
Involvement of Lipids in the Pathogenesis of Amyotrophic Lateral Sclerosis



Abstract
1. Introduction
2. Animal Models Used for Studying ALS Mechanisms
2.1. Models with Expression of Mutant SOD1 Isoforms
2.2. Models with Mutations in Proteins Involved in RNA Metabolism
2.3. Models with Mutations in Newly Discovered Genes
3. Hyperlipidemia in ALS
3.1. Triglycerides
3.2. Phospholipids
3.3. Polyunsaturated Fatty Acids
3.4. PUFAs, Diet and Nutrition
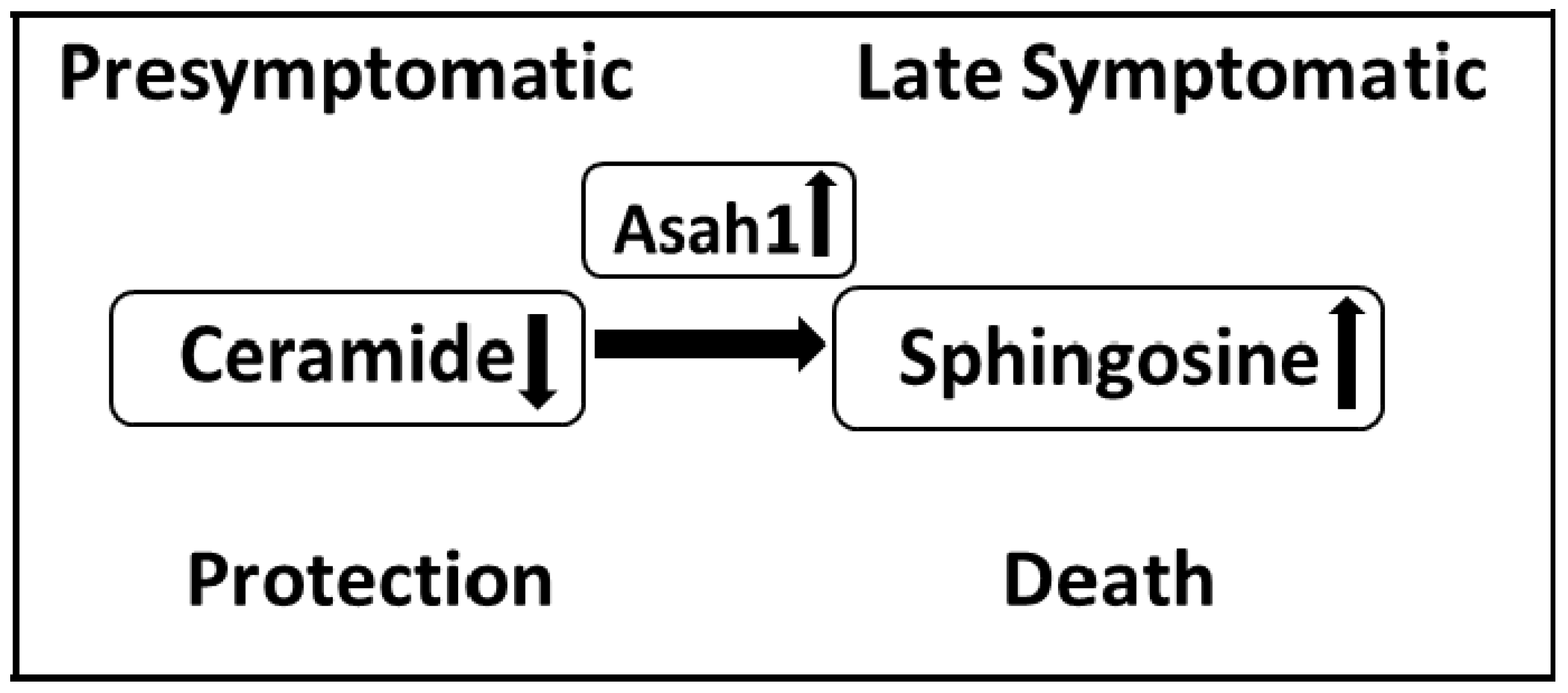
4. Sphingolipids in ALS
4.1. Ceramides

{kind=link}
{kind=link}
| Pre-Sympt | Early Sympt | Sympt | Model | References | |
|---|---|---|---|---|---|
| Ceramide–sphingosine axis | |||||
| Ceramide | ↑ | ne | ↑ | mouse SOD1G93A | [91] |
| ne | ↓ | ↑ | mouse SOD1G93A | [81] | |
| ↓↑ | ne | ↓↑ | mouse SOD1G86R | [92] | |
| ne | ne | ↑ | rat SOD1G93A | [93] | |
| ↓ | nd | nd | mouse FUS (1-359) | [94] | |
| Asah1 mRNA expression | ne | ↑ | ↑ | mouse SOD1G93A | [95] |
| nd | nd | ↑ | mouse FUS (1-359) | [94] | |
| Sphingosine | nd | nd | ↑ | mouse FUS(1-359) | [96] |
| Complex sphingolipids, ceramide sources | |||||
| Sphingomyelin | nd | ne | ↑ | mouse SOD1G93A | [91] |
| ne | ↓ | ↓ | mouse SOD1G93A | [81] | |
| ↑↓ | ne | ↑ | mouse SOD1G86R | [92] | |
| Galactosyl ceramide | ne | ↓ | ↓ | mouse SOD1G93A | [81] |
| Glucosyl ceramide | ne | nd | ↑ | mouse SOD1G93A | [81] |
| ↓ | ne | ↓↑ | mouse SOD1G86R | [92] | |
| Gangliosides GM3 | ne | ↑ | ↑ | mouse SOD1G93A | [81] |
| Gangliosides GM1 | ne | ↓ | ↓ | mouse SOD1G93A | [81] |
4.2. Sphingoid Bases
4.3. Lactosylceramides and Galactosylceramides
4.4. Glucosylceramides
4.5. Gangliosides
4.6. Cholesterol and Its Relationship with Sphingolipids in ALS
4.7. The Potential of Fingolimod as a Therapeutic Strategy in ALS
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ALS | amyotrophic lateral sclerosis |
| ALSFRS-R | amyotrophic lateral sclerosis functional rating scale revised |
| ApoE | apolipoprotein E |
| Asah1 | acid ceramidase |
| BBB | blood-brain barrier |
| BDNF | brain-derived neurotrophic factor |
| C9ORF72 | chromosome 9 open reading frame 72 |
| CNS | central nervous system |
| CSF | cerebrospinal fluid |
| FUS | fused in sarcoma |
| FVT1 | 3-ketodihydrosphingosine reductase |
| GCS | glucosylceramide synthase |
| HEX | hexosaminidase |
| HDL | high-density lipoproteins |
| IL-6 | Interleukin-6 |
| iPS cells | induced pluripotent stem cell |
| LPO | lipid peroxidation |
| LDL | low-density lipoproteins |
| MCT1 | monocarboxylate transporter 1 |
| Nrf2 | nuclear factor erythroid-derived 2-like 2 |
| PUFAs | saturated fatty acids |
| SMA | spinal muscular atrophy |
| SMS | sphingomyelin synthase |
| SOD1 | superoxide dismutase 1 |
| SPT | serine palmitoyltransferase |
| SPTLC1 | subunit 1 of the long chain serine palmitoyltransferase |
| TDP-43 | TAR DNA binding protein |
| TFIID | transcription factor II D |
| TNF-α | tumor necrosis factor |
| UGCG | UDP-glucose ceramide glycosyltransferase |
| UGT8 | galactosyltransferase |
| VCP | valosin containing protein |
References
- Van Den Bosch, L. Genetic Rodent Models of Amyotrophic Lateral Sclerosis. BioMed. Res. Int. 2011, 2011, e348765. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, M.; Brown, R.H. Genetics of Amyotrophic Lateral Sclerosis. Cold Spring Harb. Perspect. Med. 2018, 8, a024125. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, P.; Dewil, M.; Robberecht, W.; Van Den Bosch, L. Excitotoxicity and Amyotrophic Lateral Sclerosis. Neurodegener. Dis. 2005, 2, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Greco, V.; Longone, P.; Spalloni, A.; Pieroni, L.; Urbani, A. Crosstalk between oxidative stress and mitochondrial damage: Focus on amyotrophic lateral sclerosis. In Mitochondria in Health and in Sickness; Urbani, A., Babu, M., Eds.; Springer: Singapore, 2019; pp. 71–82. ISBN 9789811383670. [Google Scholar]
- Shi, P.; Gal, J.; Kwinter, D.M.; Liu, X.; Zhu, H. Mitochondrial Dysfunction in Amyotrophic Lateral Sclerosis. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2010, 1802, 45–51. [Google Scholar] [CrossRef]
- McCombe, A.P.; Henderson, D.R. The Role of Immune and Inflammatory Mechanisms in ALS. Curr. Mol. Med. 2011, 999, 1–9. [Google Scholar] [CrossRef]
- Hensley, K.; Floyd, R.A.; Gordon, B.; Mou, S.; Pye, Q.N.; Stewart, C.; West, M.; Williamson, K. Temporal Patterns of Cytokine and Apoptosis-Related Gene Expression in Spinal Cords of the G93A-SOD1 Mouse Model of Amyotrophic Lateral Sclerosis: Gene Expression Changes in ALS Mice. J. Neurochem. 2002, 82, 365–374. [Google Scholar] [CrossRef]
- Ekegren, T.; Grundström, E.; Lindholm, D.; Aquilonius, S.-M. Upregulation of Bax Protein and Increased DNA Degradation in ALS Spinal Cord Motor Neurons. Acta Neurol. Scand. 2009, 100, 317–321. [Google Scholar] [CrossRef]
- Guégan, C.; Vila, M.; Rosoklija, G.; Hays, A.P.; Przedborski, S. Recruitment of the Mitochondrial-Dependent Apoptotic Pathway in Amyotrophic Lateral Sclerosis. J. Neurosci. 2001, 21, 6569–6576. [Google Scholar] [CrossRef]
- Murugesan, V.; Chuang, W.-L.; Liu, J.; Lischuk, A.; Kacena, K.; Lin, H.; Pastores, G.M.; Yang, R.; Keutzer, J.; Zhang, K.; et al. Glucosylsphingosine Is a Key Biomarker of Gaucher Disease: Glucosylsphingosine Is a Key Biomarker of Gaucher Disease. Am. J. Hematol. 2016, 91, 1082–1089. [Google Scholar] [CrossRef]
