have proven capable of increasing CC16 expression in different cell populations [163,164,165].
The dysregulation of CC16 homeostasis has been associated with several airway and parenchymal diseases [166]. Concerning ILDs, elevated levels are found on IPF, CTD-ILD (including those associated with Sjogren’s, rheumatoid arthritis and systemic sclerosis) and HP [167,168,169,170]. Elevated serum levels in HP when compared with healthy controls [170] are relevant but unlikely sufficient to reach clinical practice. On the other hand, even though ROC analysis describes a sub-optimal discriminatory power, lower serum levels (in conjunction in CTD-ILD) vs. IPF [169] could prove useful in daily clinical practice but further studies on this matter are required.
2.2.4. AGE and RAGE
The receptor for advanced glycation end products (RAGE) is a member of the immunoglobulin superfamily, expressed mainly in the basal membrane of type 1 pneumocytes. It was first identified as the receptor for advanced glycation end products but also binds to a large variety of endogenous ligands, including ECM components such as collagen type 1 and type 4 [171]. Several effects have been attributed to this molecule. After binding, RAGE initiates intracellular cascades that lead to the transcription of proinflammatory genes [172]. It was also shown to promote leukocyte adhesion and recruitment by binding macrophage integrin MAC-1 [173] and inducing the expression of vascular cell adhesion molecule (V-CAM) in endothelial and mesothelial cells [174,175]. Finally, it is also involved in type 1 pneumocyte differentiation, epithelial adhesion to the basement membrane and lung re-epithelialization [176,177,178].
AGEs result from the reaction between monosaccharides and free amino protein groups [179]. Besides being related with oxidative stress, aging and inflammation, they have also been shown to act on ECM protein cross-links and reduce tissue viscoelasticity [180,181].
Even though distinct patterns of expression, particularly for RAGEs, have been found in different models of lung fibrosis, decreased RAGE and increased AGE expression have been described in the lungs and serum of patients with IPF when compared with controls [182,183,184], which is not completely corroborated by other studies [85]. The AGE/soluble RAGE (sRAGE) ratio was found to be different between IPF and cHP, with moderate discriminatory ability [183]. Interestingly, both serum AGE, sRAGE and AGE/sRAGE ratio distinguished patients’ cHP from fibrotic NSIP [183]. The usefulness of these biomarkers in the differential diagnosis of fibrotic HP deserves further exploring.
Table 2.
Studies exploring biomarkers of epithelial cell dysfunction in individuals with hypersensitivity pneumonitis.
Table 2.
Studies exploring biomarkers of epithelial cell dysfunction in individuals with hypersensitivity pneumonitis.
| Biomarker | Investigator | Type of Study | Population(s)/Methods | Threshold Analysis (Cutoffs)/Other Results | Conclusion(s) of Interest | Main Limitations |
|---|---|---|---|---|---|---|
| Krebs von der Lungen (KL-6) and surfactant proteins (SPs) | Takahashi et al. [137] | Cross-sectional study (case-control analysis) | Dairy farmers with and without HP (controls); comparison of serum KL-6 levels in HP, precipitin positive (Ab+) and precipitin negative (Ab−) controls | No KL-6 (U/mL): HP 1263 Ab+ controls: 328; Ab− controls: 207 | KL-6 higher in farmers with HP vs. controls and higher in seropositive vs. seronegative controls KL-6 higher in individuals after diagnosis vs. before diagnosis | Only farmers’ lung disease; Single center study; Small sample size |
| Okamoto et al. [142] | Retrospective longitudinal study (case-control analysis) | Patients with cHP, aHP, IPF, CVD-ILD; Comparison and validation of serum KL-6 and SP-D | Yes KL-6 and SP-D higher in aHP and cHP vs. IPD, CVD-ILD and sarcoidosis Not different in aHP vs. cHP Optimal cutoff for cHP vs. IPF: KL-6 1115 U/mL AUC 0.771 SP-D 209 ng/mL AUC 0.729 KL-6 and SP-D lower after 1-month steroid therapy | Higher KL-6 and SP-D shoul raise suspicion of HP Potential use in disease management | Own HP diagnostic criteria of HP; Former diagnostic criteria of IPF and sarcoidosis; Single center study. | |
| Onishi et al. [143] | Cross-sectional study (case-control analysis) | Patients with cHP and IPF; comparison of serum biomarkers | No KL-6: cHP 1506 U/mL IPF 914 U7 mL SPD cHP 235 ng/mL IPF 156 ng/mL | KL-6 and SP-D higher in cHP vs. IPF Both biomarkers included in proposed composite criteria for chronic summer-type HP | Only summer-type HP Former diagnostic/classification criteria Single center study; small sample | |
| Janssen et al. [155] | Retrospective longitudinal study (case-control analysis) | Patients with HP and healthy exposed and unexposed controls; comparison and validation of serum KL-6 and SP-D | Yes KL-6: cHP 883 U/mL E-controls 371 U/mL U-controls 177 U/mL SP-D: cHP 201 ng/mL U-controls 68 ng/mL Optimal cutoff: KL-6 275 U/mL AUC 0.98 SP-D 98 ng/mL AUC 0.96 | KL-6 and SP-D decrease with antigen avoidance; potential markers of disease | Criteria for diagnosis and classification of HP not detailed Only bird fancers’ lung disease Single center study; Small sample size | |
| Hamm et al. [157] | Cross-sectional study (case-control analysis) | Patients with HP, sarcoidosis and controls without ILD; comparsion of BAL supernatant SP-A levels | No BAL SpA: HP 9 μg/mL Sarc 8 μg/mL Controls 4.0μg/mL | BAL SP-A are elevated in sarcoidosis and HP but not specific or diagnostic | Own diagnostic criteria for HP and sarcoidosis Single center study; Small sample size | |
| Phelps et al. [159] | Cross-sectional study (case-control analysis) | Patients with IPF, HP and controls; comparison of SP-A | No BAL SP-A: HP 13.11 ug/mL IPF 7.99 μg/mL controls 4.77 μg/mL | BAL SP-A elevated in IPF (both vs. HP and controls); significance of the finding incompletely understood | IPF diagnostic criteria not detailed; Own HP criteria Only avian antigen-induced HP Single center study; Small sample size | |
| Club-cell protein (CC16) | Buendia-Roldan et al. [169] | Cross-sectional study (case-control analysis) | Patients with IPF, non-IPF ILD (CTD-ILD + cHP) and healthy controls; comparison and validation of serum and BAL CC16 | Yes. Serum CC16 higher in non-IPF vs. controls Optimal cutoff for IPF vs. non-IPF: 41 ng/mL; AUC 0.68 | High CC16 levels may increase suspicion of IPF and may complement other findings | Outdated diagnostic criteria for IPF and HP Time of blood sample collection not mentioned/Patient treatment at the time not detailed Single center study; Small sample size |
| Barnes et al. [170] | Case-control study | Patients with HP and age-matched controls; comparison of selected serum biomarkers (secondary outcome) | No CC16 HP: 36.3 ng/m controls 15.0 ng/mL | CC16 associated with increased odds of HP | HP diagnosis based on administrative data/partly on clinical unmentioned criteria Patient treatment at the time of sample collectionnot detailed | |
| Advanced glycation end products/Receptor for Advanced glycation end products (AGE/RAGE) | Machahua er al. [183] | Prospective cohort (case-control analysis) | Patients with IPF, cHP, fNSIP and controls; Comparison and validation of serum AGEs, sRAGE and AGEs/sRAGE | Yes Optimal cutoff for cHP vs. NSIP: AGEs 19.25 ug/mL (AUC 0.883); sRAGE 782.6 pg/mL (AUC 0.887); AGE/sRAGE 25.7‰ (AUC 0.882) Optimal cutoff for cHP vs. IPF: AGEs and sRAGE: NM (low AUC), AGE/sRAGE NM (AUC = 0.713) | AGE higher in cHP vs. controls; sRAGE lower in cHP vs. fNSIP and vs. controls IPF vs. cHP: AGE and sRAge not different; AGE/sRAGE as potential diagnostic tools in fibrosing ILDs | Outdated diagnostic criteria for IPF and HP Patient treatment at the time not detailed Single center study; Small sample size |
Studies comparing biomarkers of epithelial cell dysfunction in human individuals with HP vs. other conditions and/or healthy controls. BAL—bronchoalveolar lavage; cHP—chronic hypersensitivity pneumonitis; CVD-ILD—Collagen vascular disease-related interstitial lung disease; IPF—idiopathic pulmonary fibrosis fNSIP—fibrotic non-specific interstitial pneumonitis; NM: not mentioned; sarc—sarcoidosis.
2.3. Fibrogenesis and ECM Remodeling
Fibroblast recruitment and activation in the context of imbalanced regulatory cytokine milieu culminate in excessive production and deposition of extracellular matrix (ECM) [130]. These pathogenic processes have also been active areas of research, including on the potential use of the associated molecules as diagnostic tools in ILD, including HP (Table 3).
Matrix Metalloproteinases (MMPs)
Matrix metalloproteinases (also known as matrixins) are zinc-dependent endoproteases that have long been considered as the main effectors of ECM degradation, modulating the activity of ECM mediators and the behavior of smooth muscle, epithelial and immune cells [185,186]. MMPs were believed to prevent fibrotic scarring, which was understood as a dysbalance between deposition and degradation of ECM components. However, the finding of increased activity both in repair/remodeling processes and in complex fibrotic conditions as IPF challenged this concept [185,186]. Even though many of the 23 members of the MMP family play different roles in the pathogenesis of lug fibrosis, only a few have been studied as diagnostic biomarkers in ILD, mostly in IPF. MMP-1, MMP-7, MMP-3, MMP-8 and MMP-9 are overexpressed, and the combination of MMP-7 and MMP-1 appears to accurately differentiate IPF from healthy controls [85,187]. This finding was corroborated by Morais et al., who found an interesting ability for both MMP-7 and MMP-1 to discriminate IPF from other ILDs, including fibrotic HP [188]. Bruzova et al. and Maldonado et al., on the other hand, did not find a difference on the levels of MMP-7 between IPF and other fibrotic IIPs [86,189]. The latter, however, described an acceptable ability of MMP-28 to distinguish IPF from non-IPF fibrosis but not fibrotic HP from fibrotic autoimmune ILD [189]. Interestingly, in the same paper, levels of this enzyme were higher in IPF-UIP compared to non-IPF-UIP, suggesting that different mechanisms result in the expression of different molecules, even though the radiological and morphologic outcomes are similar.
Little is further known about the role of MMPs in the diagnosis of fibrotic HP. A deeper knowledge of patients’ serum and BAL levels might not only expand the short list of validated diagnostic biomarkers but also the knowledge on the common and distinct pathways of ECM deposition in fibrotic HP and other fibrotic ILDs.
Table 3.
Studies exploring biomarkers of fibrogenesis/ECM remodeling and from other sources in individuals with hypersensitivity pneumonitis.
Table 3.
