

Prostaglandin D2 Added during the Differentiation of 3T3-L1 Cells Suppresses Adipogenesis via Dysfunction of D-Prostanoid Receptor P1 and P2

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
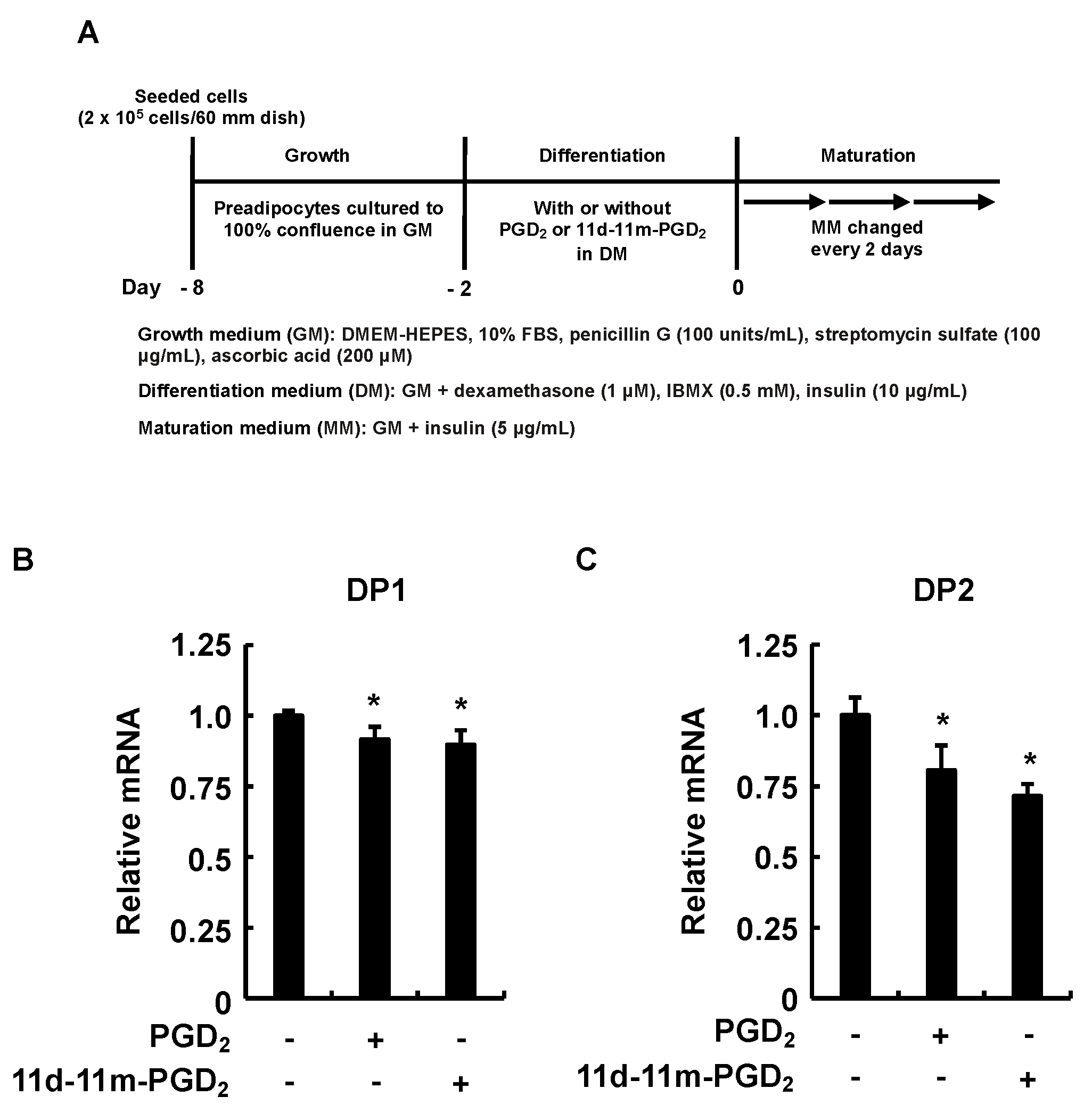
2.2. Cell Culture of 3T3-L1 Cells and Induction of Adipogenesis
2.3. Quantitation of Intracellular Triacylglycerols and Proteins
2.4. Quantitative Analysis of Gene Expression
2.5. Statistical Analyses
3. Results
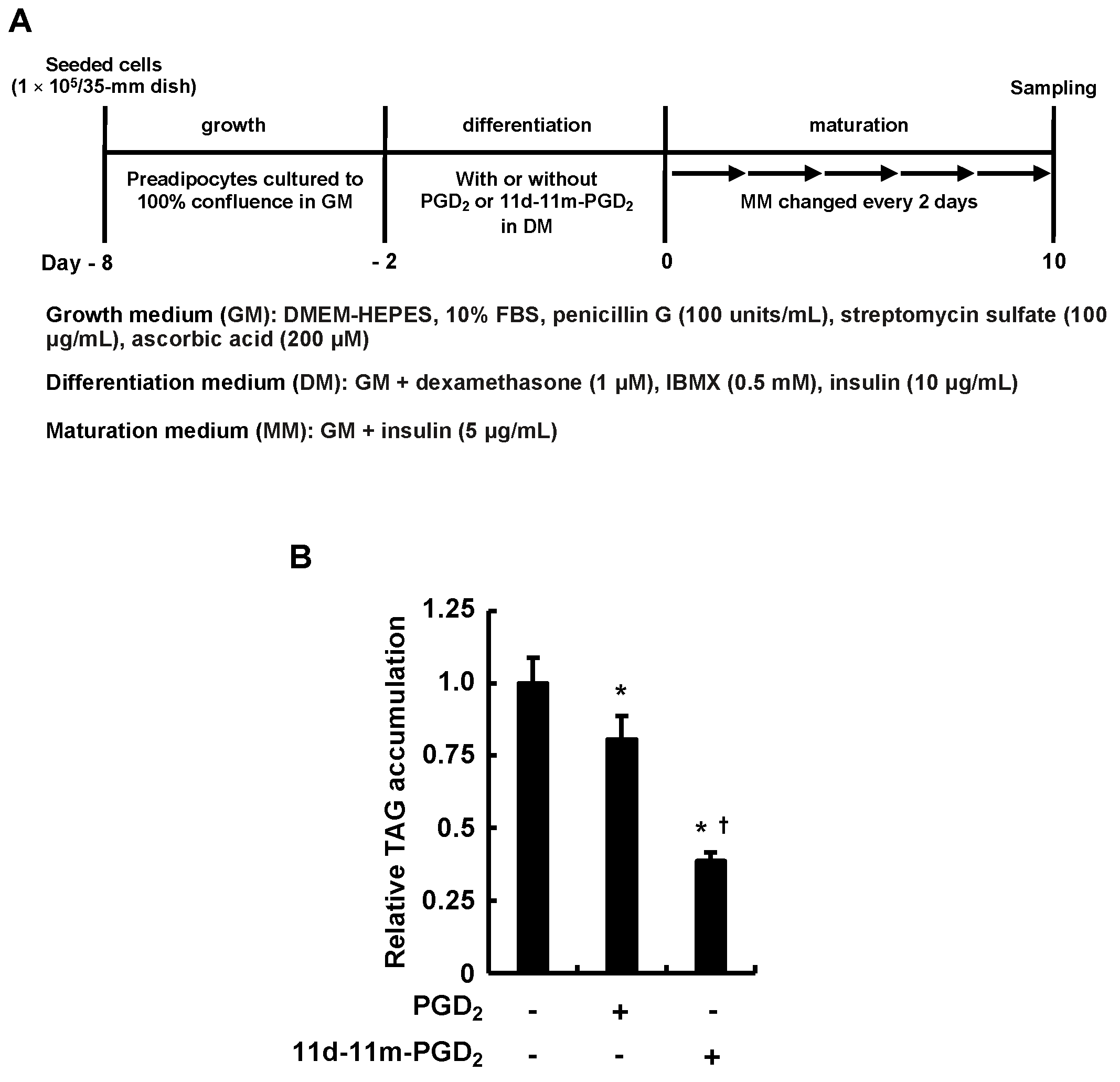
3.1. Incubating 3T3-L1 Cells with PGD2 or 11d-11m-PGD2 during the Differentiation Phase Suppressed MDI-Induced Adipogenesis
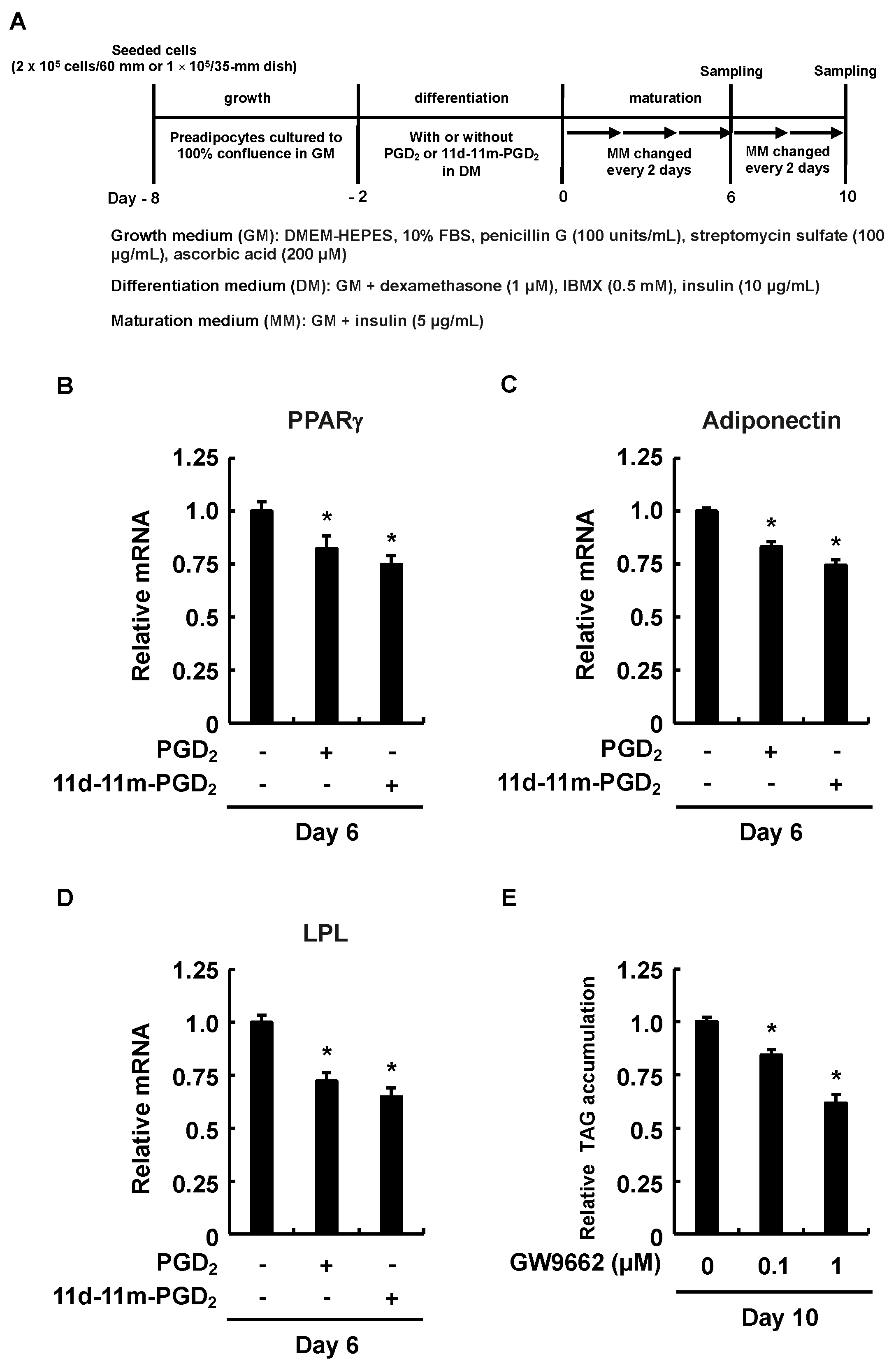
3.2. Downregulated PPARγ Expression Affected the Inhibitory Effects of Addition of PGD2 or 11d-11m-PGD2 during the Differentiation Phase on MDI-Induced Adipogenesis
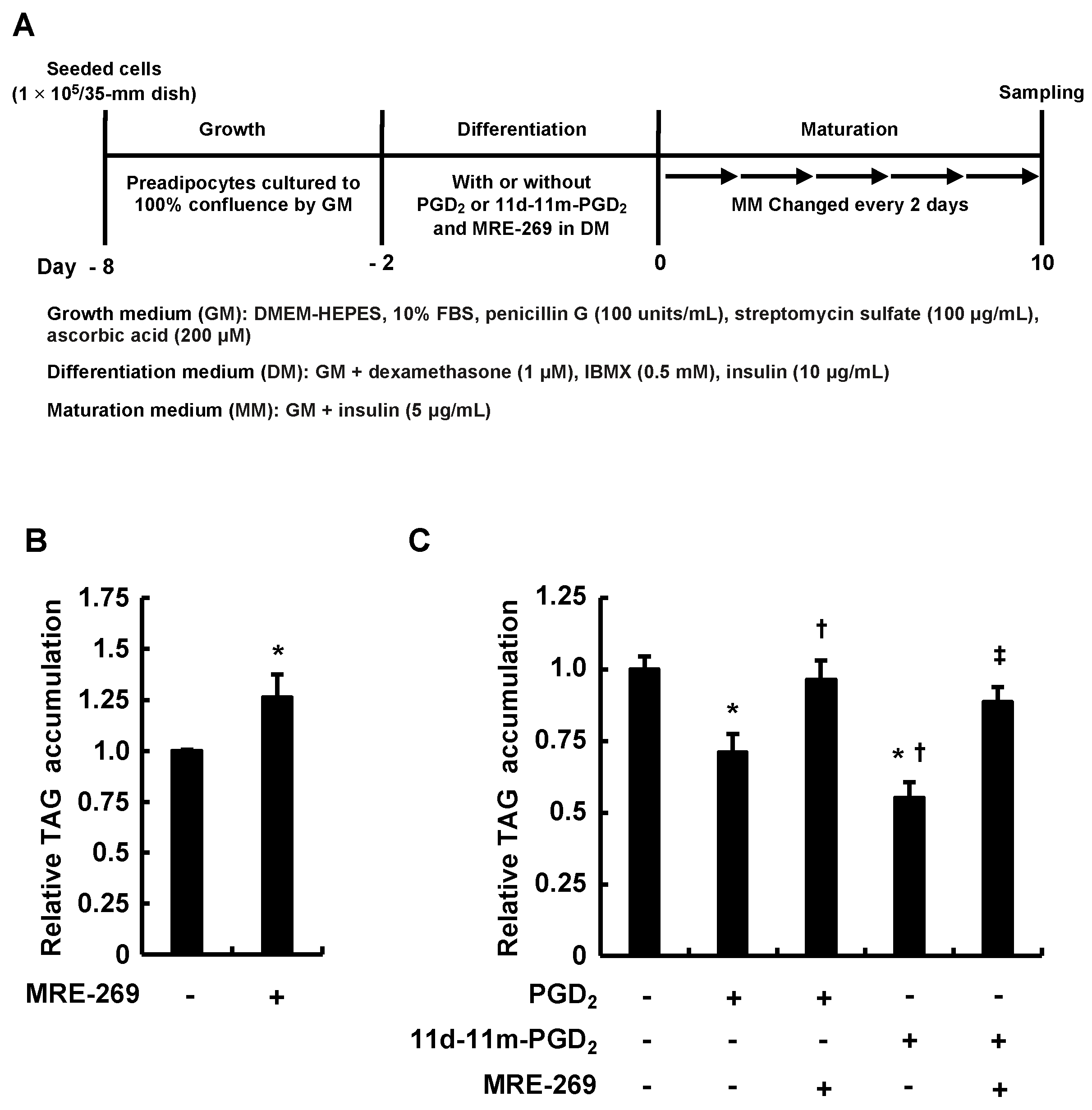
3.3. MRE-269 Attenuated the Inhibitory Effects of Addition of PGD2 or 11d-11m-PGD2 during the Differentiation Phase on MDI-Induced Adipogenesis
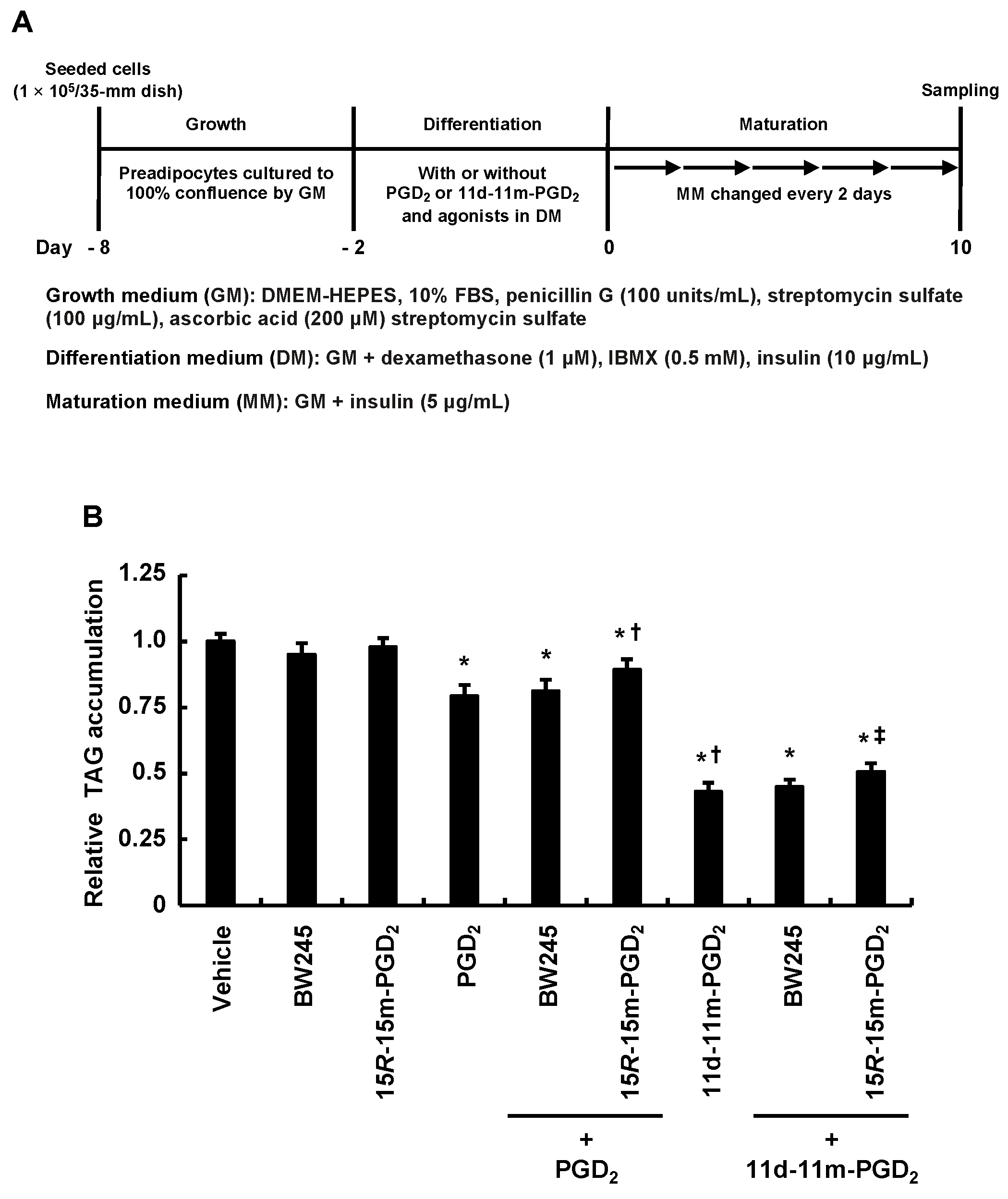
3.4. DP2 Agonist, but Not DP1 Agonist, Partially Alleviated the Inhibitory Effects of Addition of PGD2 or 11d-11m-PGD2 during the Differentiation Phase on MDI-Induced Adipogenesis
3.5. Incubation of 3T3-L1 Cells with PGD2 or 11d-11m-PGD2 during the Differentiation Phase Suppressed Expression of DP1 and DP2 during the Maturation Phase
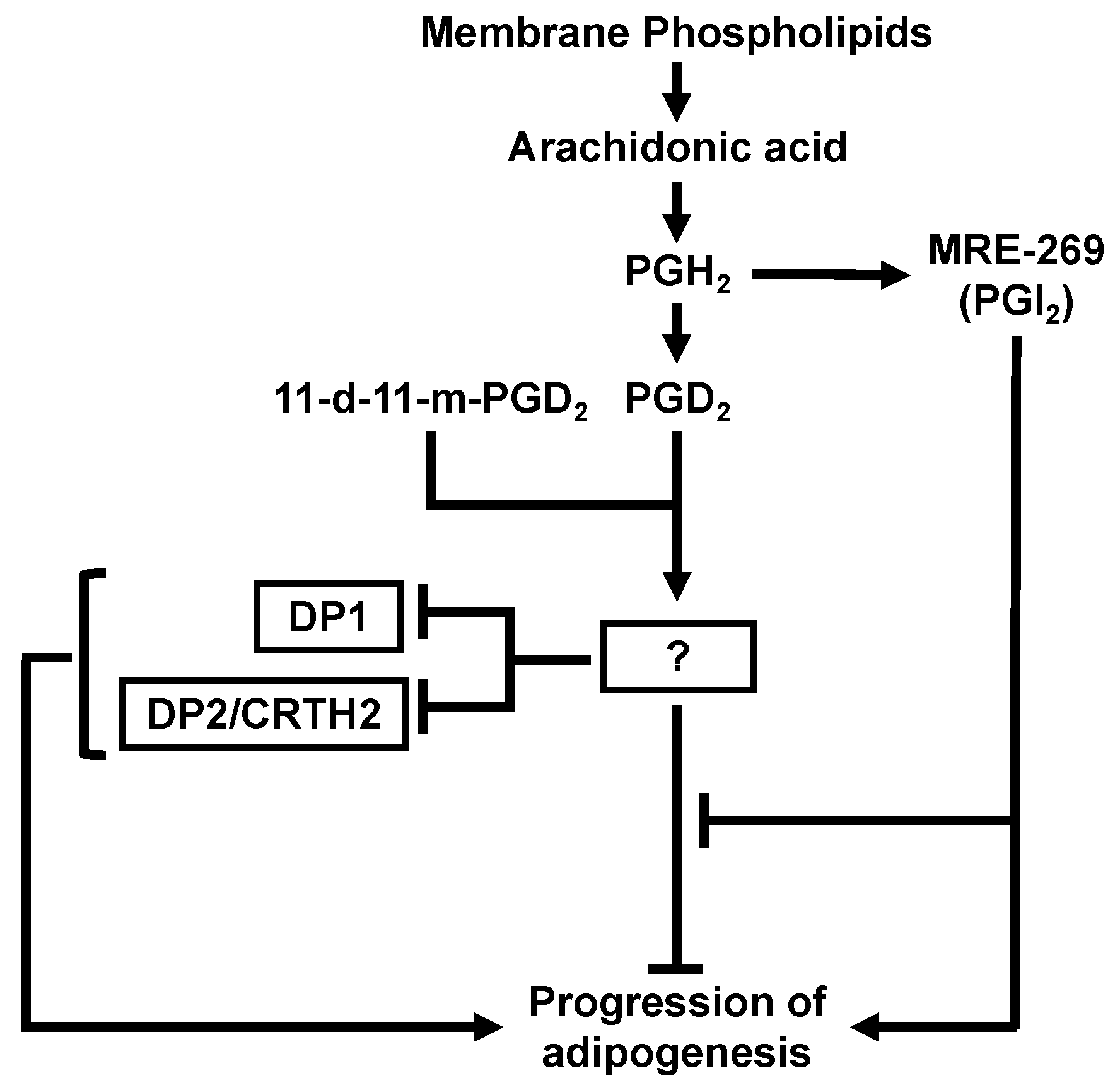
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Seidell, J.C.; Halberstadt, J. The global burden of obesity and the challenges of prevention. Ann. Nutr. Metab. 2015, 66 (Suppl. S2), 7–12. [Google Scholar] [CrossRef]
- Visscher, T.L.; Seidell, J.C. The public health impact of obesity. Annu. Rev. Public. Health 2001, 22, 355–375. [Google Scholar] [CrossRef]
- Green, H.; Kehinde, O. Sublines of mouse 3T3 cells that accumulate lipid. Cell 1974, 1, 113–116. [Google Scholar] [CrossRef]