- Blasco, H.; Veyrat-Durebex, C.; Bocca, C.; Patin, F.; Vourc’h, P.; Kouassi Nzoughet, J.; Lenaers, G.; Andres, C.R.; Simard, G.; Corcia, P.; et al. Lipidomics Reveals Cerebrospinal-Fluid Signatures of ALS. Sci. Rep. 2017, 7, 17652. [Google Scholar] [CrossRef]
- Ariga, T.; Jarvis, W.D.; Yu, R.K. Role of Sphingolipid-Mediated Cell Death in Neurodegenerative Diseases. J. Lipid Res. 1998, 39, 1–16. [Google Scholar] [CrossRef]
- Moll, T.; Marshall, J.N.G.; Soni, N.; Zhang, S.; Cooper-Knock, J.; Shaw, P.J. Membrane Lipid Raft Homeostasis Is Directly Linked to Neurodegeneration. Essays Biochem. 2021, 65, 999–1011. [Google Scholar] [CrossRef]
- Dodge, J.C. Lipid Involvement in Neurodegenerative Diseases of the Motor System: Insights from Lysosomal Storage Diseases. Front. Mol. Neurosci. 2017, 10, 356. [Google Scholar] [CrossRef]
- Chen, X.; Yazdani, S.; Piehl, F.; Magnusson, P.K.E.; Fang, F. Polygenic Link between Blood Lipids and Amyotrophic Lateral Sclerosis. Neurobiol. Aging 2018, 67, 202.e1–202.e6. [Google Scholar] [CrossRef]
- Dupuis, L.; Corcia, P.; Fergani, A.; Gonzalez De Aguilar, J.-L.; Bonnefont-Rousselot, D.; Bittar, R.; Seilhean, D.; Hauw, J.-J.; Lacomblez, L.; Loeffler, J.-P.; et al. Dyslipidemia Is a Protective Factor in Amyotrophic Lateral Sclerosis. Neurology 2008, 70, 1004–1009. [Google Scholar] [CrossRef]
- Alessenko, A.V. The potential role for sphingolipids in neuropathogenesis of Alzheimer’s disease. Biomeditsinskaya Khimiya 2013, 59, 25–50. [Google Scholar] [CrossRef]
- Alessenko, A.V.; Gurianova, S.V. Potential role for ceramides in neurodegenerative diseases. In Sphingomyelin and Ceramides: Occurrence, Biosynthesis and Role in Disease; Nova Science Publishers: Hauppage, NY, USA, 2015; pp. 21–52. [Google Scholar]
- Abbott, S.K.; Li, H.; Muñoz, S.S.; Knoch, B.; Batterham, M.; Murphy, K.E.; Halliday, G.M.; Garner, B. Altered Ceramide Acyl Chain Length and Ceramide Synthase Gene Expression in Parkinson’s Disease: Altered Ceramide in Parkinson’s Disease. Mov. Disord. 2014, 29, 518–526. [Google Scholar] [CrossRef]
- Pujol-Lereis, L.M. Alteration of Sphingolipids in Biofluids: Implications for Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 3564. [Google Scholar] [CrossRef]
- Robberecht, W.; Sapp, P.; Viaene, M.K.; Rosen, D.; McKenna-Yasek, D.; Haines, J.; Horvitz, R.; Theys, P.; Brown, R. Rapid Communication: Cu/Zn Superoxide Dismutase Activity in Familial and Sporadic Amyotrophic Lateral Sclerosis. J. Neurochem. 2008, 62, 384–387. [Google Scholar] [CrossRef]
- Peters, O.M.; Ghasemi, M.; Brown, R.H. Emerging Mechanisms of Molecular Pathology in ALS. J. Clin. Investig. 2015, 125, 1767–1779. [Google Scholar] [CrossRef]
- Ripps, M.E.; Huntley, G.W.; Hof, P.R.; Morrison, J.H.; Gordon, J.W. Transgenic Mice Expressing an Altered Murine Superoxide Dismutase Gene Provide an Animal Model of Amyotrophic Lateral Sclerosis. Proc. Natl. Acad. Sci. USA 1995, 92, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Bowling, A.C.; Schulz, J.B.; Brown, R.H.; Beal, M.F. Superoxide Dismutase Activity, Oxidative Damage, and Mitochondrial Energy Metabolism in Familial and Sporadic Amyotrophic Lateral Sclerosis. J. Neurochem. 1993, 61, 2322–2325. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, E.; Factor-Litvak, P.; Santella, R.M.; Mitsumoto, H. Clinical Perspective on Oxidative Stress in Sporadic Amyotrophic Lateral Sclerosis. Free Radic. Biol. Med. 2013, 65, 509–527. [Google Scholar] [CrossRef] [PubMed]
- Nagase, M.; Yamamoto, Y.; Miyazaki, Y.; Yoshino, H. Increased Oxidative Stress in Patients with Amyotrophic Lateral Sclerosis and the Effect of Edaravone Administration. Redox Rep. 2016, 21, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Boillée, S.; Yamanaka, K.; Lobsiger, C.S.; Copeland, N.G.; Jenkins, N.A.; Kassiotis, G.; Kollias, G.; Cleveland, D.W. Onset and Progression in Inherited ALS Determined by Motor Neurons and Microglia. Science 2006, 312, 1389–1392. [Google Scholar] [CrossRef]
- Philips, T.; Rothstein, J.D. Glial Cells in Amyotrophic Lateral Sclerosis. Exp. Neurol. 2014, 262, 111–120. [Google Scholar] [CrossRef]
- Ozdinler, P.H.; Benn, S.; Yamamoto, T.H.; Guzel, M.; Brown, R.H.; Macklis, J.D. Corticospinal Motor Neurons and Related Subcerebral Projection Neurons Undergo Early and Specific Neurodegeneration in HSOD1G93A Transgenic ALS Mice. J. Neurosci. 2011, 31, 4166–4177. [Google Scholar] [CrossRef]
- Hatzipetros, T.; Bogdanik, L.P.; Tassinari, V.R.; Kidd, J.D.; Moreno, A.J.; Davis, C.; Osborne, M.; Austin, A.; Vieira, F.G.; Lutz, C.; et al. C57BL/6J Congenic Prp-TDP43A315T Mice Develop Progressive Neurodegeneration in the Myenteric Plexus of the Colon without Exhibiting Key Features of ALS. Brain Res. 2014, 1584, 59–72. [Google Scholar] [CrossRef]
- Joyce, P.I.; Fratta, P.; Fisher, E.M.C.; Acevedo-Arozena, A. SOD1 and TDP-43 Animal Models of Amyotrophic Lateral Sclerosis: Recent Advances in Understanding Disease toward the Development of Clinical Treatments. Mamm. Genome 2011, 22, 420–448. [Google Scholar] [CrossRef]
- Kang, S.H.; Li, Y.; Fukaya, M.; Lorenzini, I.; Cleveland, D.W.; Ostrow, L.W.; Rothstein, J.D.; Bergles, D.E. Degeneration and Impaired Regeneration of Gray Matter Oligodendrocytes in Amyotrophic Lateral Sclerosis. Nat. Neurosci. 2013, 16, 571–579. [Google Scholar] [CrossRef]
- Kriz, J.; Nguyen, M.D.; Julien, J.-P. Minocycline Slows Disease Progression in a Mouse Model of Amyotrophic Lateral Sclerosis. Neurobiol. Dis. 2002, 10, 268–278. [Google Scholar] [CrossRef]