Studies exploring biomarkers of fibrogenesis/ECM remodeling and from other sources in individuals with hypersensitivity pneumonitis.
| Biomarker | Investigator | Type of Study | Population(s)/Methods | Threshold Analysis (Cutoffs)/Other Results | Conclusion(s) of Interest | Main Limitations |
|---|---|---|---|---|---|---|
| Matrix metalloproteinases (MMPs) | Rosas et al. [187] | Cross-sectional study (case-control analysis) | Patients with IPF, saHP/cHP, Sarcoidosis, COPD; Comparison of serum protein concentration (multiplex assay) | Yes. MMP7 and MMP1 higher in IPF vs. HP (2.3 and 1.31-fold, respectively) Optimal cutoffs and AUC: not mentioned Combination of high MMP1 + High MMP7: S 96.3% Sp 87.2% | Serum MMP1 and MMP7 as potential biomarkers in the differential diagnosis of IPF and HP | Outdated diagnostic criteria for IPF and HP Only avian inciting antigens/exposure Time of sample collection not mentioned/Patient treatment at the time not detailed Single center study; Small sample size |
| Morais et al. [188] | Cross-sectional study (case-control analysis) | Patients with IPF and non-IPF ILD (including HP); Comparison of serum MMP-7 and MMP-1 | Yes (IPF vs. other ILDs) MMP-1 higher in IPF vs. non-IPF-UIP Optimal cutoff for IPF vs. ther ILDs: MMP-1: 4.15 ng/mL AUC 0.63 MMP-7: 3.91 ng/mL, AUC 0.73 Combination: AUC 0.74 | Potential role of serum MMP-1 and MMP-7 as diagnostic biomarkers in IPF | HP not as independent group; Former diagnostic criteria of IPF and HP Single center study; Small sample size | |
| Maldonado et al. [189] | Cross-sectional study (case-control analysis) | Patients with IPF and non-IPF fibrosis (including fHP) and healthy controls; Comparison and validation of MMP28 concentration in two cohorts | Yes. MMP28 higher in IPF vs. non-IPF and controls; Optimal cutoff for IPF vs. non-IPF: 4.5 ng/mL AUC 0.72 and 0.69 | MMP28 as new biomarker ofr differential diagnosis of IPF with cHP and fibrotic autoimmune driven-ILD | HP not as independent group and diagnostic criteria not detailed | |
| Lipid mediators/Adipokines | D’Alessandro et al. [99] | Cross-sectional study (case-control analysis) | Patients with IPF and fHP; comparison and validation of BAL and serum multiplex lipid profiling | Yes. Optimal cutoff BAL: Apo A1 20.99 ng/mL; Apo C3 3.62 ng/mL Apo C3 3.62 ng/mL. Combined performance: AUC 81% Optimal cutoff serum: Apo A1 12.0 ng/mL; CCL2 0.88 ng/mL Apo C3 11.53 ng/mL. Combined model performance: AUC 93% | BAL Apo A1, adipsin, Apo C3 and APN higher in HP vs. IPF Serum Apo A1 higher in HP; MCP-1 (CCL2) and Apo C3 lower in HP vs. IPF Overall performance better in BAL vs. serum | Diagnostic criteria of fHP not detailed Single center study; Small sample size |
Studies comparing biomarkers of fibrogenesis/ECM remodeling and from other sources in human individuals with HP vs. other ILDs/healthy controls. Biomarkers of other classes are highlighted. APN—adiponectin; AUC—area under curve; BAL—bronchoalveolar lavage cHP—chronic hypersensitivity pneumonitis; COPD—chronic obstructive pulmonary disease IPF—idiopathic pulmonary fibrosis; MCP-1/CCL2 Monocyte Chemoattractant Protein-1/ Chemokine (CC-motif) ligand 2; saHP—subacute hypersensitivity pneumonitis.
2.4. Other Sources
2.4.1. Metabolic Biomarkers
Lipid Mediators
Apo A1 is the major component of high-density lipoprotein particles and has shown anti-inflammatory properties in models of lung injury [190,191].
Decreased levels of Apo A1 have been described after high-throughput, whole-proteome BAL analysis of patients with IPF [192]. Compared to fHP, D’Alessandro et al. recently reported significantly lower levels of this molecule in both the serum and the BAL, with an AUC of 93% for the latter [99]. This study also reports a reasonable ability of Apo C3, another lipoprotein, to discriminate IPF from fHP (AUC of 69% and 68%, respectively, for measurements in the serum and in the BAL).
Adipokines
Adipose tissue’s secretory function has been increasingly recognized. Changes to the homeostasis of adipokines in patients with ILD have been documented, including exacerbations [193,194] and these proteins appear to regulate biological activities in endothelial, fibroblast and immune cells in several tissues [195]. D’Alessandro et al. showed distinct patterns of expression of adipsin and adiponectin in the BAL of patients with IPF and fHP and created a discriminatory model using these biomarkers [99].
2.5. Future Directions
Novel categories of biomarkers (e.g., exosomes, mitochondrial DNA, microRNA, quantitative imaging, transcriptomics, microbiome related) are emerging and may one day help clinicians establish or rule out the diagnosis of HP with confidence. Moreover, high-throughput processes and analysis of the existing and upcoming data may significantly enhance biomarker research in HP, as they have in other entities [196].
The significance of biomarkers in ILD is still distant from oncology, but the achievements in this area may serve as an example of the potential that molecular research holds. Even though we are still probably far from guiding therapy according to specific molecular biomarkers in ILD, their inclusion in the protocols of most clinical trials could be the first steps in the journey toward daily clinical practice. Moreover, drugs targeting molecules in the fibrotic mechanistic pathway are already a reality [197,198] and research in new compounds affecting these pathways keeps evolving and is on the verge of providing alternative compounds (Table 4).
Table 4.
Drugs with potential use in fibrotic HP undergoing clinical trial and their effect in previously mentioned biomarkers.
3. Discussion
Fibrotic HP is associated with significant morbidity and mortality and its phenotype resembles that of other ILDs, with IPF at the forefront [19]. In most fibrotic ILDs, predicting progression is one of the most relevant issues for the clinician, as lung function decline is associated with increased mortality [201]. However, this should not be the only concern, particularly in the case of HP, where the relationship between early diagnosis, the detection of the culprit antigen and patient outcomes [21] highlight the relevance of establishing a confident diagnosis.
Biomarkers are objectively measured indicators of biologic processes and play a major role in the diagnosis and management of a wide range of diseases in different organs and systems [202]. With regard to the lung, measuring molecule concentrations and/or cell constituents in BAL fluid has the advantage of portraying more closely the pathophysiology of the organ when compared with serum sampling. However, the development and validation of useful diagnostic biomarkers has proceeded at a slow pace [44]. Even though several biomarkers have demonstrated accuracy in patients with ILD vs. individuals without ILD, this is of limited clinical relevance [44]. On the other hand, finding a marker accurately able to discriminate between fHP and different fibrotic ILDs is of the highest clinical relevance [13], but this has been limited by the overlapping mechanisms of inflammation, airway and parenchymal damage and fibrogenesis between diseases. In the case of HP, understanding its complex pathogenesis, including heterogeneous mechanisms of sensitization, the myriad of inciting antigens and diverse clinical presentations is crucial to consensually define the disease, distinguish it from other ILDs and consequently develop research [8,12], namely in diagnostic biomarkers.
We found a relatively large number of serum and BAL biomarkers that were able to discriminate between patients with HP and individuals without ILD and, less frequently, with other ILDs. However, we found several issues limiting support for clinical application of diagnostic biomarkers in fibrotic HP. These include almost invariably small sampling and, very importantly, heterogeneity in diagnostic (gold-standard) criteria across the studies, which has been aggravated by the recent changes to the classification of the disease. There is no evidence from clinical trials, lack of data on cost-benefit analysis [203] and validation with threshold/accuracy analysis has been performed only in a minority of studies. Thus, the existing evidence was overall deemed insufficient to justify the incorporation of the reviewed biomarkers into current clinical guidelines and daily practice, with some exceptions. BAL lymphocyte counts and specific serum IgGs are, perhaps, the two more relevant ones, and their input is widely considered in the context of multidisciplinary discussion, even though their performance appears lower than desirable [12] and their clinical utility is repeatedly a subject of discussion. Larger prospective, multicentric studies using current criteria for fibrotic and non-fibrotic HP would probably enlighten their performance in the current practice and perhaps increase the confidence In their role in establishing the diagnosis in the multidisciplinary setting. On the other hand, even though biomarkers such as KL-6, SP-D, YKL-40 and apolipoproteins have not made their way to the day-to-day diagnostic approach to fibrotic HP in most tertiary centers, they have shown, in our opinion, promising results on research contexts and have the potential to translate into clinical practice once reproducibility is further confirmed.
4. Conclusions
Even though the development and validation of biomarkers are of particular interest and may be crucial to the simplicity of diagnosing and treating interstitial lung disease, they have been running at a slower pace than desired. As we demonstrated, the journey in the search for diagnostic biomarkers in fibrotic HP has been hindered by the delay in reaching consensus on diagnostic criteria, by heterogeneous classification of the disease and by the conduction of studies with small samples. With current evidence limiting clinical application, the future may lie in the application of combined panels of existing molecules or in the development of emerging biomarker categories. Still, the key to success could simply be in validating the existing ones, alone or in combination, in prospective, multicentric and well-characterized cohorts.