- Green, H.; Kehinde, O. An established preadipose cell line and its differentiation in culture. II. Factors affecting the adipose conversion. Cell 1975, 5, 19–27. [Google Scholar] [CrossRef]
- Rangwala, S.M.; Lazar, M.A. Transcriptional control of adipogenesis. Annu. Rev. Nutr. 2000, 20, 535–559. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.D.; Spiegelman, B.M. Molecular regulation of adipogenesis. Annu. Rev. Cell Dev. Biol. 2000, 16, 145–171. [Google Scholar] [CrossRef]
- Zhu, Y.; Qi, C.; Korenberg, J.R.; Chen, X.N.; Noya, D.; Rao, M.S.; Reddy, J.K. Structural organization of mouse peroxisome proliferator-activated receptor γ (mPPARγ) gene: Alternative promoter use and different splicing yield two mPPARγ isoforms. Proc. Natl. Acad. Sci. USA 1995, 92, 7921–7925. [Google Scholar] [CrossRef]
- Fajas, L.; Auboeuf, D.; Raspé, E.; Schoonjans, K.; Lefebvre, A.M.; Saladin, R.; Najib, J.; Laville, M.; Fruchart, J.C.; Deeb, S.; et al. The organization, promoter analysis, and expression of the human PPARγ gene. J. Biol. Chem. 1997, 272, 18779–18789. [Google Scholar] [CrossRef]
- Tontonoz, P.; Hu, E.; Spiegelman, B.M. Stimulation of adipogenesis in fibroblasts by PPARγ2, a lipid-activated transcription factor. Cell 1994, 79, 1147–1156. [Google Scholar] [CrossRef]
- Wright, H.M.; Clish, C.B.; Mikami, T.; Hauser, S.; Yanagi, K.; Hiramatsu, R.; Serhan, C.N.; Spiegelman, B.M. A synthetic antagonist for the peroxisome proliferator-activated receptor γ inhibits adipocyte differentiation. J. Biol. Chem. 2000, 275, 1873–1877. [Google Scholar] [CrossRef]
- Camp, H.S.; Chaudhry, A.; Leff, T. A novel potent antagonist of peroxisome proliferator-activated receptor γ blocks adipocyte differentiation but does not revert the phenotype of terminally differentiated adipocytes. Endocrinology 2001, 142, 3207–3213. [Google Scholar] [CrossRef]