- Van Den Bosch, L.; Tilkin, P.; Lemmens, G.; Robberecht, W. Minocycline Delays Disease Onset and Mortality in a Transgenic Model of ALS. Neuroreport 2002, 13, 1067–1070. [Google Scholar] [CrossRef]
- Kwong, L.K.; Neumann, M.; Sampathu, D.M.; Lee, V.M.-Y.; Trojanowski, J.Q. TDP-43 Proteinopathy: The Neuropathology Underlying Major Forms of Sporadic and Familial Frontotemporal Lobar Degeneration and Motor Neuron Disease. Acta Neuropathol. 2007, 114, 63–70. [Google Scholar] [CrossRef]
- Rutherford, N.J.; Zhang, Y.-J.; Baker, M.; Gass, J.M.; Finch, N.A.; Xu, Y.-F.; Stewart, H.; Kelley, B.J.; Kuntz, K.; Crook, R.J.P.; et al. Novel Mutations in TARDBP (TDP-43) in Patients with Familial Amyotrophic Lateral Sclerosis. PLoS Genet. 2008, 4, e1000193. [Google Scholar] [CrossRef]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Polymenidou, M.; Hutt, K.R.; Vu, A.Q.; Baughn, M.; Huelga, S.C.; Clutario, K.M.; Ling, S.-C.; Liang, T.Y.; Mazur, C.; et al. Divergent Roles of ALS-Linked Proteins FUS/TLS and TDP-43 Intersect in Processing Long Pre-MRNAs. Nat. Neurosci. 2012, 15, 1488–1497. [Google Scholar] [CrossRef]
- Polymenidou, M.; Lagier-Tourenne, C.; Hutt, K.R.; Bennett, C.F.; Cleveland, D.W.; Yeo, G.W. Misregulated RNA Processing in Amyotrophic Lateral Sclerosis. Brain Res. 2012, 1462, 3–15. [Google Scholar] [CrossRef]
- Sun, Z.; Diaz, Z.; Fang, X.; Hart, M.P.; Chesi, A.; Shorter, J.; Gitler, A.D. Molecular Determinants and Genetic Modifiers of Aggregation and Toxicity for the ALS Disease Protein FUS/TLS. PLoS Biol. 2011, 9, e1000614. [Google Scholar] [CrossRef]
- Thompson, V.F.; Victor, R.A.; Morera, A.A.; Moinpour, M.; Liu, M.N.; Kisiel, C.C.; Pickrel, K.; Springhower, C.E.; Schwartz, J.C. Transcription-Dependent Formation of Nuclear Granules Containing FUS and RNA Pol II. Biochemistry 2018, 57, 7021–7032. [Google Scholar] [CrossRef]
- De Santis, R.; Santini, L.; Colantoni, A.; Peruzzi, G.; De Turris, V.; Alfano, V.; Bozzoni, I.; Rosa, A. FUS Mutant Human Motoneurons Display Altered Transcriptome and MicroRNA Pathways with Implications for ALS Pathogenesis. Stem Cell Rep. 2017, 9, 1450–1462. [Google Scholar] [CrossRef]
- Alexander, E.J.; Ghanbari Niaki, A.; Zhang, T.; Sarkar, J.; Liu, Y.; Nirujogi, R.S.; Pandey, A.; Myong, S.; Wang, J. Ubiquilin 2 Modulates ALS/FTD-Linked FUS–RNA Complex Dynamics and Stress Granule Formation. Proc. Natl. Acad. Sci. USA 2018, 115, E11485–E11494. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Guo, W.; Mitra, J.; Hegde, P.M.; Vandoorne, T.; Eckelmann, B.J.; Mitra, S.; Tomkinson, A.E.; Van Den Bosch, L.; Hegde, M.L. Mutant FUS Causes DNA Ligation Defects to Inhibit Oxidative Damage Repair in Amyotrophic Lateral Sclerosis. Nat. Commun. 2018, 9, 3683. [Google Scholar] [CrossRef] [PubMed]
- Delva, L.; Gallais, I.; Guillouf, C.; Denis, N.; Orvain, C.; Moreau-Gachelin, F. Multiple Functional Domains of the Oncoproteins Spi-1/PU.1 and TLS Are Involved in Their Opposite Splicing Effects in Erythroleukemic Cells. Oncogene 2004, 23, 4389–4399. [Google Scholar] [CrossRef] [PubMed]
- Uranishi, H.; Tetsuka, T.; Yamashita, M.; Asamitsu, K.; Shimizu, M.; Itoh, M.; Okamoto, T. Involvement of the Pro-Oncoprotein TLS (Translocated in Liposarcoma) in Nuclear Factor-ΚB P65-Mediated Transcription as a Coactivator. J. Biol. Chem. 2001, 276, 13395–13401. [Google Scholar] [CrossRef] [PubMed]
- Immanuel, D.; Zinszner, H.; Ron, D. Association of SARFH (Sarcoma-Associated RNA-Binding Fly Homolog) with Regions of Chromatin Transcribed by RNA Polymerase II. Mol. Cell. Biol. 1995, 15, 4562–4571. [Google Scholar] [CrossRef]
- Shelkovnikova, T.A.; Peters, O.M.; Deykin, A.V.; Connor-Robson, N.; Robinson, H.; Ustyugov, A.A.; Bachurin, S.O.; Ermolkevich, T.G.; Goldman, I.L.; Sadchikova, E.R.; et al. Fused in Sarcoma (FUS) Protein Lacking Nuclear Localization Signal (NLS) and Major RNA Binding Motifs Triggers Proteinopathy and Severe Motor Phenotype in Transgenic Mice. J. Biol. Chem. 2013, 288, 25266–25274. [Google Scholar] [CrossRef]
- Robinson, H.K.; Deykin, A.V.; Bronovitsky, E.V.; Ovchinnikov, R.K.; Ustyugov, A.A.; Shelkovnikova, T.A.; Kukharsky, M.S.; Ermolkevich, T.G.; Goldman, I.L.; Sadchikova, E.R.; et al. Early Lethality and Neuronal Proteinopathy in Mice Expressing Cytoplasm-Targeted FUS That Lacks the RNA Recognition Motif. Amyotroph. Lateral Scler. Front. Degener. 2015, 16, 402–409. [Google Scholar] [CrossRef]
- Deng, H.-X.; Chen, W.; Hong, S.-T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in UBQLN2 Cause Dominant X-Linked Juvenile and Adult-Onset ALS and ALS/Dementia. Nature 2011, 477, 211–215. [Google Scholar] [CrossRef]
- Watts, G.D.J.; Wymer, J.; Kovach, M.J.; Mehta, S.G.; Mumm, S.; Darvish, D.; Pestronk, A.; Whyte, M.P.; Kimonis, V.E. Inclusion Body Myopathy Associated with Paget Disease of Bone and Frontotemporal Dementia Is Caused by Mutant Valosin-Containing Protein. Nat. Genet. 2004, 36, 377–381. [Google Scholar] [CrossRef]
- Weihl, C.C.; Miller, S.E.; Hanson, P.I.; Pestronk, A. Transgenic Expression of Inclusion Body Myopathy Associated Mutant P97/VCP Causes Weakness and Ubiquitinated Protein Inclusions in Mice. Hum. Mol. Genet. 2007, 16, 919–928. [Google Scholar] [CrossRef]
- Nalbandian, A.; Nguyen, C.; Katheria, V.; Llewellyn, K.J.; Badadani, M.; Caiozzo, V.; Kimonis, V.E. Exercise Training Reverses Skeletal Muscle Atrophy in an Experimental Model of VCP Disease. PLoS ONE 2013, 8, e76187. [Google Scholar] [CrossRef]