Author Contributions
Conceptualization, J.O.P.: data curating and writing—original draft preparation, reviewing and editing; figure reviewing and editing; V.F.: data curating and writing—figure preparation, reviewing and editing; T.M.A.: conceptualization and writing—reviewing and editing; S.F.—reviewing and editing C.R.C.: conceptualization and writing—reviewing and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
Figure 1 and Figure 2 were created with Biorender.com (accessed on 20 December 2022). Figure 2 was adapted from “Respiratory Epithelium”, by Biorender.com (2022)—retrieved from https://app.biorender.com/biorender-templates (accessed on 20th December 2022).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Selman, M.; Pardo, A.; King, T.E., Jr. Hypersensitivity pneumonitis: Insights in diagnosis and pathobiology. Am. J. Respir. Crit. Care Med. 2012, 186, 314–324. [Google Scholar] [CrossRef]
- Riario Sforza, G.G.; Marinou, A. Hypersensitivity pneumonitis: A complex lung disease. Clin. Mol. Allergy 2017, 15, 6. [Google Scholar] [CrossRef]
- Pepys, J.; Riddell, R.W.; Citron, K.M.; Clayton, Y.M. Precipitins Against Extracts of Hay and Moulds in the Serum of Patients with Farmer’s Lung, Aspergillosis, Asthma, and Sarcoidosis. Thorax 1962, 17, 366–374. [Google Scholar] [CrossRef]
- Kobayashi, M.; Stahmann, M.A.; Rankin, J.; Dickie, H.A. Antigens in moldy hay as the cause of farmer’s lung. Proc. Soc. Exp. Biol. Med. 1963, 113, 472–476. [Google Scholar] [CrossRef]
- Bishop, J.M.; Melnick, S.C.; Raine, J. Farmer’s Lung: Studies of Pulmonary Function and Aetiology. Q. J. Med. 1963, 32, 257–278. [Google Scholar]
- Arranz, I.O. Diagnosis and Follow up of Chronic Hypersensitivity Pneumonitis: Utility of Non-Invasive Measurement of Airway Inflammation. Ph.D. Thesis, Universitat Autonoma de Barcelona, Barcelona, Spain, 2016. [Google Scholar]
- Salvaggio, J.E. Robert A. Cooke memorial lecture. Hypersensitivity pneumonitis. J. Allergy Clin. Immunol. 1987, 79, 558–571. [Google Scholar] [CrossRef]
- Vasakova, M.; Selman, M.; Morell, F.; Sterclova, M.; Molina-Molina, M.; Raghu, G. Hypersensitivity Pneumonitis: Current Concepts of Pathogenesis and Potential Targets for Treatment. Am. J. Respir. Crit. Care Med. 2019, 200, 301–308. [Google Scholar] [CrossRef]
- Barrera, L.; Mendoza, F.; Zuniga, J.; Estrada, A.; Zamora, A.C.; Melendro, E.I.; Ramirez, R.; Pardo, A.; Selman, M. Functional diversity of T-cell subpopulations in subacute and chronic hypersensitivity pneumonitis. Am. J. Respir. Crit. Care Med. 2008, 177, 44–55. [Google Scholar] [CrossRef]
- Mitaka, K.; Miyazaki, Y.; Yasui, M.; Furuie, M.; Miyake, S.; Inase, N.; Yoshizawa, Y. Th2-biased immune responses are important in a murine model of chronic hypersensitivity pneumonitis. Int. Arch. Allergy Immunol. 2011, 154, 264–274. [Google Scholar] [CrossRef]
- Kishi, M.; Miyazaki, Y.; Jinta, T.; Furusawa, H.; Ohtani, Y.; Inase, N.; Yoshizawa, Y. Pathogenesis of cBFL in common with IPF? Correlation of IP-10/TARC ratio with histological patterns. Thorax 2008, 63, 810–816. [Google Scholar] [CrossRef]
- Raghu, G.; Remy-Jardin, M.; Ryerson, C.J.; Myers, J.L.; Kreuter, M.; Vasakova, M.; Bargagli, E.; Chung, J.H.; Collins, B.F.; Bendstrup, E.; et al. Diagnosis of Hypersensitivity Pneumonitis in Adults. An Official ATS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2020, 202, e36–e69. [Google Scholar] [CrossRef]
- Fernandez Perez, E.R.; Travis, W.D.; Lynch, D.A.; Brown, K.K.; Johannson, K.A.; Selman, M.; Ryu, J.H.; Wells, A.U.; Tony Huang, Y.C.; Pereira, C.A.C.; et al. Diagnosis and Evaluation of Hypersensitivity Pneumonitis: CHEST Guideline and Expert Panel Report. Chest 2021, 160, e97–e156. [Google Scholar] [CrossRef]
- Richerson, H.B.; Bernstein, I.L.; Fink, J.N.; Hunninghake, G.W.; Novey, H.S.; Reed, C.E.; Salvaggio, J.E.; Schuyler, M.R.; Schwartz, H.J.; Stechschulte, D.J. Guidelines for the clinical evaluation of hypersensitivity pneumonitis. Report of the Subcommittee on Hypersensitivity Pneumonitis. J. Allergy Clin. Immunol. 1989, 84, 839–844. [Google Scholar] [CrossRef]
- Lacasse, Y.; Selman, M.; Costabel, U.; Dalphin, J.C.; Ando, M.; Morell, F.; Erkinjuntti-Pekkanen, R.; Muller, N.; Colby, T.V.; Schuyler, M.; et al. Clinical diagnosis of hypersensitivity pneumonitis. Am. J. Respir. Crit. Care Med. 2003, 168, 952–958. [Google Scholar] [CrossRef]
- Vasakova, M.; Morell, F.; Walsh, S.; Leslie, K.; Raghu, G. Hypersensitivity Pneumonitis: Perspectives in Diagnosis and Management. Am. J. Respir. Crit. Care Med. 2017, 196, 680–689. [Google Scholar] [CrossRef]
- Walsh, S.L.F.; Wells, A.U.; Desai, S.R.; Poletti, V.; Piciucchi, S.; Dubini, A.; Nunes, H.; Valeyre, D.; Brillet, P.Y.; Kambouchner, M.; et al. Multicentre evaluation of multidisciplinary team meeting agreement on diagnosis in diffuse parenchymal lung disease: A case-cohort study. Lancet Respir. Med. 2016, 4, 557–565. [Google Scholar] [CrossRef]
- Travis, W.D.; Costabel, U.; Hansell, D.M.; King, T.E., Jr.; Lynch, D.A.; Nicholson, A.G.; Ryerson, C.J.; Ryu, J.H.; Selman, M.; Wells, A.U.; et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am. J. Respir. Crit. Care Med. 2013, 188, 733–748. [Google Scholar] [CrossRef]
- Morell, F.; Villar, A.; Montero, M.A.; Munoz, X.; Colby, T.V.; Pipvath, S.; Cruz, M.J.; Raghu, G. Chronic hypersensitivity pneumonitis in patients diagnosed with idiopathic pulmonary fibrosis: A prospective case-cohort study. Lancet Respir. Med. 2013, 1, 685–694. [Google Scholar] [CrossRef]
- Mohr, L.C. Hypersensitivity pneumonitis. Curr. Opin. Pulm. Med. 2004, 10, 401–411. [Google Scholar] [CrossRef]
- Fernandez Perez, E.R.; Swigris, J.J.; Forssen, A.V.; Tourin, O.; Solomon, J.J.; Huie, T.J.; Olson, A.L.; Brown, K.K. Identifying an inciting antigen is associated with improved survival in patients with chronic hypersensitivity pneumonitis. Chest 2013, 144, 1644–1651. [Google Scholar] [CrossRef]
- Terho, E.O.; Heinonen, O.P.; Lammi, S.; Laukkanen, V. Incidence of clinically confirmed farmer’s lung in Finland and its relation to meteorological factors. Eur. J. Respir. Dis. Suppl. 1987, 152, 47–56. [Google Scholar] [PubMed]
- Thomeer, M.J.; Costabe, U.; Rizzato, G.; Poletti, V.; Demedts, M. Comparison of registries of interstitial lung diseases in three European countries. Eur. Respir. J. Suppl. 2001, 32, 114s–118s. [Google Scholar]
- Okamoto, T.; Miyazaki, Y.; Ogura, T.; Chida, K.; Kohno, N.; Kohno, S.; Taniguchi, H.; Akagawa, S.; Mochizuki, Y.; Yamauchi, K.; et al. Nationwide epidemiological survey of chronic hypersensitivity pneumonitis in Japan. Respir. Investig. 2013, 51, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Rittig, A.H.; Hilberg, O.; Ibsen, R.; Lokke, A. Incidence, comorbidity and survival rate of hypersensitivity pneumonitis: A national population-based study. ERJ Open Res. 2019, 5, 00259-2018. [Google Scholar] [CrossRef] [PubMed]
- Solaymani-Dodaran, M.; West, J.; Smith, C.; Hubbard, R. Extrinsic allergic alveolitis: Incidence and mortality in the general population. QJM 2007, 100, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Hyldgaard, C.; Hilberg, O.; Muller, A.; Bendstrup, E. A cohort study of interstitial lung diseases in central Denmark. Respir. Med. 2014, 108, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Fernandez Perez, E.R.; Kong, A.M.; Raimundo, K.; Koelsch, T.L.; Kulkarni, R.; Cole, A.L. Epidemiology of Hypersensitivity Pneumonitis among an Insured Population in the United States: A Claims-based Cohort Analysis. Ann. Am. Thorac. Soc. 2018, 15, 460–469. [Google Scholar] [CrossRef]
- King, T.E., Jr. Hypersensitivity Pneumonitis (Extrinsic Allergic Alveolitis): Epidemiology, Causes, and Pathogenesis; Hollingsworth, H., Ed.; UpToDate: Waltham, MA, USA, 2019. [Google Scholar]
- Hilberg, O.; Hoffmann-Vold, A.M.; Smith, V.; Bouros, D.; Kilpeläinen, M.; Guiot, J.; Morais, A.; Clemente, S.; Daniil, Z.; Papakosta, D.; et al. Epidemiology of interstitial lung diseases and their progressive-fibrosing behaviour in six European countries. ERJ Open Res. 2022, 8, 00597-2021. [Google Scholar] [CrossRef]
- Singh, S.; Collins, B.F.; Sharma, B.B.; Joshi, J.M.; Talwar, D.; Katiyar, S.; Singh, N.; Ho, L.; Samaria, J.K.; Bhattacharya, P.; et al. Interstitial Lung Disease in India. Results of a Prospective Registry. Am. J. Respir. Crit. Care Med. 2017, 195, 801–813. [Google Scholar] [CrossRef]
- Costabel, U.; Miyazaki, Y.; Pardo, A.; Koschel, D.; Bonella, F.; Spagnolo, P.; Guzman, J.; Ryerson, C.J.; Selman, M. Hypersensitivity pneumonitis. Nat. Rev. Dis. Primers 2020, 6, 65. [Google Scholar] [CrossRef]