- Gurnell, M.; Wentworth, J.M.; Agostini, M.; Adams, M.; Collingwood, T.N.; Provenzano, C.; Browne, P.O.; Rajanayagam, O.; Burris, T.P.; Schwabe, J.W.; et al. A dominant-negative peroxisome proliferator-activated receptor γ (PPARγ) mutant is a constitutive repressor and inhibits PPARγ-mediated adipogenesis. J. Biol. Chem. 2000, 275, 5754–5759. [Google Scholar] [CrossRef]
- Barak, Y.; Nelson, M.C.; Ong, E.S.; Jones, Y.Z.; Ruiz-Lozano, P.; Chien, K.R.; Koder, A.; Evans, R.M. PPARγ is required for placental, cardiac, and adipose tissue development. Mol. Cell 1999, 4, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.D.; Sarraf, P.; Troy, A.E.; Bradwin, G.; Moore, K.; Milstone, D.S.; Spiegelman, B.M.; Mortensen, R.M. PPARγ is required for the differentiation of adipose tissue in vivo and in vitro. Mol. Cell 1999, 4, 611–617. [Google Scholar] [CrossRef]
- Iwaki, M.; Matsuda, M.; Maeda, N.; Funahashi, T.; Matsuzawa, Y.; Makishima, M.; Shimomura, I. Induction of adiponectin, a fat-derived antidiabetic and antiatherogenic factor, by nuclear receptors. Diabetes 2003, 52, 1655–1663. [Google Scholar] [CrossRef] [PubMed]
- Schoonjans, K.; Peinado-Onsurbe, J.; Lefebvre, A.M.; Heyman, R.A.; Briggs, M.; Deeb, S.; Staels, B.; Auwerx, J. PPARα and PPARγ activators direct a distinct tissue-specific transcriptional response via a PPRE in the lipoprotein lipase gene. EMBO J. 1996, 15, 5336–5348. [Google Scholar] [CrossRef]
- Kondo, H.; Shimomura, I.; Matsukawa, Y.; Kumada, M.; Takahashi, M.; Matsuda, M.; Ouchi, N.; Kihara, S.; Kawamoto, T.; Sumitsuji, S.; et al. Association of adiponectin mutation with type 2 diabetes: A candidate gene for the insulin resistance syndrome. Diabetes 2002, 51, 2325–2328. [Google Scholar] [CrossRef] [PubMed]
- Auwerx, J.; Leroy, P.; Schoonjans, K. Lipoprotein lipase: Recent contributions from molecular biology. Crit. Rev. Clin. Lab. Sci. 1992, 29, 243–268. [Google Scholar] [CrossRef]
- Wise, L.S.; Green, H. Studies of lipoprotein lipase during the adipose conversion of 3T3 cells. Cell 1978, 13, 233–242. [Google Scholar] [CrossRef]