- Huang, R.; Guo, X.; Chen, X.; Zheng, Z.; Wei, Q.; Cao, B.; Zeng, Y.; Shang, H. The Serum Lipid Profiles of Amyotrophic Lateral Sclerosis Patients: A Study from South-West China and a Meta-Analysis. Amyotroph. Lateral Scler. Front. Degener. 2015, 16, 359–365. [Google Scholar] [CrossRef]
- Rafiq, M.K.; Lee, E.; Bradburn, M.; McDermott, C.J.; Shaw, P.J. Effect of Lipid Profile on Prognosis in the Patients with Amyotrophic Lateral Sclerosis: Insights from the Olesoxime Clinical Trial. Amyotroph. Lateral Scler. Front. Degener. 2015, 16, 478–484. [Google Scholar] [CrossRef]
- Chio, A.; Calvo, A.; Ilardi, A.; Cavallo, E.; Moglia, C.; Mutani, R.; Palmo, A.; Galletti, R.; Marinou, K.; Papetti, L.; et al. Lower Serum Lipid Levels Are Related to Respiratory Impairment in Patients with ALS. Neurology 2009, 73, 1681–1685. [Google Scholar] [CrossRef]
- Dorst, J.; Kühnlein, P.; Hendrich, C.; Kassubek, J.; Sperfeld, A.D.; Ludolph, A.C. Patients with Elevated Triglyceride and Cholesterol Serum Levels Have a Prolonged Survival in Amyotrophic Lateral Sclerosis. J. Neurol. 2011, 258, 613–617. [Google Scholar] [CrossRef]
- Arima, H.; Omura, T.; Hayasaka, T.; Masaki, N.; Hanada, M.; Xu, D.; Banno, T.; Kobayashi, K.; Takeuchi, H.; Kadomatsu, K.; et al. Reductions of Docosahexaenoic Acid-Containing Phosphatidylcholine Levels in the Anterior Horn of an ALS Mouse Model. Neuroscience 2015, 297, 127–136. [Google Scholar] [CrossRef]
- Henriques, A.; Blasco, H.; Fleury, M.-C.; Corcia, P.; Echaniz-Laguna, A.; Robelin, L.; Rudolf, G.; Lequeu, T.; Bergaentzle, M.; Gachet, C.; et al. Blood Cell Palmitoleate-Palmitate Ratio Is an Independent Prognostic Factor for Amyotrophic Lateral Sclerosis. PLoS ONE 2015, 10, e0131512. [Google Scholar] [CrossRef]
- Vejux, A.; Namsi, A.; Nury, T.; Moreau, T.; Lizard, G. Biomarkers of Amyotrophic Lateral Sclerosis: Current Status and Interest of Oxysterols and Phytosterols. Front. Mol. Neurosci. 2018, 11, 12. [Google Scholar] [CrossRef]
- Wuolikainen, A.; Acimovic, J.; Lövgren-Sandblom, A.; Parini, P.; Andersen, P.M.; Björkhem, I. Cholesterol, Oxysterol, Triglyceride, and Coenzyme Q Homeostasis in ALS. Evidence against the Hypothesis That Elevated 27-Hydroxycholesterol Is a Pathogenic Factor. PLoS ONE 2014, 9, e113619. [Google Scholar] [CrossRef]
- Farooqui, A.A.; Horrocks, L.A. Excitotoxicity and neurological disorders: Involvement of membrane phospholipids. In International Review of Neurobiology; Elsevier: Amsterdam, The Netherlands, 1994; Volume 36, pp. 267–323. ISBN 9780123668363. [Google Scholar]
- Fitzgerald, K.C.; O’Reilly, É.J.; Falcone, G.J.; McCullough, M.L.; Park, Y.; Kolonel, L.N.; Ascherio, A. Dietary ω-3 Polyunsaturated Fatty Acid Intake and Risk for Amyotrophic Lateral Sclerosis. JAMA Neurol. 2014, 71, 1102. [Google Scholar] [CrossRef]
- Cacabelos, D.; Ayala, V.; Granado-Serrano, A.B.; Jové, M.; Torres, P.; Boada, J.; Cabré, R.; Ramírez-Núñez, O.; Gonzalo, H.; Soler-Cantero, A.; et al. Interplay between TDP-43 and Docosahexaenoic Acid-Related Processes in Amyotrophic Lateral Sclerosis. Neurobiol. Dis. 2016, 88, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Fiala, M.; Mizwicki, M.T.; Sayre, J.; Magpantay, L.; Siani, A.; Mahanian, M.; Chattopadhyay, M.; La Cava, A.; Wiedau-Pazos, M. Neuronal Phagocytosis by Inflammatory Macrophages in ALS Spinal Cord: Inhibition of Inflammation by Resolvin D1. Am. J. Neurodegener. Dis. 2012, 1, 60–74. [Google Scholar] [PubMed]
- Iłżecka, J. Prostaglandin E2 Is Increased in Amyotrophic Lateral Sclerosis Patients: Prostaglandin E2 in Amyotrophic Lateral Sclerosis. Acta Neurol. Scand. 2003, 108, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Miyagishi, H.; Kosuge, Y.; Takano, A.; Endo, M.; Nango, H.; Yamagata-Murayama, S.; Hirose, D.; Kano, R.; Tanaka, Y.; Ishige, K.; et al. Increased Expression of 15-Hydroxyprostaglandin Dehydrogenase in Spinal Astrocytes During Disease Progression in a Model of Amyotrophic Lateral Sclerosis. Cell. Mol. Neurobiol. 2017, 37, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lee, J.J.; Park, N.Y.; Dubey, S.K.; Kim, T.; Ruan, K.; Lim, S.B.; Park, S.-H.; Ha, S.; Kovlyagina, I.; et al. Multi-Omic Analysis of Selectively Vulnerable Motor Neuron Subtypes Implicates Altered Lipid Metabolism in ALS. Nat. Neurosci. 2021, 24, 1673–1685. [Google Scholar] [CrossRef]
- O’Reilly, É.J.; Bjornevik, K.; Furtado, J.D.; Kolonel, L.N.; Le Marchand, L.; McCullough, M.L.; Stevens, V.L.; Shadyab, A.H.; Snetselaar, L.; Manson, J.E.; et al. Prediagnostic Plasma Polyunsaturated Fatty Acids and the Risk of Amyotrophic Lateral Sclerosis. Neurology 2020, 94, e811–e819. [Google Scholar] [CrossRef]
- Yip, P.K.; Pizzasegola, C.; Gladman, S.; Biggio, M.L.; Marino, M.; Jayasinghe, M.; Ullah, F.; Dyall, S.C.; Malaspina, A.; Bendotti, C.; et al. The Omega-3 Fatty Acid Eicosapentaenoic Acid Accelerates Disease Progression in a Model of Amyotrophic Lateral Sclerosis. PLoS ONE 2013, 8, e61626. [Google Scholar] [CrossRef]
- Diaz-Amarilla, P.; Miquel, E.; Trostchansky, A.; Trias, E.; Ferreira, A.M.; Freeman, B.A.; Cassina, P.; Barbeito, L.; Vargas, M.R.; Rubbo, H. Electrophilic Nitro-Fatty Acids Prevent Astrocyte-Mediated Toxicity to Motor Neurons in a Cell Model of Familial Amyotrophic Lateral Sclerosis via Nuclear Factor Erythroid 2-Related Factor Activation. Free Radic. Biol. Med. 2016, 95, 112–120. [Google Scholar] [CrossRef]