- Nasser, M.; Larrieu, S.; Boussel, L.; Si-Mohamed, S.; Bazin, F.; Marque, S.; Massol, J.; Thivolet-Bejui, F.; Chalabreysse, L.; Maucort-Boulch, D.; et al. Estimates of epidemiology, mortality and disease burden associated with progressive fibrosing interstitial lung disease in France (the PROGRESS study). Respir. Res. 2021, 22, 162. [Google Scholar] [CrossRef] [PubMed]
- Quaresma, M.; Coleman, M.P.; Rachet, B. 40-year trends in an index of survival for all cancers combined and survival adjusted for age and sex for each cancer in England and Wales, 1971–2011: A population-based study. Lancet 2015, 385, 1206–1218. [Google Scholar] [CrossRef] [PubMed]
- Fernandez Perez, E.R.; Sprunger, D.B.; Ratanawatkul, P.; Maier, L.A.; Huie, T.J.; Swigris, J.J.; Solomon, J.J.; Mohning, M.P.; Keith, R.C.; Brown, K.K. Increasing Hypersensitivity Pneumonitis-related Mortality in the United States from 1988 to 2016. Am. J. Respir. Crit. Care Med. 2019, 199, 1284–1287. [Google Scholar] [CrossRef] [PubMed]
- Biomarkers Definitions Working, G. Biomarkers and surrogate endpoints: Preferred definitions and conceptual framework. Clin. Pharmacol. Ther. 2001, 69, 89–95. [Google Scholar]
- Califf, R.M. Biomarker definitions and their applications. Exp. Biol. Med. 2018, 243, 213–221. [Google Scholar] [CrossRef]
- Kern, S.E. Why your new cancer biomarker may never work: Recurrent patterns and remarkable diversity in biomarker failures. Cancer Res. 2012, 72, 6097–6101. [Google Scholar] [CrossRef]
- Alfaro, T.M.; Robalo Cordeiro, C. Comorbidity in idiopathic pulmonary fibrosis—What can biomarkers tell us? Ther. Adv. Respir. Dis. 2020, 14, 1753466620910092. [Google Scholar] [CrossRef]
- Crespo, A.; Alfaro, T.; Somogyi, V.; Kreuter, M. Updates in using a molecular classifier to identify usual interstitial pneumonia in conventional transbronchial lung biopsy samples. Breathe 2020, 16, 200067. [Google Scholar] [CrossRef]
- Ohnishi, H.; Yokoyama, A.; Kondo, K.; Hamada, H.; Abe, M.; Nishimura, K.; Hiwada, K.; Kohno, N. Comparative study of KL-6, surfactant protein-A, surfactant protein-D, and monocyte chemoattractant protein-1 as serum markers for interstitial lung diseases. Am. J. Respir. Crit. Care Med. 2002, 165, 378–381. [Google Scholar] [CrossRef]
- Ohtsuki, Y.; Nakanishi, N.; Fujita, J.; Yoshinouchi, T.; Kobayashi, M.; Ueda, N.; Lee, G.H.; Furihata, M. Immunohistochemical distribution of SP-D, compared with that of SP-A and KL-6, in interstitial pneumonias. Med. Mol. Morphol. 2007, 40, 163–167. [Google Scholar] [CrossRef]
- Prasse, A.; Muller-Quernheim, J. Non-invasive biomarkers in pulmonary fibrosis. Respirology 2009, 14, 788–795. [Google Scholar] [CrossRef] [PubMed]
- Tzouvelekis, A.; Kouliatsis, G.; Anevlavis, S.; Bouros, D. Serum biomarkers in interstitial lung diseases. Respir. Res. 2005, 6, 78. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Kaner, R.J.; Guiot, J.; Maher, T.M.; Tomassetti, S.; Moiseev, S.; Kuwana, M.; Brown, K.K. Diagnostic and Prognostic Biomarkers for Chronic Fibrosing Interstitial Lung Diseases With a Progressive Phenotype. Chest 2020, 158, 646–659. [Google Scholar] [CrossRef] [PubMed]
- Campo, I.; Zorzetto, M.; Bonella, F. Facts and promises on lung biomarkers in interstitial lung diseases. Expert. Rev. Respir. Med. 2015, 9, 437–457. [Google Scholar] [CrossRef] [PubMed]
- Guiot, J.; Moermans, C.; Henket, M.; Corhay, J.L.; Louis, R. Blood Biomarkers in Idiopathic Pulmonary Fibrosis. Lung 2017, 195, 273–280. [Google Scholar] [CrossRef]
- Bonella, F.; Costabel, U. Biomarkers in connective tissue disease-associated interstitial lung disease. Semin. Respir. Crit. Care Med. 2014, 35, 181–200. [Google Scholar] [CrossRef]
- Hwang, S.J.; Kim, H.S.; Chung, D.H. Fas/Fas ligand-mediated apoptosis promotes hypersensitivity pneumonitis in mice by enhancing maturation of dendritic cells. Am. J. Respir. Crit. Care Med. 2010, 181, 1250–1261. [Google Scholar] [CrossRef]
- Takemura, T.; Akashi, T.; Kamiya, H.; Ikushima, S.; Ando, T.; Oritsu, M.; Sawahata, M.; Ogura, T. Pathological differentiation of chronic hypersensitivity pneumonitis from idiopathic pulmonary fibrosis/usual interstitial pneumonia. Histopathology 2012, 61, 1026–1035. [Google Scholar] [CrossRef]
- Espoladore, L.M.; Gregorio, B.B.; Lima, M.S.; de Pereira, C.A.; Soares, M.R.; Coletta, E.N. Cytological analysis of bronchoalveolar lavage in patients with interstitial lung diseases and the relation of cytological analysis to fibrosis in high-resolution computed tomography. Anal. Quant. Cytopathol. Histpathol. 2014, 36, 206–212. [Google Scholar]
- Adams, T.N.; Newton, C.A.; Batra, K.; Abu-Hijleh, M.; Barbera, T.; Torrealba, J.; Glazer, C.S. Utility of Bronchoalveolar Lavage and Transbronchial Biopsy in Patients with Hypersensitivity Pneumonitis. Lung 2018, 196, 617–622. [Google Scholar] [CrossRef]
- Gaxiola, M.; Buendia-Roldan, I.; Mejia, M.; Carrillo, G.; Estrada, A.; Navarro, M.C.; Rojas-Serrano, J.; Selman, M. Morphologic diversity of chronic pigeon breeder’s disease: Clinical features and survival. Respir. Med. 2011, 105, 608–614. [Google Scholar] [CrossRef]
- Hill, M.; Petnak, T.; Moua, T. Bronchoalveolar lavage lymphocytosis in hypersensitivity pneumonitis: A retrospective cohort analysis with elimination of incorporation bias. BMC Pulm. Med. 2022, 22, 49. [Google Scholar] [CrossRef]
- Salvaggio, J.E.; Karr, R.M. Hypersensitivity pneumonitis; state of the art. Chest 1979, 75, 270–274. [Google Scholar] [CrossRef]
- Schuyler, M.; Cormier, Y. The diagnosis of hypersensitivity pneumonitis. Chest 1997, 111, 534–536. [Google Scholar] [CrossRef] [PubMed]
- American Thoracic, S.; European Respiratory, S. American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am. J. Respir. Crit. Care Med. 2002, 165, 277–304. [Google Scholar]
- Bonella, F.; Costabel, U. The perpetual enigma of bronchoalveolar lavage fluid lymphocytosis in chronic hypersensitivity pneumonitis: Is it of diagnostic value? Eur. Respir. J. 2020, 56, 2001534. [Google Scholar] [CrossRef]
- Tzilas, V.; Tzouvelekis, A.; Bouros, E.; Karampitsakos, T.; Ntasiou, M.; Katsaras, M.; Costabel, U.; Wells, A.; Bouros, D. Diagnostic value of BAL lymphocytosis in patients with indeterminate for usual interstitial pneumonia imaging pattern. Eur. Respir. J. 2019, 54, 1901144. [Google Scholar] [CrossRef] [PubMed]
- Patolia, S.; Tamae Kakazu, M.; Chami, H.A.; Chua, A.; Diaz-Mendoza, J.; Duggal, A.; Jenkins, A.R.; Knight, S.L.; Raghu, G.; Wilson, K.C. Bronchoalveolar Lavage Lymphocytes in the Diagnosis of Hypersensitivity Pneumonitis among Patients with Interstitial Lung Disease. Ann. Am. Thorac. Soc. 2020, 17, 1455–1467. [Google Scholar] [CrossRef]
- Adderley, N.; Humphreys, C.J.; Barnes, H.; Ley, B.; Premji, Z.A.; Johannson, K.A. Bronchoalveolar lavage fluid lymphocytosis in chronic hypersensitivity pneumonitis: A systematic review and meta-analysis. Eur. Respir. J. 2020, 56, 2000206. [Google Scholar] [CrossRef]
- Yoshizawa, Y.; Ohtani, Y.; Hayakawa, H.; Sato, A.; Suga, M.; Ando, M. Chronic hypersensitivity pneumonitis in Japan: A nationwide epidemiologic survey. J. Allergy Clin. Immunol. 1999, 103, 315–320. [Google Scholar] [CrossRef]
- Caillaud, D.M.; Vergnon, J.M.; Madroszyk, A.; Melloni, B.M.; Murris, M.; Dalphin, J.C.; French Group of Environmental Immunoallergic Bronchopulmonary Diseases. Bronchoalveolar lavage in hypersensitivity pneumonitis: A series of 139 patients. Inflamm. Allergy Drug Targets 2012, 11, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Santos, V.; Martins, N.; Sousa, C.; Jacob, M.; Padrao, E.; Melo, N.; Mota, P.C.; Bastos, H.N.; Guimaraes, S.; Moura, C.S.; et al. Hypersensitivity pneumonitis: Main features characterization in a Portuguese cohort. Pulmonology 2020, 26, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Morell, F.; Roger, A.; Reyes, L.; Cruz, M.J.; Murio, C.; Munoz, X. Bird fancier’s lung: A series of 86 patients. Medicine 2008, 87, 110–130. [Google Scholar] [CrossRef] [PubMed]
- Ratjen, F.; Costabel, U.; Griese, M.; Paul, K. Bronchoalveolar lavage fluid findings in children with hypersensitivity pneumonitis. Eur. Respir. J. 2003, 21, 144–148. [Google Scholar] [CrossRef]
- Ando, M.; Konishi, K.; Yoneda, R.; Tamura, M. Difference in the phenotypes of bronchoalveolar lavage lymphocytes in patients with summer-type hypersensitivity pneumonitis, farmer’s lung, ventilation pneumonitis, and bird fancier’s lung: Report of a nationwide epidemiologic study in Japan. J. Allergy Clin. Immunol. 1991, 87, 1002–1009. [Google Scholar] [CrossRef]
- Cordeiro, C.R.; Jones, J.C.; Alfaro, T.; Ferreira, A.J. Bronchoalveolar lavage in occupational lung diseases. Semin. Respir. Crit. Care Med. 2007, 28, 504–513. [Google Scholar] [CrossRef]
- Alfaro, T.; Robalo Cordeiro, C. Bronchoalveolar Lavage. In Idiopathic Pulmonary Fibrosis: ERS Monograph; Costabel, U., Crestani, B., Wells, A.U., Eds.; European Respiratory Society: Sheffield, UK, 2016; Volume 71, pp. 74–81. [Google Scholar]