- Vane, J.R.; Bakhle, Y.S.; Botting, R.M. Cyclooxygenases 1 and 2. Annu. Rev. Pharmacol. Toxicol. 1998, 38, 97–120. [Google Scholar] [CrossRef]
- Smith, W.L.; DeWitt, D.L.; Garavito, R.M. Cyclooxygenases: Structural, cellular, and molecular biology. Annu. Rev. Biochem. 2000, 69, 145–182. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, Y.; Tsuboi, H.; Okuno, Y.; Tamba, S.; Tsuchiya, S.; Tsujimoto, G.; Ichikawa, A. Microarray evaluation of EP4 recep-tor-mediated prostaglandin E2 suppression of 3T3-L1 adipocyte differentiation. Biochem. Biophys. Res. Commun. 2004, 322, 911–917. [Google Scholar] [CrossRef]
- Tsuboi, H.; Sugimoto, Y.; Kainoh, T.; Ichikawa, A. Prostanoid EP4 receptor is involved in suppression of 3T3-L1 adipocyte differentiation. Biochem. Biophys. Res. Commun. 2004, 322, 1066–1072. [Google Scholar] [CrossRef] [PubMed]
- Inazumi, T.; Shirata, N.; Morimoto, K.; Takano, H.; Segi-Nishida, E.; Sugimoto, Y. Prostaglandin E2-EP4 signaling suppresses adipocyte differentiation in mouse embryonic fibroblasts via an autocrine mechanism. J. Lipid Res. 2011, 52, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.W.; Casimir, D.A.; Ntambi, J.M. The mechanism of inhibition of 3T3-L1 preadipocyte differentiation by prosta-glandin F2α. Endocrinology 1996, 137, 5641–5650. [Google Scholar] [CrossRef]
- Fujimori, K.; Ueno, T.; Nagata, N.; Kashiwagi, K.; Aritake, K.; Amano, F.; Urade, Y. Suppression of adipocyte differentiation by aldo-keto reductase 1B3 acting as prostaglandin F2α synthase. J. Biol. Chem. 2010, 285, 8880–8886. [Google Scholar] [CrossRef]
- Forman, B.M.; Tontonoz, P.; Chen, J.; Brun, R.P.; Spiegelman, B.M.; Evans, R.M. 15-Deoxy-Δ12,14-prostaglandin J2 is a ligand for the adipocyte determination factor PPARγ. Cell 1995, 83, 803–812. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Lenhard, J.M.; Willson, T.M.; Patel, I.; Morris, D.C.; Lehmann, J.M. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor γ and promotes adipocyte differentiation. Cell 1995, 83, 813–819. [Google Scholar] [CrossRef]