- Rubbo, H. Nitro-Fatty Acids: Novel Anti-Inflammatory Lipid Mediators. Braz. J. Med. Biol. Res. 2013, 46, 728–734. [Google Scholar] [CrossRef]
- Trostchansky, A.; Mastrogiovanni, M.; Miquel, E.; Rodríguez-Bottero, S.; Martínez-Palma, L.; Cassina, P.; Rubbo, H. Profile of Arachidonic Acid-Derived Inflammatory Markers and Its Modulation by Nitro-Oleic Acid in an Inherited Model of Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2018, 11, 131. [Google Scholar] [CrossRef]
- Veldink, J.H.; Kalmijn, S.; Groeneveld, G.-J.; Wunderink, W.; Koster, A.; De Vries, J.H.M.; Van Der Luyt, J.; Wokke, J.H.J.; Van Den Berg, L.H. Intake of Polyunsaturated Fatty Acids and Vitamin E Reduces the Risk of Developing Amyotrophic Lateral Sclerosis. J. Neurol. Neurosurg. Psychiatry 2006, 78, 367–371. [Google Scholar] [CrossRef]
- Pape, J.A.; Grose, J.H. The Effects of Diet and Sex in Amyotrophic Lateral Sclerosis. Rev. Neurol. 2020, 176, 301–315. [Google Scholar] [CrossRef]
- Zhao, W.; Varghese, M.; Vempati, P.; Dzhun, A.; Cheng, A.; Wang, J.; Lange, D.; Bilski, A.; Faravelli, I.; Pasinetti, G.M. Caprylic Triglyceride as a Novel Therapeutic Approach to Effectively Improve the Performance and Attenuate the Symptoms Due to the Motor Neuron Loss in ALS Disease. PLoS ONE 2012, 7, e49191. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Principles of Bioactive Lipid Signalling: Lessons from Sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef]
- Phillips, G.R.; Saville, J.T.; Hancock, S.E.; Brown, S.H.J.; Jenner, A.M.; McLean, C.; Fuller, M.; Newell, K.A.; Mitchell, T.W. The Long and the Short of Huntington’s Disease: How the Sphingolipid Profile Is Shifted in the Caudate of Advanced Clinical Cases. Brain Commun. 2022, 4, fcab303. [Google Scholar] [CrossRef]
- Alessenko, A.V.; Bugrova, A.E.; Dudnik, L.B. Connection of Lipid Peroxide Oxidation with the Sphingomyelin Pathway in the Development of Alzheimer’s Disease. Biochem. Soc. Trans. 2004, 32, 144–146. [Google Scholar] [CrossRef]
- Brodowicz, J.; Przegaliński, E.; Müller, C.P.; Filip, M. Ceramide and Its Related Neurochemical Networks as Targets for Some Brain Disorder Therapies. Neurotox. Res. 2018, 33, 474–484. [Google Scholar] [CrossRef]
- Dodge, J.C.; Treleaven, C.M.; Pacheco, J.; Cooper, S.; Bao, C.; Abraham, M.; Cromwell, M.; Sardi, S.P.; Chuang, W.-L.; Sidman, R.L.; et al. Glycosphingolipids Are Modulators of Disease Pathogenesis in Amyotrophic Lateral Sclerosis. Proc. Natl. Acad. Sci. USA 2015, 112, 8100–8105. [Google Scholar] [CrossRef]
- Godoy-Corchuelo, J.M.; Fernández-Beltrán, L.C.; Ali, Z.; Gil-Moreno, M.J.; López-Carbonero, J.I.; Guerrero-Sola, A.; Larrad-Sainz, A.; Matias-Guiu, J.; Matias-Guiu, J.A.; Cunningham, T.J.; et al. Lipid Metabolic Alterations in the ALS–FTD Spectrum of Disorders. Biomedicines 2022, 10, 1105. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Many Ceramides. J. Biol. Chem. 2011, 286, 27855–27862. [Google Scholar] [CrossRef]
- Gomez-Larrauri, A.; Presa, N.; Dominguez-Herrera, A.; Ouro, A.; Trueba, M.; Gomez-Muñoz, A. Role of Bioactive Sphingolipids in Physiology and Pathology. Essays Biochem. 2020, 64, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Mao, C. Ceramidases: Regulators of Cellular Responses Mediated by Ceramide, Sphingosine, and Sphingosine-1-Phosphate. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2008, 1781, 424–434. [Google Scholar] [CrossRef] [PubMed]
- Chaurasia, B.; Summers, S.A. Ceramides in Metabolism: Key Lipotoxic Players. Annu. Rev. Physiol. 2021, 83, 303–330. [Google Scholar] [CrossRef] [PubMed]
- Dawkins, J.L.; Hulme, D.J.; Brahmbhatt, S.B.; Auer-Grumbach, M.; Nicholson, G.A. Mutations in SPTLC1, Encoding Serine Palmitoyltransferase, Long Chain Base Subunit-1, Cause Hereditary Sensory Neuropathy Type I. Nat. Genet. 2001, 27, 309–312. [Google Scholar] [CrossRef]
- Krebs, S.; Medugorac, I.; Röther, S.; Strässer, K.; Förster, M. A Missense Mutation in the 3-Ketodihydrosphingosine Reductase FVT1 as Candidate Causal Mutation for Bovine Spinal Muscular Atrophy. Proc. Natl. Acad. Sci. USA 2007, 104, 6746–6751. [Google Scholar] [CrossRef]
- Zhou, J.; Tawk, M.; Tiziano, F.D.; Veillet, J.; Bayes, M.; Nolent, F.; Garcia, V.; Servidei, S.; Bertini, E.; Castro-Giner, F.; et al. Spinal Muscular Atrophy Associated with Progressive Myoclonic Epilepsy Is Caused by Mutations in ASAH1. Am. J. Hum. Genet. 2012, 91, 5–14. [Google Scholar] [CrossRef]
- Mohassel, P.; Donkervoort, S.; Lone, M.A.; Nalls, M.; Gable, K.; Gupta, S.D.; Foley, A.R.; Hu, Y.; Saute, J.A.M.; Moreira, A.L.; et al. Childhood Amyotrophic Lateral Sclerosis Caused by Excess Sphingolipid Synthesis. Nat. Med. 2021, 27, 1197–1204. [Google Scholar] [CrossRef]
- Cutler, R.G.; Pedersen, W.A.; Camandola, S.; Rothstein, J.D.; Mattson, M.P. Evidence That Accumulation of Ceramides and Cholesterol Esters Mediates Oxidative Stress-Induced Death of Motor Neurons in Amyotrophic Lateral Sclerosis. Ann. Neurol. 2002, 52, 448–457. [Google Scholar] [CrossRef]
- Henriques, A.; Croixmarie, V.; Priestman, D.A.; Rosenbohm, A.; Dirrig-Grosch, S.; D’Ambra, E.; Huebecker, M.; Hussain, G.; Boursier-Neyret, C.; Echaniz-Laguna, A.; et al. Amyotrophic Lateral Sclerosis and Denervation Alter Sphingolipids and Up-Regulate Glucosylceramide Synthase. Hum. Mol. Genet. 2015, 24, 7390–7405. [Google Scholar] [CrossRef]