- Murayama, J.; Yoshizawa, Y.; Ohtsuka, M.; Hasegawa, S. Lung fibrosis in hypersensitivity pneumonitis. Association with CD4+ but not CD8+ cell dominant alveolitis and insidious onset. Chest 1993, 104, 38–43. [Google Scholar] [CrossRef]
- Drent, M.; Jacobs, J.A.; Cobben, N.A.; Costabel, U.; Wouters, E.F.; Mulder, P.G. Computer program supporting the diagnostic accuracy of cellular BALF analysis: A new release. Respir. Med. 2001, 95, 781–786. [Google Scholar] [CrossRef][Green Version]
- Crouser, E.D.; Maier, L.A.; Wilson, K.C.; Bonham, C.A.; Morgenthau, A.S.; Patterson, K.C.; Abston, E.; Bernstein, R.C.; Blankstein, R.; Chen, E.S.; et al. Diagnosis and Detection of Sarcoidosis. An Official American Thoracic Society Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2020, 201, e26–e51. [Google Scholar] [CrossRef]
- Meyer, K.C.; Raghu, G.; Baughman, R.P.; Brown, K.K.; Costabel, U.; du Bois, R.M.; Drent, M.; Haslam, P.L.; Kim, D.S.; Nagai, S.; et al. An official American Thoracic Society clinical practice guideline: The clinical utility of bronchoalveolar lavage cellular analysis in interstitial lung disease. Am. J. Respir. Crit. Care Med. 2012, 185, 1004–1014. [Google Scholar] [CrossRef]
- Ye, Q.; Nakamura, S.; Sarria, R.; Costabel, U.; Guzman, J. Interleukin 12, interleukin 18, and tumor necrosis factor alpha release by alveolar macrophages: Acute and chronic hypersensitivity pneumonitis. Ann. Allergy Asthma Immunol. 2009, 102, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Gudmundsson, G.; Hunninghake, G.W. Interferon-gamma is necessary for the expression of hypersensitivity pneumonitis. J. Clin. Investig. 1997, 99, 2386–2390. [Google Scholar] [CrossRef] [PubMed]
- Nance, S.; Cross, R.; Yi, A.K.; Fitzpatrick, E.A. IFN-gamma production by innate immune cells is sufficient for development of hypersensitivity pneumonitis. Eur. J. Immunol. 2005, 35, 1928–1938. [Google Scholar] [CrossRef] [PubMed]
- Raymond, T.; Schaller, M.; Hogaboam, C.M.; Lukacs, N.W.; Rochford, R.; Kunkel, S.L. Toll-like receptors, Notch ligands, and cytokines drive the chronicity of lung inflammation. Proc. Am. Thorac. Soc. 2007, 4, 635–641. [Google Scholar] [CrossRef]
- Fong, D.J.; Hogaboam, C.M.; Matsuno, Y.; Akira, S.; Uematsu, S.; Joshi, A.D. Toll-like receptor 6 drives interleukin-17A expression during experimental hypersensitivity pneumonitis. Immunology 2010, 130, 125–136. [Google Scholar] [CrossRef]
- Joshi, A.D.; Fong, D.J.; Oak, S.R.; Trujillo, G.; Flaherty, K.R.; Martinez, F.J.; Hogaboam, C.M. Interleukin-17-mediated immunopathogenesis in experimental hypersensitivity pneumonitis. Am. J. Respir. Crit. Care Med. 2009, 179, 705–716. [Google Scholar] [CrossRef]
- Simonian, P.L.; Roark, C.L.; Wehrmann, F.; Lanham, A.K.; Diaz del Valle, F.; Born, W.K.; O’Brien, R.L.; Fontenot, A.P. Th17-polarized immune response in a murine model of hypersensitivity pneumonitis and lung fibrosis. J. Immunol. 2009, 182, 657–665. [Google Scholar] [CrossRef]
- Matěj, R.; Smětáková, M.; Vašáková, M.; Nováková, J.; Sterclová, M.; Kukal, J.; Olejár, T. PAR-2, IL-4R, TGF-β and TNF-α in bronchoalveolar lavage distinguishes extrinsic allergic alveolitis from sarcoidosis. Exp. Ther. Med. 2014, 8, 533–538. [Google Scholar] [CrossRef]
- Sterclova, M.; Matej, R.; Mandakova, P.; Skibova, J.; Vasakova, M. Role of interleukin 4 and its receptor in clinical presentation of chronic extrinsic allergic alveolitis: A pilot study. Multidiscip. Respir. Med. 2013, 8, 35. [Google Scholar] [CrossRef]
- Simonian, P.L.; Roark, C.L.; Wehrmann, F.; Lanham, A.M.; Born, W.K.; O’Brien, R.L.; Fontenot, A.P. IL-17A-expressing T cells are essential for bacterial clearance in a murine model of hypersensitivity pneumonitis. J. Immunol. 2009, 182, 6540–6549. [Google Scholar] [CrossRef]
- Zakaria, M.W.; El-Korashy, R.I.; Selim, S.; Badawy, I.; Amum, K.J. Serum level of transforming growth factor-beta1 in major idiopathic interstitial pneumonia. Egypt. J. Bronchol. 2020, 14, 22. [Google Scholar] [CrossRef]
- Willems, S.; Verleden, S.E.; Vanaudenaerde, B.M.; Wynants, M.; Dooms, C.; Yserbyt, J.; Somers, J.; Verbeken, E.K.; Verleden, G.M.; Wuyts, W.A. Multiplex protein profiling of bronchoalveolar lavage in idiopathic pulmonary fibrosis and hypersensitivity pneumonitis. Ann. Thorac. Med. 2013, 8, 38–45. [Google Scholar] [PubMed]
- Bruzova, M.; Pavlova, M.; Matej, R.; Sterclova, M.; Vasakova, M. Interstitial Score and Concentrations of IL-4Rα, PAR-2, and MMP-7 in Bronchoalveolar Lavage Fluid Could Be Useful Markers for Distinguishing Idiopathic Interstitial Pneumonias. Diagnostics 2021, 11, 693. [Google Scholar] [CrossRef] [PubMed]
- Martina, S.; Martina, V.; Monika, M.; Jan, P.; Libor, K.; Ilja, S. Angiostatic versus angiogenic chemokines in IPF and EAA. Respir. Med. 2009, 103, 1651–1656. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sugiyama, Y.; Kasahara, T.; Mukaida, N.; Matsushima, K.; Kitamura, S. Chemokines in bronchoalveolar lavage fluid in summer-type hypersensitivity pneumonitis. Eur. Respir. J. 1995, 8, 1084–1090. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Ishizuka, M.; Furusawa, H.; Honda, T.; Kawahara, T.; Tateishi, T.; Miyazaki, Y. Acute inflammatory and immunologic responses against antigen in chronic bird-related hypersensitivity pneumonitis. Allergol. Int. 2019, 68, 321–328. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Unoura, K.; Tateishi, T.; Akashi, T.; Takemura, T.; Tomita, M.; Inase, N.; Yoshizawa, Y. Higher serum CCL17 may be a promising predictor of acute exacerbations in chronic hypersensitivity pneumonitis. Respir. Res. 2013, 14, 57. [Google Scholar] [CrossRef]
- Watanabe, M.; Horimasu, Y.; Iwamoto, H.; Yamaguchi, K.; Sakamoto, S.; Masuda, T.; Nakashima, T.; Miyamoto, S.; Ohshimo, S.; Fujitaka, K.; et al. C-C Motif Chemokine Ligand 15 May Be a Useful Biomarker for Predicting the Prognosis of Patients with Chronic Hypersensitivity Pneumonitis. Respiration 2019, 98, 212–220. [Google Scholar] [CrossRef]
- Inoue, T.; Fujishima, S.; Ikeda, E.; Yoshie, O.; Tsukamoto, N.; Aiso, S.; Aikawa, N.; Kubo, A.; Matsushima, K.; Yamaguchi, K. CCL22 and CCL17 in rat radiation pneumonitis and in human idiopathic pulmonary fibrosis. Eur. Respir. J. 2004, 24, 49–56. [Google Scholar] [CrossRef]
- Nukui, Y.; Yamana, T.; Masuo, M.; Tateishi, T.; Kishino, M.; Tateishi, U.; Tomita, M.; Hasegawa, T.; Aritsu, T.; Miyazaki, Y. Serum CXCL9 and CCL17 as biomarkers of declining pulmonary function in chronic bird-related hypersensitivity pneumonitis. PLoS ONE 2019, 14, e0220462. [Google Scholar] [CrossRef]
- Pardo, A.; Smith, K.M.; Abrams, J.; Coffman, R.; Bustos, M.; McClanahan, T.K.; Grein, J.; Murphy, E.E.; Zlotnik, A.; Selman, M. CCL18/DC-CK-1/PARC up-regulation in hypersensitivity pneumonitis. J. Leukoc. Biol. 2001, 70, 610–616. [Google Scholar] [CrossRef]
- Cai, M.; Bonella, F.; He, X.; Sixt, S.U.; Sarria, R.; Guzman, J.; Costabel, U. CCL18 in serum, BAL fluid and alveolar macrophage culture supernatant in interstitial lung diseases. Respir. Med. 2013, 107, 1444–1452. [Google Scholar] [CrossRef] [PubMed]
- Iyonaga, K.; Takeya, M.; Saita, N.; Sakamoto, O.; Yoshimura, T.; Ando, M.; Takahashi, K. Monocyte chemoattractant protein-1 in idiopathic pulmonary fibrosis and other interstitial lung diseases. Hum. Pathol. 1994, 25, 455–463. [Google Scholar] [CrossRef] [PubMed]
- García de Alba, C.; Buendia-Roldán, I.; Salgado, A.; Becerril, C.; Ramírez, R.; González, Y.; Checa, M.; Navarro, C.; Ruiz, V.; Pardo, A.; et al. Fibrocytes contribute to inflammation and fibrosis in chronic hypersensitivity pneumonitis through paracrine effects. Am. J. Respir. Crit. Care Med. 2015, 191, 427–436. [Google Scholar] [CrossRef]
- Suga, M.; Iyonaga, K.; Ichiyasu, H.; Saita, N.; Yamasaki, H.; Ando, M. Clinical significance of MCP-1 levels in BALF and serum in patients with interstitial lung diseases. Eur. Respir. J. 1999, 14, 376–382. [Google Scholar] [CrossRef] [PubMed]
- d’Alessandro, M.; Bergantini, L.; Cameli, P.; Lanzarone, N.; Perillo, F.; Perrone, A.; Bargagli, E. BAL and serum multiplex lipid profiling in idiopathic pulmonary fibrosis and fibrotic hypersensitivity pneumonitis. Life Sci. 2020, 256, 117995. [Google Scholar] [CrossRef]
- Panina-Bordignon, P.; Papi, A.; Mariani, M.; Di Lucia, P.; Casoni, G.; Bellettato, C.; Buonsanti, C.; Miotto, D.; Mapp, C.; Villa, A.; et al. The C-C chemokine receptors CCR4 and CCR8 identify airway T cells of allergen-challenged atopic asthmatics. J. Clin. Investig. 2001, 107, 1357–1364. [Google Scholar] [CrossRef]
- Miyazaki, E.; Nureki, S.; Fukami, T.; Shigenaga, T.; Ando, M.; Ito, K.; Ando, H.; Sugisaki, K.; Kumamoto, T.; Tsuda, T. Elevated levels of thymus- and activation-regulated chemokine in bronchoalveolar lavage fluid from patients with eosinophilic pneumonia. Am. J. Respir. Crit. Care Med. 2002, 165, 1125–1131. [Google Scholar] [CrossRef]