- Mazid, M.A.; Chowdhury, A.A.; Nagao, K.; Nishimura, K.; Jisaka, M.; Nagaya, T.; Yokota, K. Endogenous 15-deoxy-Δ12,14-prostaglandin J2 synthesized by adipocytes during maturation phase contributes to upregulation of fat storage. FEBS Lett. 2006, 580, 6885–6890. [Google Scholar] [CrossRef]
- Hossain, M.S.; Chowdhury, A.A.; Rahman, M.S.; Nishimura, K.; Jisaka, M.; Nagaya, T.; Shono, F.; Yokota, K. Development of enzyme-linked immunosorbent assay for Δ12-prostaglandin J2 and its application to the measurement of endogenous product generated by cultured adipocytes during the maturation phase. Prostaglandins Other Lipid Mediat. 2011, 94, 73–80. [Google Scholar] [CrossRef]
- Massiera, F.; Saint-Marc, P.; Seydoux, J.; Murata, T.; Kobayashi, T.; Narumiya, S.; Guesnet, P.; Amri, E.Z.; Negrel, R.; Ailhaud, G. Arachidonic acid and prostacyclin signaling promote adipose tissue development: A human health concern? J. Lipid Res. 2003, 44, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.S. Prostacyclin: A major prostaglandin in the regulation of adipose tissue development. J. Cell Physiol. 2019, 234, 3254–3262. [Google Scholar] [CrossRef] [PubMed]
- Falcetti, E.; Flavell, D.M.; Staels, B.; Tinker, A.; Haworth, S.G.; Clapp, L.H. IP receptor-dependent activation of PPARγ by stable prostacyclin analogues. Biochem. Biophys. Res. Commun. 2007, 360, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Mazid, M.A.; Nishimura, K.; Nagao, K.; Jisaka, M.; Nagaya, T.; Yokota, K. Development of enzyme-linked immunosorbent assay for prostaglandin D2 using the stable isosteric analogue as a hapten mimic and its application. Prostaglandins Other Lipid Mediat. 2007, 83, 219–224. [Google Scholar] [CrossRef]