- Chaves-Filho, A.B.; Pinto, I.F.D.; Dantas, L.S.; Xavier, A.M.; Inague, A.; Faria, R.L.; Medeiros, M.H.G.; Glezer, I.; Yoshinaga, M.Y.; Miyamoto, S. Alterations in Lipid Metabolism of Spinal Cord Linked to Amyotrophic Lateral Sclerosis. Sci. Rep. 2019, 9, 11642. [Google Scholar] [CrossRef]
- Shupik, M.A.; Gutner, U.A.; Ustyugov, A.A.; Rezvykh, A.P.; Funikov, S.Y.; Maloshitskaya, O.A.; Sokolov, S.A.; Lebedev, A.T.; Alessenko, A.V. Changes in the metabolism of sphingomyelin and ceramide in the brain structures and spinal cord of transgenic mice (FUS (1-359)) modeling amyotrophic lateral sclerosis. Russ. J. Bioorganic Chem. 2022, 48, 115–126. [Google Scholar] [CrossRef]
- Fernández-Beltrán, L.C.; Godoy-Corchuelo, J.M.; Losa-Fontangordo, M.; Williams, D.; Matias-Guiu, J.; Corrochano, S. A Transcriptomic Meta-Analysis Shows Lipid Metabolism Dysregulation as an Early Pathological Mechanism in the Spinal Cord of SOD1 Mice. Int. J. Mol. Sci. 2021, 22, 9553. [Google Scholar] [CrossRef]
- Gutner, U.A.; Shupik, M.A.; Maloshitskaya, O.A.; Sokolov, S.A.; Rezvykh, A.P.; Funikov, S.Y.; Lebedev, A.T.; Ustyugov, A.A.; Alessenko, A.V. Changes in the Metabolism of Sphingoid Bases in the Brain and Spinal Cord of Transgenic FUS (1-359) Mice, a Model of Amyotrophic Lateral Sclerosis. Biochem. Biokhimiia 2019, 84, 1166–1176. [Google Scholar] [CrossRef]
- Kirby, J.; Ning, K.; Ferraiuolo, L.; Heath, P.R.; Ismail, A.; Kuo, S.-W.; Valori, C.F.; Cox, L.; Sharrack, B.; Wharton, S.B.; et al. Phosphatase and Tensin Homologue/Protein Kinase B Pathway Linked to Motor Neuron Survival in Human Superoxide Dismutase 1-Related Amyotrophic Lateral Sclerosis. Brain 2011, 134, 506–517. [Google Scholar] [CrossRef]
- Pehar, M.; Cassina, P.; Vargas, M.R.; Castellanos, R.; Viera, L.; Beckman, J.S.; Estévez, A.G.; Barbeito, L. Astrocytic Production of Nerve Growth Factor in Motor Neuron Apoptosis: Implications for Amyotrophic Lateral Sclerosis. J. Neurochem. 2004, 89, 464–473. [Google Scholar] [CrossRef]
- Alessenko, A.V.; Khrenov, A.V. Role of Sphingosine in Induced Apoptosis. Lipids 1999, 34, S75–S76. [Google Scholar] [CrossRef]
- Tamiya-Koizumi, K.; Murate, T.; Suzuki, M.; Simbulan, C.M.; Nakagawa, M.; Takemura, M.; Furuta, K.; Izuta, S.; Yoshida, S. Inhibition of DNA Primase by Sphingosine and Its Analogues Parallels with Their Growth Suppression of Cultured Human Leukemic Cells. IUBMB Life 1997, 41, 1179–1189. [Google Scholar] [CrossRef]
- Koiv, A.; Palvimo, J.; Kinnunen, P.K.J. Evidence for Ternary Complex Formation by Histone H1, DNA, and Liposomes. Biochemistry 1995, 34, 8018–8027. [Google Scholar] [CrossRef]
- Burg, T.; Rossaert, E.; Moisse, M.; Van Damme, P.; Van Den Bosch, L. Histone Deacetylase Inhibition Regulates Lipid Homeostasis in a Mouse Model of Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2021, 22, 11224. [Google Scholar] [CrossRef]
- Taha, T.A.; Mullen, T.D.; Obeid, L.M. A House Divided: Ceramide, Sphingosine, and Sphingosine-1-Phosphate in Programmed Cell Death. Biochim. Biophys. Acta BBA Biomembr. 2006, 1758, 2027–2036. [Google Scholar] [CrossRef]
- Rohrbach, T.; Maceyka, M.; Spiegel, S. Sphingosine Kinase and Sphingosine-1-Phosphate in Liver Pathobiology. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Henriques, A.; Croixmarie, V.; Bouscary, A.; Mosbach, A.; Keime, C.; Boursier-Neyret, C.; Walter, B.; Spedding, M.; Loeffler, J.-P. Sphingolipid Metabolism Is Dysregulated at Transcriptomic and Metabolic Levels in the Spinal Cord of an Animal Model of Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2018, 10, 433. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Pandey, A. The Yin and Yang of Lactosylceramide Metabolism: Implications in Cell Function. Biochim. Biophys. Acta BBA Gen. Subj. 2008, 1780, 370–382. [Google Scholar] [CrossRef] [PubMed]
- Won, J.-S.; Singh, A.K.; Singh, I. Lactosylceramide: A Lipid Second Messenger in Neuroinflammatory Disease. J. Neurochem. 2007, 103, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Frakes, A.E.; Ferraiuolo, L.; Haidet-Phillips, A.M.; Schmelzer, L.; Braun, L.; Miranda, C.J.; Ladner, K.J.; Bevan, A.K.; Foust, K.D.; Godbout, J.P.; et al. Microglia Induce Motor Neuron Death via the Classical NF-ΚB Pathway in Amyotrophic Lateral Sclerosis. Neuron 2014, 81, 1009–1023. [Google Scholar] [CrossRef]
- Zhuang, S.; Kong, L.; Li, C.; Chen, L.; Zhang, T. GALC Mutations in Chinese Patients with Late-Onset Krabbe Disease: A Case Report. BMC Neurol. 2019, 19, 122. [Google Scholar] [CrossRef]
- Ishibashi, Y.; Kohyama-Koganeya, A.; Hirabayashi, Y. New Insights on Glucosylated Lipids: Metabolism and Functions. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2013, 1831, 1475–1485. [Google Scholar] [CrossRef]
- Kohyama-Koganeya, A.; Sasamura, T.; Oshima, E.; Suzuki, E.; Nishihara, S.; Ueda, R.; Hirabayashi, Y. Drosophila Glucosylceramide Synthase. J. Biol. Chem. 2004, 279, 35995–36002. [Google Scholar] [CrossRef]
- Henriques, A.; Huebecker, M.; Blasco, H.; Keime, C.; Andres, C.R.; Corcia, P.; Priestman, D.A.; Platt, F.M.; Spedding, M.; Loeffler, J.-P. Inhibition of β-Glucocerebrosidase Activity Preserves Motor Unit Integrity in a Mouse Model of Amyotrophic Lateral Sclerosis. Sci. Rep. 2017, 7, 5235. [Google Scholar] [CrossRef]
- Inokuchi, J. Chapter 22 Neurotrophic and neuroprotective actions of an enhancer of ganglioside biosynthesis. In International Review of Neurobiology; Elsevier: Amsterdam, The Netherlands, 2009; Volume 85, pp. 319–336. ISBN 9780123748935. [Google Scholar]