- Shanley, T.P.; Peters, J.L.; Jones, M.L.; Chensue, S.W.; Kunkel, S.L.; Ward, P.A. Regulatory effects of endogenous interleukin-1 receptor antagonist protein in immunoglobulin G immune complex-induced lung injury. J. Clin. Investig. 1996, 97, 963–970. [Google Scholar] [CrossRef][Green Version]
- Costabel, U.; Bonella, F.; Guzman, J. Chronic hypersensitivity pneumonitis. Clin. Chest Med. 2012, 33, 151–163. [Google Scholar] [CrossRef]
- Jenkins, A.R.; Chua, A.; Chami, H.; Diaz-Mendoza, J.; Duggal, A.; Knight, S.; Patolia, S.; Tamae-Kakazu, M.; Raghu, G.; Wilson, K.C. Questionnaires or Serum Immunoglobulin G Testing in the Diagnosis of Hypersensitivity Pneumonitis among Patients with Interstitial Lung Disease. Ann. Am. Thorac. Soc. 2021, 18, 130–147. [Google Scholar] [CrossRef] [PubMed]
- Morell, F. Idiopathic pulmonary fibrosis: Importance of accurate diagnosis and treatment. Arch. Bronconeumol. 2013, 49, 319–320. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.G.; Da Silva, C.A.; Dela Cruz, C.S.; Ahangari, F.; Ma, B.; Kang, M.J.; He, C.H.; Takyar, S.; Elias, J.A. Role of chitin and chitinase/chitinase-like proteins in inflammation, tissue remodeling, and injury. Annu. Rev. Physiol. 2011, 73, 479–501. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.G.; Dela Cruz, C.S.; Herzog, E.; Rosenberg, S.M.; Ahangari, F.; Elias, J.A. YKL-40, a chitinase-like protein at the intersection of inflammation and remodeling. Am. J. Respir. Crit. Care Med. 2012, 185, 692–694. [Google Scholar] [CrossRef]
- Liu, C.; Li, Q.; Zhou, X.; Kolosov, V.P.; Perelman, J.M. The chitinase-like protein YKL-40 increases mucin5AC production in human bronchial epithelial cells. Exp. Cell Res. 2013, 319, 2866–2873. [Google Scholar] [CrossRef]
- Johansen, J.S. Studies on serum YKL-40 as a biomarker in diseases with inflammation, tissue remodelling, fibroses and cancer. Dan. Med. Bull. 2006, 53, 172–209. [Google Scholar]
- Johansen, J.S.; Milman, N.; Hansen, M.; Garbarsch, C.; Price, P.A.; Graudal, N. Increased serum YKL-40 in patients with pulmonary sarcoidosis--a potential marker of disease activity? Respir. Med. 2005, 99, 396–402. [Google Scholar] [CrossRef]
- Furuhashi, K.; Suda, T.; Nakamura, Y.; Inui, N.; Hashimoto, D.; Miwa, S.; Hayakawa, H.; Kusagaya, H.; Nakano, Y.; Nakamura, H.; et al. Increased expression of YKL-40, a chitinase-like protein, in serum and lung of patients with idiopathic pulmonary fibrosis. Respir. Med. 2010, 104, 1204–1210. [Google Scholar] [CrossRef]
- Korthagen, N.M.; van Moorsel, C.H.; Barlo, N.P.; Ruven, H.J.; Kruit, A.; Heron, M.; van den Bosch, J.M.; Grutters, J.C. Serum and BALF YKL-40 levels are predictors of survival in idiopathic pulmonary fibrosis. Respir. Med. 2011, 105, 106–113. [Google Scholar] [CrossRef]
- Tong, X.; Ma, Y.; Liu, T.; Li, Z.; Liu, S.; Wu, G.; Fan, H. Can YKL-40 be used as a biomarker for interstitial lung disease?: A systematic review and meta-analysis. Medicine 2021, 100, e25631. [Google Scholar] [CrossRef]
- Long, X.; He, X.; Ohshimo, S.; Griese, M.; Sarria, R.; Guzman, J.; Costabel, U.; Bonella, F. Serum YKL-40 as predictor of outcome in hypersensitivity pneumonitis. Eur. Respir. J. 2017, 49, 1501924. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Diez, S.; Munoz, X.; Ojanguren, I.; Romero-Mesones, C.; Espejo, D.; Villar, A.; Gomez-Olles, S.; Cruz, M.J. YKL-40 and KL-6 Levels in Serum and Sputum of Patients Diagnosed With Hypersensitivity Pneumonitis. J. Allergy Clin. Immunol. Pract. 2022, 10, 2414–2423. [Google Scholar] [CrossRef] [PubMed]
- Mandrekar, J.N. Receiver operating characteristic curve in diagnostic test assessment. J. Thorac. Oncol. 2010, 5, 1315–1316. [Google Scholar] [CrossRef] [PubMed]
- Vietri, L.; Fui, A.; Bergantini, L.; d’Alessandro, M.; Cameli, P.; Sestini, P.; Rottoli, P.; Bargagli, E. Serum amyloid A: A potential biomarker of lung disorders. Respir. Investig. 2020, 58, 21–27. [Google Scholar] [CrossRef]
- Chen, E.S.; Song, Z.; Willett, M.H.; Heine, S.; Yung, R.C.; Liu, M.C.; Groshong, S.D.; Zhang, Y.; Tuder, R.M.; Moller, D.R. Serum amyloid A regulates granulomatous inflammation in sarcoidosis through Toll-like receptor-2. Am. J. Respir. Crit. Care Med. 2010, 181, 360–373. [Google Scholar] [CrossRef]
- Rubinstein, I.; Knecht, A.; de Beer, F.C.; Baum, G.L.; Pras, M. Serum amyloid-A protein concentrations in sarcoidosis. Isr. J. Med. Sci. 1989, 25, 461–462. [Google Scholar]
- Salazar, A.; Mana, J.; Fiol, C.; Hurtado, I.; Argimon, J.M.; Pujol, R.; Pinto, X. Influence of serum amyloid A on the decrease of high density lipoprotein-cholesterol in active sarcoidosis. Atherosclerosis 2000, 152, 497–502. [Google Scholar] [CrossRef]
- Miyoshi, S.; Hamada, H.; Kadowaki, T.; Hamaguchi, N.; Ito, R.; Irifune, K.; Higaki, J. Comparative evaluation of serum markers in pulmonary sarcoidosis. Chest 2010, 137, 1391–1397. [Google Scholar] [CrossRef]
- Bargagli, E.; Magi, B.; Olivieri, C.; Bianchi, N.; Landi, C.; Rottoli, P. Analysis of serum amyloid A in sarcoidosis patients. Respir. Med. 2011, 105, 775–780. [Google Scholar] [CrossRef]
- Beijer, E.; Roodenburg-Benschop, C.; Schimmelpennink, M.C.; Grutters, J.C.; Meek, B.; Veltkamp, M. Elevated Serum Amyloid a Levels Are not Specific for Sarcoidosis but Associate with a Fibrotic Pulmonary Phenotype. Cells 2021, 10, 585. [Google Scholar] [CrossRef]
- Lakota, K.; Carns, M.; Podlusky, S.; Mrak-Poljsak, K.; Hinchcliff, M.; Lee, J.; Tomsic, M.; Sodin-Semrl, S.; Varga, J. Serum amyloid A is a marker for pulmonary involvement in systemic sclerosis. PLoS ONE 2015, 10, e0110820. [Google Scholar] [CrossRef] [PubMed]
- Migita, K.; Kawabe, Y.; Tominaga, M.; Origuchi, T.; Aoyagi, T.; Eguchi, K. Serum amyloid A protein induces production of matrix metalloproteinases by human synovial fibroblasts. Lab. Investig. 1998, 78, 535–539. [Google Scholar] [PubMed]
- Vietri, L.; d’Alessandro, M.; Bergantini, L.; Carleo, A.; Cameli, P.; Mazzei, M.A.; Sestini, P.; Bargagli, E. Specificity of serum amyloid A as a biomarker of idiopathic pulmonary fibrosis. Intern. Med. J. 2020, 50, 1571–1574. [Google Scholar] [CrossRef] [PubMed]
- Vietri, L.; Bennett, D.; Cameli, P.; Bergantini, L.; Cillis, G.; Sestini, P.; Bargagli, E.; Rottoli, P. Serum amyloid A in patients with idiopathic pulmonary fibrosis. Respir. Investig. 2019, 57, 430–434. [Google Scholar] [CrossRef] [PubMed]
- Bergantini, L.; d’Alessandro, M.; Vietri, L.; Rana, G.D.; Cameli, P.; Acerra, S.; Sestini, P.; Bargagli, E. Utility of serological biomarker’ panels for diagnostic accuracy of interstitial lung diseases. Immunol. Res. 2020, 68, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Hermans, C.; Bernard, A. Lung epithelium-specific proteins: Characteristics and potential applications as markers. Am. J. Respir. Crit. Care Med. 1999, 159, 646–678. [Google Scholar] [CrossRef]
- Jinta, T.; Miyazaki, Y.; Kishi, M.; Akashi, T.; Takemura, T.; Inase, N.; Yoshizawa, Y. The pathogenesis of chronic hypersensitivity pneumonitis in common with idiopathic pulmonary fibrosis: Expression of apoptotic markers. Am. J. Clin. Pathol. 2010, 134, 613–620. [Google Scholar] [CrossRef]
- Kohno, N.; Kyoizumi, S.; Awaya, Y.; Fukuhara, H.; Yamakido, M.; Akiyama, M. New serum indicator of interstitial pneumonitis activity. Sialylated carbohydrate antigen KL-6. Chest 1989, 96, 68–73. [Google Scholar]
- Kohno, N.; Awaya, Y.; Oyama, T.; Yamakido, M.; Akiyama, M.; Inoue, Y.; Yokoyama, A.; Hamada, H.; Fujioka, S.; Hiwada, K. KL-6, a mucin-like glycoprotein, in bronchoalveolar lavage fluid from patients with interstitial lung disease. Am. Rev. Respir. Dis. 1993, 148, 637–642. [Google Scholar] [CrossRef]
- Ishikawa, N.; Hattori, N.; Yokoyama, A.; Kohno, N. Utility of KL-6/MUC1 in the clinical management of interstitial lung diseases. Respir. Investig. 2012, 50, 3–13. [Google Scholar] [CrossRef]
- Hanzawa, S.; Tateishi, T.; Ishizuka, M.; Inoue, Y.; Honda, T.; Kawahara, T.; Tomita, M.; Miyazaki, Y. Changes in serum KL-6 levels during short-term strict antigen avoidance are associated with the prognosis of patients with fibrotic hypersensitivity pneumonitis caused by avian antigens. Respir. Investig. 2020, 58, 457–464. [Google Scholar] [CrossRef]