- Wakai, E.; Aritake, K.; Urade, Y.; Fujimori, K. Prostaglandin D2 enhances lipid accumulation through suppression of lipolysis via DP2 (CRTH2) receptors in adipocytes. Biochem. Biophys. Res. Commun. 2017, 490, 393–399. [Google Scholar] [CrossRef]
- Rahman, M.S.; Syeda, P.K.; Nartey, M.N.N.; Chowdhury, M.M.I.; Shimizu, H.; Nishimura, K.; Jisaka, M.; Shono, F.; Yokota. Comparison of pro-adipogenic effects between prostaglandin (PG)D2 and its stable, isosteric analogue, 11-deoxy-11-methylene-PGD2, during the maturation phase of cultured adipocytes. Prostaglandins Other Lipid Mediat. 2018, 139, 71–79. [Google Scholar] [CrossRef]
- Chowdhury, A.A.; Hossain, M.S.; Rahman, M.S.; Nishimura, K.; Jisaka, M.; Nagaya, T.; Shono, F.; Yokota, K. Sustained expression of lipocalin-type prostaglandin D synthase in the antisense direction positively regulates adipogenesis in cloned cultured preadipocytes. Biochem. Biophys. Res. Commun. 2011, 411, 287–292. [Google Scholar] [CrossRef]
- Hossain, M.S.; Chowdhury, A.A.; Rahman, M.S.; Nishimura, K.; Jisaka, M.; Nagaya, T.; Shono, F.; Yokota, K. Stable expression of lipocalin-type prostaglandin D synthase in cultured preadipocytes impairs adipogenesis program independently of endogenous prostanoids. Exp. Cell Res. 2012, 318, 408–415. [Google Scholar] [CrossRef]
- Lowell, B.B. PPARγ: An essential regulator of adipogenesis and modulator of fat cell function. Cell 1999, 99, 239–242. [Google Scholar] [CrossRef]
- Khan, F.; Syeda, P.K.; Nartey, M.N.N.; Rahman, M.S.; Islam, M.S.; Nishimura, K.; Jisaka, M.; Shono, F.; Yokota, K. Pretreatment of cultured preadipocytes with arachidonic acid during the differentiation phase without a cAMP-elevating agent enhances fat storage after the maturation phase. Prostaglandins Other Lipid Mediat. 2016, 123, 16–27. [Google Scholar] [CrossRef]
- Wu, Z.; Rosen, E.D.; Brun, R.; Hauser, S.; Adelmant, G.; Troy, A.E.; McKeon, C.; Darlington, G.J.; Spiegelman, B.M. Cross-regulation of C/EBPα and PPARγ controls the transcriptional pathway of adipogenesis and insulin sensitivity. Mol. Cell 1999, 3, 151–158. [Google Scholar] [CrossRef]