- Dawson, G.; Stefansson, K. Gangliosides of Human Spinal Cord: Aberrant Composition of Cords from Patients with Amyotrophic Lateral Sclerosis. J. Neurosci. Res. 1984, 12, 213–220. [Google Scholar] [CrossRef]
- Salazar-Grueso, E.F.; Routbort, M.J.; Martin, J.; Dawson, G.; Roos, R.P. Polyclonal IgM Anti-GM1 Ganglioside Antibody in Patients with Motor Neuron Disease and Variants. Ann. Neurol. 1990, 27, 558–563. [Google Scholar] [CrossRef]
- Stevens, A.; Weller, M.; Wietholter, H. A Characteristic Ganglioside Antibody Pattern in the CSF of Patients with Amyotrophic Lateral Sclerosis. J. Neurol. Neurosurg. Psychiatry 1993, 56, 361–364. [Google Scholar] [CrossRef]
- Rapport, M.M.; Donnenfeld, H.; Brunner, W.; Hungund, B.; Bartfeld, H. Ganglioside Patterns in Amyotrophic Lateral Sclerosis Brain Regions. Ann. Neurol. 1985, 18, 60–67. [Google Scholar] [CrossRef]
- Yim, S.H.; Farrer, R.G.; Hammer, J.A.; Yavin, E.; Quarles, R.H. Differentiation of Oligodendrocytes Cultured from Developing Rat Brain Is Enhanced by Exogenous GM3 Ganglioside. J. Neurosci. Res. 1994, 38, 268–281. [Google Scholar] [CrossRef]
- Ferraiuolo, L.; Meyer, K.; Sherwood, T.W.; Vick, J.; Likhite, S.; Frakes, A.; Miranda, C.J.; Braun, L.; Heath, P.R.; Pineda, R.; et al. Oligodendrocytes Contribute to Motor Neuron Death in ALS via SOD1-Dependent Mechanism. Proc. Natl. Acad. Sci. USA 2016, 113, E6496–E6505. [Google Scholar] [CrossRef]
- Olsen, M.K.; Roberds, S.L.; Ellerbrock, B.R.; Fleck, T.J.; McKinley, D.K.; Gurney, M.E. Disease Mechanisms Revealed by Transcription Profiling in SOD1-G93A Transgenic Mouse Spinal Cord. Ann. Neurol. 2001, 50, 730–740. [Google Scholar] [CrossRef]
- Lobsiger, C.S.; Boillée, S.; Cleveland, D.W. Toxicity from Different SOD1 Mutants Dysregulates the Complement System and the Neuronal Regenerative Response in ALS Motor Neurons. Proc. Natl. Acad. Sci. USA 2007, 104, 7319–7326. [Google Scholar] [CrossRef]
- Baker, D.J.; Blackburn, D.J.; Keatinge, M.; Sokhi, D.; Viskaitis, P.; Heath, P.R.; Ferraiuolo, L.; Kirby, J.; Shaw, P.J. Lysosomal and Phagocytic Activity Is Increased in Astrocytes during Disease Progression in the SOD1 G93A Mouse Model of Amyotrophic Lateral Sclerosis. Front. Cell. Neurosci. 2015, 9, 410. [Google Scholar] [CrossRef]
- Sutedja, N.A.; Van Der Schouw, Y.T.; Fischer, K.; Sizoo, E.M.; Huisman, M.H.B.; Veldink, J.H.; Van Den Berg, L.H. Beneficial Vascular Risk Profile Is Associated with Amyotrophic Lateral Sclerosis. J. Neurol. Neurosurg. Psychiatry 2011, 82, 638–642. [Google Scholar] [CrossRef]
- Russell, D.W.; Halford, R.W.; Ramirez, D.M.O.; Shah, R.; Kotti, T. Cholesterol 24-Hydroxylase: An Enzyme of Cholesterol Turnover in the Brain. Annu. Rev. Biochem. 2009, 78, 1017–1040. [Google Scholar] [CrossRef]
- Barenholz, Y. Sphingomyelin and cholesterol: From membrane biophysics and rafts to potential medical applications. In Membrane Dynamics and Domains; Quinn, P.J., Ed.; Springer: Boston, MA, USA, 2004; Volume 37, pp. 167–215. ISBN 9781441934475. [Google Scholar]
- Slotte, J.P.; Bierman, E.L. Depletion of Plasma-Membrane Sphingomyelin Rapidly Alters the Distribution of Cholesterol between Plasma Membranes and Intracellular Cholesterol Pools in Cultured Fibroblasts. Biochem. J. 1988, 250, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Marmillot, P.; Patel, S.; Lakshman, M.R. Reverse Cholesterol Transport Is Regulated by Varying Fatty Acyl Chain Saturation and Sphingomyelin Content in Reconstituted High-Density Lipoproteins. Metabolism 2007, 56, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Stranahan, A.M.; Cutler, R.G.; Button, C.; Telljohann, R.; Mattson, M.P. Diet-Induced Elevations in Serum Cholesterol Are Associated with Alterations in Hippocampal Lipid Metabolism and Increased Oxidative Stress: Elevated Serum Cholesterol Alters Brain Lipids. J. Neurochem. 2011, 118, 611–615. [Google Scholar] [CrossRef]
- Stein, O.; Ben-Naim, M.; Dabach, Y.; Hollander, G.; Stein, Y. Modulation of Sphingomyelinase-Induced Cholesterol Esterification in Fibroblasts, CaCo2 Cells, Macrophages and Smooth Muscle Cells. Biochim. Biophys. Acta BBA Lipids Lipid Metab. 1992, 1126, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Härmälä, A.; Pärn, M.I.; Slotte, J.P. Sphingosine Inhibits Sphingomyelinase-Induced Cholesteryl Ester Formation in Cultured Fibroblasts. Biochim. Biophys. Acta BBA Lipids Lipid Metab. 1993, 1210, 97–104. [Google Scholar] [CrossRef]
- Chitnis, T.; Weiner, H.L. CNS Inflammation and Neurodegeneration. J. Clin. Investig. 2017, 127, 3577–3587. [Google Scholar] [CrossRef]
- Alexianu, M.E.; Kozovska, M.; Appel, S.H. Immune Reactivity in a Mouse Model of Familial ALS Correlates with Disease Progression. Neurology 2001, 57, 1282–1289. [Google Scholar] [CrossRef]
- Malaspina, A.; Puentes, F.; Amor, S. Disease Origin and Progression in Amyotrophic Lateral Sclerosis: An Immunology Perspective. Int. Immunol. 2015, 27, 117–129. [Google Scholar] [CrossRef]
- Mantovani, S.; Garbelli, S.; Pasini, A.; Alimonti, D.; Perotti, C.; Melazzini, M.; Bendotti, C.; Mora, G. Immune System Alterations in Sporadic Amyotrophic Lateral Sclerosis Patients Suggest an Ongoing Neuroinflammatory Process. J. Neuroimmunol. 2009, 210, 73–79. [Google Scholar] [CrossRef]