- Fathi, M.; Barbasso Helmers, S.; Lundberg, I.E. KL-6: A serological biomarker for interstitial lung disease in patients with polymyositis and dermatomyositis. J. Intern. Med. 2012, 271, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, J.; Kitamura, S. KL-6: A serum marker for interstitial pneumonia. Chest 1995, 108, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Munakata, M.; Ohtsuka, Y.; Satoh-Kamachi, A.; Sato, R.; Homma, Y.; Kawakami, Y. Serum KL-6 concentrations in dairy farmers. Chest 2000, 118, 445–450. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yoshikawa, S.; Tsushima, K.; Yasuo, M.; Fujimoto, K.; Kubo, K.; Kumagai, T.; Yamazaki, Y. Hypersensitivity pneumonitis caused by Penicillium citrinum, not Enoki spores. Am. J. Ind. Med. 2007, 50, 1010–1017. [Google Scholar] [CrossRef] [PubMed]
- d’Alessandro, M.; Bergantini, L.; Cameli, P.; Vietri, L.; Lanzarone, N.; Alonzi, V.; Pieroni, M.; Refini, M.R.; Sestini, P.; Bonella, F.; et al. Krebs von den Lungen-6 as a biomarker for disease severity assessment in interstitial lung disease: A comprehensive review. Biomark. Med. 2020, 14, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Lanzarone, N.; Gentili, F.; Alonzi, V.; Bergantini, L.; d’Alessandro, M.; Rottoli, P.; Refini, R.M.; Pieroni, M.; Vietri, L.; Bianchi, F.; et al. Bronchoalveolar lavage and serum KL-6 concentrations in chronic hypersensitivity pneumonitis: Correlations with radiological and immunological features. Intern. Emerg. Med. 2020, 15, 1247–1254. [Google Scholar] [CrossRef]
- Ji, Y.; Bourke, S.J.; Spears, M.; Wain, L.V.; Boyd, G.; Lynch, P.P.; Cunningham, M.; Boyd, K.; Donnelly, I.; Kohno, N.; et al. Krebs von den Lungen-6 (KL-6) is a pathophysiological biomarker of early-stage acute hypersensitivity pneumonitis among pigeon fanciers. Clin. Exp. Allergy 2020, 50, 1391–1399. [Google Scholar] [CrossRef]
- Okamoto, T.; Fujii, M.; Furusawa, H.; Tsuchiya, K.; Miyazaki, Y.; Inase, N. The usefulness of KL-6 and SP-D for the diagnosis and management of chronic hypersensitivity pneumonitis. Respir. Med. 2015, 109, 1576–1581. [Google Scholar] [CrossRef]
- Onishi, Y.; Kawamura, T.; Higashino, T.; Kagami, R.; Hirata, N.; Miyake, K. Clinical features of chronic summer-type hypersensitivity pneumonitis and proposition of diagnostic criteria. Respir. Investig. 2020, 58, 59–67. [Google Scholar] [CrossRef]
- Sakamoto, K.; Taniguchi, H.; Kondoh, Y.; Johkoh, T.; Sumikawa, H.; Kimura, T.; Nishiyama, O.; Kato, K.; Kataoka, K.; Ono, K.; et al. Serum KL-6 in fibrotic NSIP: Correlations with physiologic and radiologic parameters. Respir. Med. 2010, 104, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Chiba, S.; Ohta, H.; Abe, K.; Hisata, S.; Ohkouchi, S.; Hoshikawa, Y.; Kondo, T.; Ebina, M. The Diagnostic Value of the Interstitial Biomarkers KL-6 and SP-D for the Degree of Fibrosis in Combined Pulmonary Fibrosis and Emphysema. Pulm. Med. 2012, 2012, 492960. [Google Scholar] [CrossRef][Green Version]
- Moll, S.A.; Wiertz, I.A.; Vorselaars, A.D.M.; Ruven, H.J.T.; van Moorsel, C.H.M.; Grutters, J.C. Change in Serum Biomarker CA 15-3 as an Early Predictor of Response to Treatment and Survival in Hypersensitivity Pneumonitis. Lung 2020, 198, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Kruit, A.; Gerritsen, W.B.; Pot, N.; Grutters, J.C.; van den Bosch, J.M.; Ruven, H.J. CA 15-3 as an alternative marker for KL-6 in fibrotic lung diseases. Sarcoidosis Vasc. Diffuse Lung Dis. 2010, 27, 138–146. [Google Scholar] [PubMed]
- Gomes, P.S.; Soares, M.R.; Marchenta, M.; Meirelles, G.S.P.; Ferreira, R.G.; Botelho, A.B.; Martins, R.B.; Pereira, C.A.C. Carbohydrate antigen 15-3 as a marker of disease severity in patients with chronic hypersensitivity pneumonitis. J. Bras. Pneumol. 2021, 47, e20200589. [Google Scholar] [CrossRef]
- Ricci, A.; Mariotta, S.; Bronzetti, E.; Bruno, P.; Vismara, L.; De Dominicis, C.; Lagana, B.; Paone, G.; Mura, M.; Rogliani, P.; et al. Serum CA 15-3 is increased in pulmonary fibrosis. Sarcoidosis Vasc. Diffuse Lung Dis. 2009, 26, 54–63. [Google Scholar]
- Okada, M.; Suzuki, K.; Nakanishi, T.; Nakashima, M. Serum levels of KL-6 are positively correlated with those of CA15-3 in patients with interstitial pneumonia associated with collagen diseases. Respirology 2006, 11, 509–510. [Google Scholar] [CrossRef]
- Sorensen, G.L. Surfactant Protein D in Respiratory and Non-Respiratory Diseases. Front. Med. 2018, 5, 18. [Google Scholar] [CrossRef]
- Wright, J.R. Immunoregulatory functions of surfactant proteins. Nat. Rev. Immunol. 2005, 5, 58–68. [Google Scholar] [CrossRef]
- Kishore, U.; Greenhough, T.J.; Waters, P.; Shrive, A.K.; Ghai, R.; Kamran, M.F.; Bernal, A.L.; Reid, K.B.; Madan, T.; Chakraborty, T. Surfactant proteins SP-A and SP-D: Structure, function and receptors. Mol. Immunol. 2006, 43, 1293–1315. [Google Scholar] [CrossRef]
- Tanaka, H.; Sugawara, H.; Saikai, T.; Tsunematsu, K.; Takahashi, H.; Abe, S. Mushroom worker’s lung caused by spores of Hypsizigus marmoreus (Bunashimeji): Elevated serum surfactant protein D levels. Chest 2000, 118, 1506–1509. [Google Scholar] [CrossRef] [PubMed]
- Janssen, R.; Grutters, J.C.; Sato, H.; van Velzen-Blad, H.; Zanen, P.; Kohno, N.; Welsh, K.I.; du Bois, R.M.; van den Bosch, J.M. Analysis of KL-6 and SP-D as disease markers in bird fancier’s lung. Sarcoidosis Vasc. Diffuse Lung Dis. 2005, 22, 51–57. [Google Scholar]
- Guzman, J.; Wang, Y.M.; Kalaycioglu, O.; Schoenfeld, B.; Hamm, H.; Bartsch, W.; Costabel, U. Increased surfactant protein A content in human alveolar macrophages in hypersensitivity pneumonitis. Acta Cytol. 1992, 36, 668–673. [Google Scholar] [PubMed]
- Hamm, H.; Lührs, J.; Guzman y Rotaeche, J.; Costabel, U.; Fabel, H.; Bartsch, W. Elevated surfactant protein A in bronchoalveolar lavage fluids from sarcoidosis and hypersensitivity pneumonitis patients. Chest 1994, 106, 1766–1770. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cormier, Y.; Israël-Assayag, E.; Desmeules, M.; Lesur, O. Effect of contact avoidance or treatment with oral prednisolone on bronchoalveolar lavage surfactant protein A levels in subjects with farmer’s lung. Thorax 1996, 51, 1210–1215. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Phelps, D.S.; Umstead, T.M.; Mejia, M.; Carrillo, G.; Pardo, A.; Selman, M. Increased surfactant protein-A levels in patients with newly diagnosed idiopathic pulmonary fibrosis. Chest 2004, 125, 617–625. [Google Scholar] [CrossRef]
- Lee, Y.C.; Zhang, Z.; Mukherjee, A.B. Mice lacking uteroglobin are highly susceptible to developing pulmonary fibrosis. FEBS Lett. 2006, 580, 4515–4520. [Google Scholar] [CrossRef]
- Laucho-Contreras, M.E.; Polverino, F.; Gupta, K.; Taylor, K.L.; Kelly, E.; Pinto-Plata, V.; Divo, M.; Ashfaq, N.; Petersen, H.; Stripp, B.; et al. Protective role for club cell secretory protein-16 (CC16) in the development of COPD. Eur. Respir. J. 2015, 45, 1544–1556. [Google Scholar] [CrossRef]
- Miller, T.L.; Shashikant, B.N.; Pilon, A.L.; Pierce, R.A.; Shaffer, T.H.; Wolfson, M.R. Effects of recombinant Clara cell secretory protein (rhCC10) on inflammatory-related matrix metalloproteinase activity in a preterm lamb model of neonatal respiratory distress. Pediatr. Crit. Care Med. 2007, 8, 40–46. [Google Scholar] [CrossRef]
- Yao, X.L.; Ikezono, T.; Cowan, M.; Logun, C.; Angus, C.W.; Shelhamer, J.H. Interferon-gamma stimulates human Clara cell secretory protein production by human airway epithelial cells. Am. J. Physiol. 1998, 274, L864–L869. [Google Scholar]
- Magdaleno, S.M.; Wang, G.; Jackson, K.J.; Ray, M.K.; Welty, S.; Costa, R.H.; DeMayo, F.J. Interferon-gamma regulation of Clara cell gene expression: In vivo and in vitro. Am. J. Physiol. 1997, 272, L1142–L1151. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.M.; Lee, K.H.; Lee, S.M.; Lim, J.J.; Yang, S.C.; Yoo, C.G.; Lee, C.T.; Han, S.K.; Shim, Y.S.; Kim, Y.W. The immune modulation of Clara cell-10 in human peripheral monocytes and dendritic cells. Int. J. Mol. Med. 2010, 26, 415–423. [Google Scholar] [PubMed]
- Almuntashiri, S.; Zhu, Y.; Han, Y.; Wang, X.; Somanath, P.R.; Zhang, D. Club Cell Secreted Protein CC16: Potential Applications in Prognosis and Therapy for Pulmonary Diseases. J. Clin. Med. 2020, 9, 4039. [Google Scholar] [CrossRef]
- Doubkova, M.; Karpisek, M.; Mazoch, J.; Skrickova, J.; Doubek, M. Prognostic significance of surfactant protein A, surfactant protein D, Clara cell protein 16, S100 protein, trefoil factor 3, and prostatic secretory protein 94 in idiopathic pulmonary fibrosis, sarcoidosis, and chronic pulmonary obstructive disease. Sarcoidosis Vasc. Diffuse Lung Dis. 2016, 33, 224–234. [Google Scholar]
- Tsoumakidou, M.; Bouloukaki, I.; Thimaki, K.; Tzanakis, N.; Siafakas, N.M. Innate immunity proteins in chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Exp. Lung Res. 2010, 36, 373–380. [Google Scholar] [CrossRef]
- Buendia-Roldan, I.; Ruiz, V.; Sierra, P.; Montes, E.; Ramirez, R.; Vega, A.; Salgado, A.; Vargas, M.H.; Mejia, M.; Pardo, A.; et al. Increased Expression of CC16 in Patients with Idiopathic Pulmonary Fibrosis. PLoS ONE 2016, 11, e0168552. [Google Scholar] [CrossRef] [PubMed]