- Ochsner, S.A.; Abraham, D.; Martin, K.; Ding, W.; McOwiti, A.; Kankanamge, W.; Wang, Z.; Andreano, K.; Hamilton, R.A.; Chen, Y.; et al. The Signaling Pathways Project, an integrated ’omics knowledgebase for mammalian cellular signaling pathways. Sci. Data 2019, 6, 252. [Google Scholar] [CrossRef]
- Reginato, M.J.; Krakow, S.L.; Bailey, S.T.; Lazar, M.A. Prostaglandins promote and block adipogenesis through opposing effects on peroxisome proliferator-activated receptor γ. J. Biol. Chem. 1998, 273, 1855–1858. [Google Scholar] [CrossRef]
- Fujimori, K.; Yano, M.; Ueno, T. Synergistic suppression of early phase of adipogenesis by microsomal PGE synthase-1 (PTGES1)-produced PGE2 and aldo-keto reductase 1B3-produced PGF2α. PLoS ONE 2012, 7, e44698. [Google Scholar] [CrossRef]
- Smith, P.J.; Wise, L.S.; Berkowitz, R.; Wan, C.; Rubin, C.S. Insulin-like growth factor-I is an essential regulator of the differentiation of 3T3-L1 adipocytes. J. Biol. Chem. 1988, 263, 9402–9408. [Google Scholar] [CrossRef]
- White, M.F.; Kahn, C.R. The insulin signaling system. J. Biol. Chem. 1994, 269, 1–4. [Google Scholar] [CrossRef]
- Schmitz-Peiffer, C.; Whitehead, J.P. IRS-1 regulation in health and disease. IUBMB Life 2003, 55, 367–374. [Google Scholar] [CrossRef]
- Kim, W.K.; Jung, H.; Kim, D.H.; Kim, E.Y.; Chung, J.W.; Cho, Y.S.; Park, S.G.; Park, B.C.; Ko, Y.; Bae, K.H.; et al. Regulation of adipogenic differentiation by LAR tyrosine phosphatase in human mesenchymal stem cells and 3T3-L1 preadipocytes. J. Cell Sci. 2009, 122, 4160–4167. [Google Scholar] [CrossRef]
- Welsh, C.L.; Pandey, P.; Ahuja, L.G. Protein Tyrosine Phosphatases: A new paradigm in an old signaling system? Adv. Cancer Res. 2021, 152, 263–303. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GenBank Accession No. | Target Genes | Primers (5→3′) | Length (bp) | Tm (°C) | Product Length (bp) |