- Turner, M.R.; Cagnin, A.; Turkheimer, F.E.; Miller, C.C.J.; Shaw, C.E.; Brooks, D.J.; Leigh, P.N.; Banati, R.B. Evidence of Widespread Cerebral Microglial Activation in Amyotrophic Lateral Sclerosis: An [11C](R)-PK11195 Positron Emission Tomography Study. Neurobiol. Dis. 2004, 15, 601–609. [Google Scholar] [CrossRef]
- Potenza, R.L.; De Simone, R.; Armida, M.; Mazziotti, V.; Pèzzola, A.; Popoli, P.; Minghetti, L. Fingolimod: A Disease-Modifier Drug in a Mouse Model of Amyotrophic Lateral Sclerosis. Neurotherapeutics 2016, 13, 918–927. [Google Scholar] [CrossRef]
- Hooten, K.G.; Beers, D.R.; Zhao, W.; Appel, S.H. Protective and Toxic Neuroinflammation in Amyotrophic Lateral Sclerosis. Neurotherapeutics 2015, 12, 364–375. [Google Scholar] [CrossRef]
- Appel, S.H.; Stewart, S.S.; Appel, V.; Harati, Y.; Mietlowski, W.; Weiss, W.; Belendiuk, G.W. A Double-Blind Study of the Effectiveness of Cyclosporine in Amyotrophic Lateral Sclerosis. Arch. Neurol. 1988, 45, 381–386. [Google Scholar] [CrossRef]
- Meucci, N.; Nobile-Orazio, E. Intravenous Immunoglobulin Therapy in Amyotrophic Lateral Sclerosis. J. Neurol. 1996, 243, 117–120. [Google Scholar] [CrossRef]
- Berry, J.D.; Paganoni, S.; Atassi, N.; Macklin, E.A.; Goyal, N.; Rivner, M.; Simpson, E.; Appel, S.; Grasso, D.L.; Mejia, N.I.; et al. Phase IIa Trial of Fingolimod for Amyotrophic Lateral Sclerosis Demonstrates Acceptable Acute Safety and Tolerability: Fingolimod in ALS. Muscle Nerve 2017, 56, 1077–1084. [Google Scholar] [CrossRef]
- Constantinescu, V.; Akgün, K.; Ziemssen, T. Current Status and New Developments in Sphingosine-1-Phosphate Receptor Antagonism: Fingolimod and More. Expert Opin. Drug Metab. Toxicol. 2022, 18, 675–693. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Loomis, C.R.; Merrill, A.H.; Bell, R.M. Sphingosine Inhibition of Protein Kinase C Activity and of Phorbol Dibutyrate Binding in Vitro and in Human Platelets. J. Biol. Chem. 1986, 261, 12604–12609. [Google Scholar] [CrossRef] [PubMed]
- Bartke, N.; Hannun, Y.A. Bioactive Sphingolipids: Metabolism and Function. J. Lipid Res. 2009, 50, S91–S96. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, S.; Dev, K.K. Sphingosine-1-Phosphate Receptor Therapies: Advances in Clinical Trials for CNS-Related Diseases. Neuropharmacology 2017, 113, 597–607. [Google Scholar] [CrossRef]
- Pinschewer, D.D.; Ochsenbein, A.F.; Odermatt, B.; Brinkmann, V.; Hengartner, H.; Zinkernagel, R.M. FTY720 Immunosuppression Impairs Effector T Cell Peripheral Homing Without Affecting Induction, Expansion, and Memory. J. Immunol. 2000, 164, 5761–5770. [Google Scholar] [CrossRef] [PubMed]
- Kahan, B.D. FTY720: From Bench to Bedside. Transplant. Proc. 2004, 36, S531–S543. [Google Scholar] [CrossRef] [PubMed]
- Serpero, L.D.; Filaci, G.; Parodi, A.; Battaglia, F.; Kalli, F.; Brogi, D.; Mancardi, G.L.; Uccelli, A.; Fenoglio, D. Fingolimod Modulates Peripheral Effector and Regulatory T Cells in MS Patients. J. Neuroimmune Pharmacol. 2013, 8, 1106–1113. [Google Scholar] [CrossRef]
- Anastasiadou, S.; Knöll, B. The Multiple Sclerosis Drug Fingolimod (FTY720) Stimulates Neuronal Gene Expression, Axonal Growth and Regeneration. Exp. Neurol. 2016, 279, 243–260. [Google Scholar] [CrossRef]
- Henkel, J.S.; Beers, D.R.; Wen, S.; Rivera, A.L.; Toennis, K.M.; Appel, J.E.; Zhao, W.; Moore, D.H.; Powell, S.Z.; Appel, S.H. Regulatory T-lymphocytes Mediate Amyotrophic Lateral Sclerosis Progression and Survival. EMBO Mol. Med. 2013, 5, 64–79. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.J.; Park, K.H.; Park, M.H.; Huang, E.J.; Kim, S.H.; Bae, J.-S.; Jin, H.K. Acid Sphingomyelinase Inhibition Improves Motor Behavioral Deficits and Neuronal Loss in an Amyotrophic Lateral Sclerosis Mouse Model. BMB Rep. 2022, 55, 621–626. [Google Scholar] [CrossRef]


| Lipid Type | Tissue | Alterations | References | |
|---|---|---|---|---|
| Total lipids | Human plasma (ALS patients) | Dis- or hyper-metabolism (2/3 patiens-dislipidemia) | ↓↑ | [15,16,54] |
| Triglycerides | Human plasma (ALS patients) | elevated triglyceride had a longer life expectancy | ↑ | [54,57] |
| Human cerebrospinal fluid (ALS patients) | long-chain triglycerides | ↓ | [11] | |
| Phospholipids | Human cerebrospinal fluid (ALS patients) | PC (20:4), SPM (22:0) PC (36:4p), PC (36:4e) | ↑ | [11] |
| Model mice brain | PC (36:2), PC (36:4), PC (40:6) | ↑ | [11] | |
| The spinal cord of transgenic SOD 1G93A mice | PC (diacyl-16:0/22:6), PC (diacyl-18:0/22:6), PC (18:1/22:6) | ↓ | [58] | |
| Polyunsaturated fatty acids | Human plasma (ALS patients) | Palmitoleate (16:1), oleate (18:1) | ↑ | [59] |
| Cholesterol | Human plasma (ALS patients) | Total cholesterol | ↑ nd | [55,57,60] [54] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alessenko, A.V.; Gutner, U.A.; Shupik, M.A. Involvement of Lipids in the Pathogenesis of Amyotrophic Lateral Sclerosis. Life 2023, 13, 510. https://doi.org/10.3390/life13020510
Alessenko AV, Gutner UA, Shupik MA. Involvement of Lipids in the Pathogenesis of Amyotrophic Lateral Sclerosis. Life. 2023; 13(2):510. https://doi.org/10.3390/life13020510
Chicago/Turabian StyleAlessenko, Alisa V., Uliana A. Gutner, and Maria A. Shupik. 2023. "Involvement of Lipids in the Pathogenesis of Amyotrophic Lateral Sclerosis" Life 13, no. 2: 510. https://doi.org/10.3390/life13020510
APA StyleAlessenko, A. V., Gutner, U. A., & Shupik, M. A. (2023). Involvement of Lipids in the Pathogenesis of Amyotrophic Lateral Sclerosis. Life, 13(2), 510. https://doi.org/10.3390/life13020510