- Barnes, H.; Olin, A.C.; Toren, K.; McSharry, C.; Donnelly, I.; Larstad, M.; Iribarren, C.; Quinlan, P.; Blanc, P.D. Occupation versus environmental factors in hypersensitivity pneumonitis: Population attributable fraction. ERJ Open Res. 2020, 6, 00374–2020. [Google Scholar] [CrossRef]
- Milutinovic, P.S.; Englert, J.M.; Crum, L.T.; Mason, N.S.; Ramsgaard, L.; Enghild, J.J.; Sparvero, L.J.; Lotze, M.T.; Oury, T.D. Clearance kinetics and matrix binding partners of the receptor for advanced glycation end products. PLoS ONE 2014, 9, e88259. [Google Scholar] [CrossRef]
- Oczypok, E.A.; Perkins, T.N.; Oury, T.D. All the “RAGE” in lung disease: The receptor for advanced glycation endproducts (RAGE) is a major mediator of pulmonary inflammatory responses. Paediatr. Respir. Rev. 2017, 23, 40–49. [Google Scholar] [CrossRef]
- Chavakis, T.; Bierhaus, A.; Al-Fakhri, N.; Schneider, D.; Witte, S.; Linn, T.; Nagashima, M.; Morser, J.; Arnold, B.; Preissner, K.T.; et al. The pattern recognition receptor (RAGE) is a counterreceptor for leukocyte integrins: A novel pathway for inflammatory cell recruitment. J. Exp. Med. 2003, 198, 1507–1515. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Hori, O.; Chen, J.X.; Li, J.F.; Crandall, J.; Zhang, J.; Cao, R.; Yan, S.D.; Brett, J.; Stern, D. Advanced glycation endproducts interacting with their endothelial receptor induce expression of vascular cell adhesion molecule-1 (VCAM-1) in cultured human endothelial cells and in mice. A potential mechanism for the accelerated vasculopathy of diabetes. J. Clin. Investig. 1995, 96, 1395–1403. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, E.; Wautier, M.P.; Wautier, J.L.; Boval, B.; Panis, Y.; Wernert, N.; Danze, P.M.; Dequiedt, P. AGEs bind to mesothelial cells via RAGE and stimulate VCAM-1 expression. Kidney Int. 2002, 61, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Shirasawa, M.; Fujiwara, N.; Hirabayashi, S.; Ohno, H.; Iida, J.; Makita, K.; Hata, Y. Receptor for advanced glycation end-products is a marker of type I lung alveolar cells. Genes Cells 2004, 9, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Demling, N.; Ehrhardt, C.; Kasper, M.; Laue, M.; Knels, L.; Rieber, E.P. Promotion of cell adherence and spreading: A novel function of RAGE, the highly selective differentiation marker of human alveolar epithelial type I cells. Cell Tissue Res. 2006, 323, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Buckley, S.T.; Ehrhardt, C. The receptor for advanced glycation end products (RAGE) and the lung. J. Biomed. Biotechnol. 2010, 2010, 917108. [Google Scholar] [CrossRef]
- Ott, C.; Jacobs, K.; Haucke, E.; Navarrete Santos, A.; Grune, T.; Simm, A. Role of advanced glycation end products in cellular signaling. Redox Biol. 2014, 2, 411–429. [Google Scholar] [CrossRef]
- Avery, N.C.; Bailey, A.J. The effects of the Maillard reaction on the physical properties and cell interactions of collagen. Pathol. Biol. 2006, 54, 387–395. [Google Scholar] [CrossRef]
- Gautieri, A.; Passini, F.S.; Silván, U.; Guizar-Sicairos, M.; Carimati, G.; Volpi, P.; Moretti, M.; Schoenhuber, H.; Redaelli, A.; Berli, M.; et al. Advanced glycation end-products: Mechanics of aged collagen from molecule to tissue. Matrix Biol. 2017, 59, 95–108. [Google Scholar] [CrossRef]
- Machahua, C.; Montes-Worboys, A.; Llatjos, R.; Escobar, I.; Dorca, J.; Molina-Molina, M.; Vicens-Zygmunt, V. Increased AGE-RAGE ratio in idiopathic pulmonary fibrosis. Respir. Res. 2016, 17, 144. [Google Scholar] [CrossRef]
- Machahua, C.; Montes-Worboys, A.; Planas-Cerezales, L.; Buendia-Flores, R.; Molina-Molina, M.; Vicens-Zygmunt, V. Serum AGE/RAGEs as potential biomarker in idiopathic pulmonary fibrosis. Respir. Res. 2018, 19, 215. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Iwamoto, H.; Horimasu, Y.; Ohshimo, S.; Fujitaka, K.; Hamada, H.; Mazur, W.; Kohno, N.; Hattori, N. AGER gene polymorphisms and soluble receptor for advanced glycation end product in patients with idiopathic pulmonary fibrosis. Respirology 2017, 22, 965–971. [Google Scholar] [CrossRef]
- Pardo, A.; Cabrera, S.; Maldonado, M.; Selman, M. Role of matrix metalloproteinases in the pathogenesis of idiopathic pulmonary fibrosis. Respir. Res. 2016, 17, 23. [Google Scholar] [CrossRef]
- Craig, V.J.; Zhang, L.; Hagood, J.S.; Owen, C.A. Matrix metalloproteinases as therapeutic targets for idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2015, 53, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Rosas, I.O.; Richards, T.J.; Konishi, K.; Zhang, Y.; Gibson, K.; Lokshin, A.E.; Lindell, K.O.; Cisneros, J.; Macdonald, S.D.; Pardo, A.; et al. MMP1 and MMP7 as potential peripheral blood biomarkers in idiopathic pulmonary fibrosis. PLoS Med. 2008, 5, e93. [Google Scholar] [CrossRef]
- Morais, A.; Beltrão, M.; Sokhatska, O.; Costa, D.; Melo, N.; Mota, P.; Marques, A.; Delgado, L. Serum metalloproteinases 1 and 7 in the diagnosis of idiopathic pulmonary fibrosis and other interstitial pneumonias. Respir. Med. 2015, 109, 1063–1068. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, M.; Buendía-Roldán, I.; Vicens-Zygmunt, V.; Planas, L.; Molina-Molina, M.; Selman, M.; Pardo, A. Identification of MMP28 as a biomarker for the differential diagnosis of idiopathic pulmonary fibrosis. PLoS ONE 2018, 13, e0203779. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Dong, J.B.; Wu, M.P. Human ApoA-I overexpression diminishes LPS-induced systemic inflammation and multiple organ damage in mice. Eur. J. Pharmacol. 2008, 590, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.J.; Li, Y.; Lou, B.; Wu, M.P. Beneficial effects of ApoA-I on LPS-induced acute lung injury and endotoxemia in mice. Life Sci. 2006, 79, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Lee, Y.H.; Kim, K.H.; Lee, S.H.; Cha, J.Y.; Shin, E.K.; Jung, S.; Jang, A.S.; Park, S.W.; Uh, S.T.; et al. Role of lung apolipoprotein A-I in idiopathic pulmonary fibrosis: Antiinflammatory and antifibrotic effect on experimental lung injury and fibrosis. Am. J. Respir. Crit. Care Med. 2010, 182, 633–642. [Google Scholar] [CrossRef]
- Masui, Y.; Asano, Y.; Takahashi, T.; Shibata, S.; Akamata, K.; Aozasa, N.; Noda, S.; Taniguchi, T.; Ichimura, Y.; Toyama, T.; et al. Clinical significance of monitoring serum adiponectin levels during intravenous pulse cyclophosphamide therapy in interstitial lung disease associated with systemic sclerosis. Mod. Rheumatol. 2013, 23, 323–329. [Google Scholar] [CrossRef]
- Enomoto, N.; Oyama, Y.; Yasui, H.; Karayama, M.; Hozumi, H.; Suzuki, Y.; Kono, M.; Furuhashi, K.; Fujisawa, T.; Inui, N.; et al. Analysis of serum adiponectin and leptin in patients with acute exacerbation of idiopathic pulmonary fibrosis. Sci. Rep. 2019, 9, 10484. [Google Scholar] [CrossRef] [PubMed]
- Haley, S.; Shah, D.; Romero, F.; Summer, R. Scleroderma-related lung disease: Are adipokines involved pathogenically? Curr. Rheumatol. Rep. 2013, 15, 381. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Mo, L.; Feng, X.; Huang, M.; Li, L. Using bioinformatics approach identifies key genes and pathways in idiopathic pulmonary fibrosis. Medicine 2020, 99, e22099. [Google Scholar] [CrossRef] [PubMed]
- Ruwanpura, S.M.; Thomas, B.J.; Bardin, P.G. Pirfenidone: Molecular Mechanisms and Potential Clinical Applications in Lung Disease. Am. J. Respir. Cell Mol. Biol. 2020, 62, 413–422. [Google Scholar] [CrossRef]
- Wollin, L.; Wex, E.; Pautsch, A.; Schnapp, G.; Hostettler, K.E.; Stowasser, S.; Kolb, M. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1434–1445. [Google Scholar] [CrossRef]
- Herrmann, F.E.; Hesslinger, C.; Wollin, L.; Nickolaus, P. BI 1015550 is a PDE4B Inhibitor and a Clinical Drug Candidate for the Oral Treatment of Idiopathic Pulmonary Fibrosis. Front. Pharmacol. 2022, 13, 838449. [Google Scholar] [CrossRef]
- Decato, B.E.; Leeming, D.J.; Sand, J.M.B.; Fischer, A.; Du, S.; Palmer, S.M.; Karsdal, M.; Luo, Y.; Minnich, A. LPA 1 antagonist BMS-986020 changes collagen dynamics and exerts antifibrotic effects in vitro and in patients with idiopathic pulmonary fibrosis. Respir. Res. 2022, 23, 61. [Google Scholar] [CrossRef]
- Brown, K.K.; Inoue, Y.; Flaherty, K.R.; Martinez, F.J.; Cottin, V.; Bonella, F.; Cerri, S.; Danoff, S.K.; Jouneau, S.; Goeldner, R.G.; et al. Predictors of mortality in subjects with progressive fibrosing interstitial lung diseases. Respirology 2022, 27, 294–300. [Google Scholar] [CrossRef]
- Chen, X.H.; Huang, S.; Kerr, D. Biomarkers in clinical medicine. IARC Sci. Publ. 2011, 163, 303–322. [Google Scholar]
- Sellarés, J.; Molina-Molina, M. Serum Biomarkers in Diffuse Interstitial Lung Diseases. Arch. Bronconeumol. (Engl. Ed.) 2020, 56, 349–350. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).