|---|---|---|---|---|---|
| NM_011146.4 | PPARγ | F: CTTCGCTGATGCACTGCCTAT | 21 | 60.81 | 216 |
| R: GGGTCAGCTCTTGTGAATGGA | 21 | 60.00 | |||
| NM_009605.5 | Adiponectin | F: AGCCGCTTATGTGTATCGCT | 20 | 59.61 | 154 |
| R: GAGTCCCGGAATGTTGCAGT | 20 | 60.32 | |||
| NM_008509.2 | LPL | F: TTGCAGAGAGAGGACTCGGA | 20 | 59.96 | 125 |
| R: GGAGTTGCACCTGTATGCCT | 20 | 60.04 | |||
| NM_008962.4 | DP1 | F: GAGTCCTATCGCTGTCAGA | 19 | 52.63 | 100 |
| R: CCAGAAGATTGCCCAGAAG | 19 | 52.63 | |||
| XM_006526696.5 | DP2 | F: GCGCTATCCGACTTGTTAG | 19 | 55.94 | 100 |
| R: GTAGCTTGCAGAAGGTAGTG | 20 | 55.88 | |||
| NM_007393.5 | β-Actin | F: GCGGGCGACGATGCT | 15 | 59.84 | 197 |
| R: TGCCAGATCTTCTCCATGTCG | 21 | 59.86 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nartey, M.N.N.; Jisaka, M.; Syeda, P.K.; Nishimura, K.; Shimizu, H.; Yokota, K. Prostaglandin D2 Added during the Differentiation of 3T3-L1 Cells Suppresses Adipogenesis via Dysfunction of D-Prostanoid Receptor P1 and P2. Life 2023, 13, 370. https://doi.org/10.3390/life13020370
Nartey MNN, Jisaka M, Syeda PK, Nishimura K, Shimizu H, Yokota K. Prostaglandin D2 Added during the Differentiation of 3T3-L1 Cells Suppresses Adipogenesis via Dysfunction of D-Prostanoid Receptor P1 and P2. Life. 2023; 13(2):370. https://doi.org/10.3390/life13020370
Chicago/Turabian StyleNartey, Michael N. N., Mitsuo Jisaka, Pinky Karim Syeda, Kohji Nishimura, Hidehisa Shimizu, and Kazushige Yokota. 2023. "Prostaglandin D2 Added during the Differentiation of 3T3-L1 Cells Suppresses Adipogenesis via Dysfunction of D-Prostanoid Receptor P1 and P2" Life 13, no. 2: 370. https://doi.org/10.3390/life13020370
APA StyleNartey, M. N. N., Jisaka, M., Syeda, P. K., Nishimura, K., Shimizu, H., & Yokota, K. (2023). Prostaglandin D2 Added during the Differentiation of 3T3-L1 Cells Suppresses Adipogenesis via Dysfunction of D-Prostanoid Receptor P1 and P2. Life, 13(2), 370. https://doi.org/10.3390/life13020370