

Role of ZNF143 and Its Association with Gene Expression Patterns, Noncoding Mutations, and the Immune System in Human Breast Cancer



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
3. Results
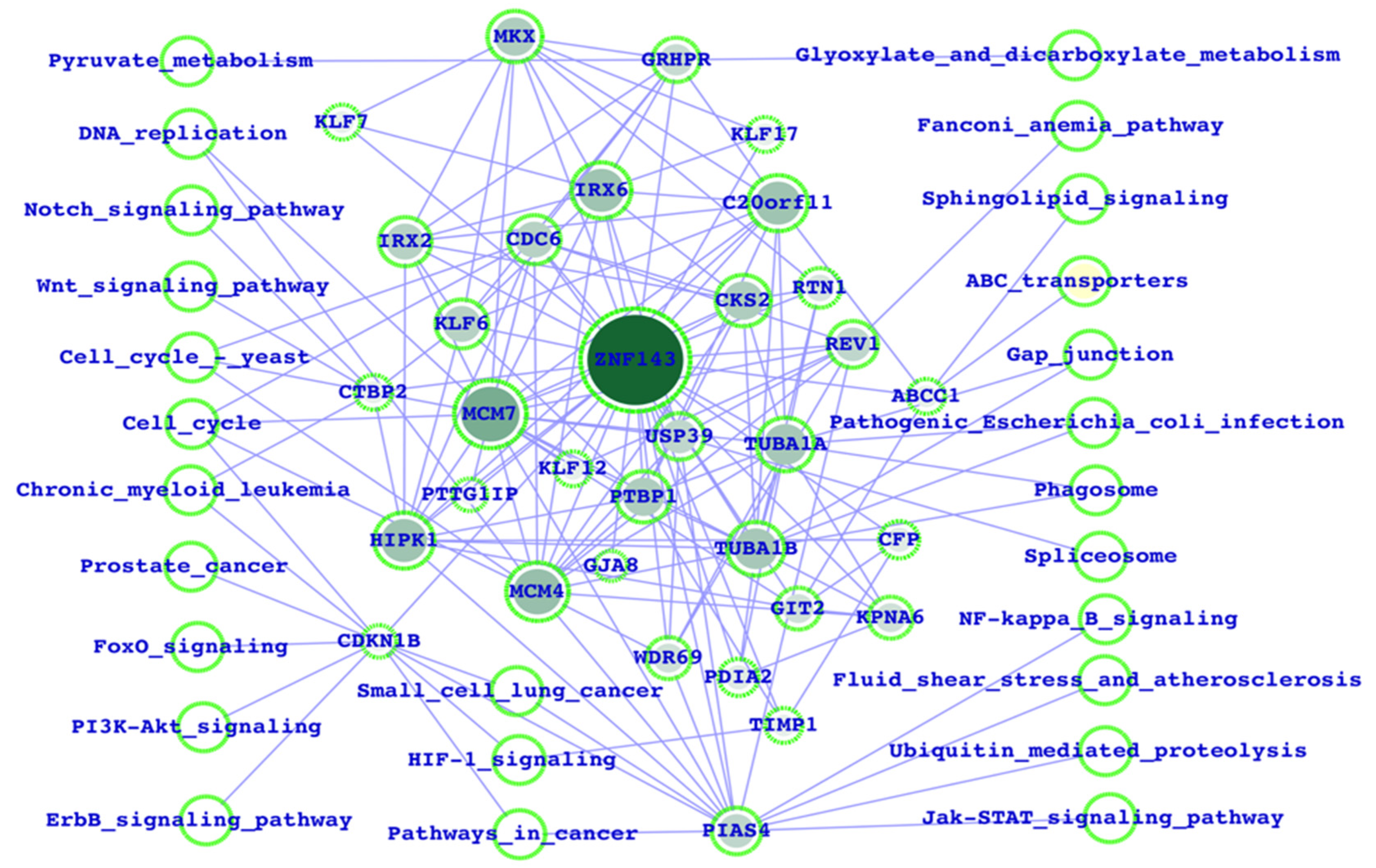
3.1. Potential ZNF143 Interactors and the Biological Functions
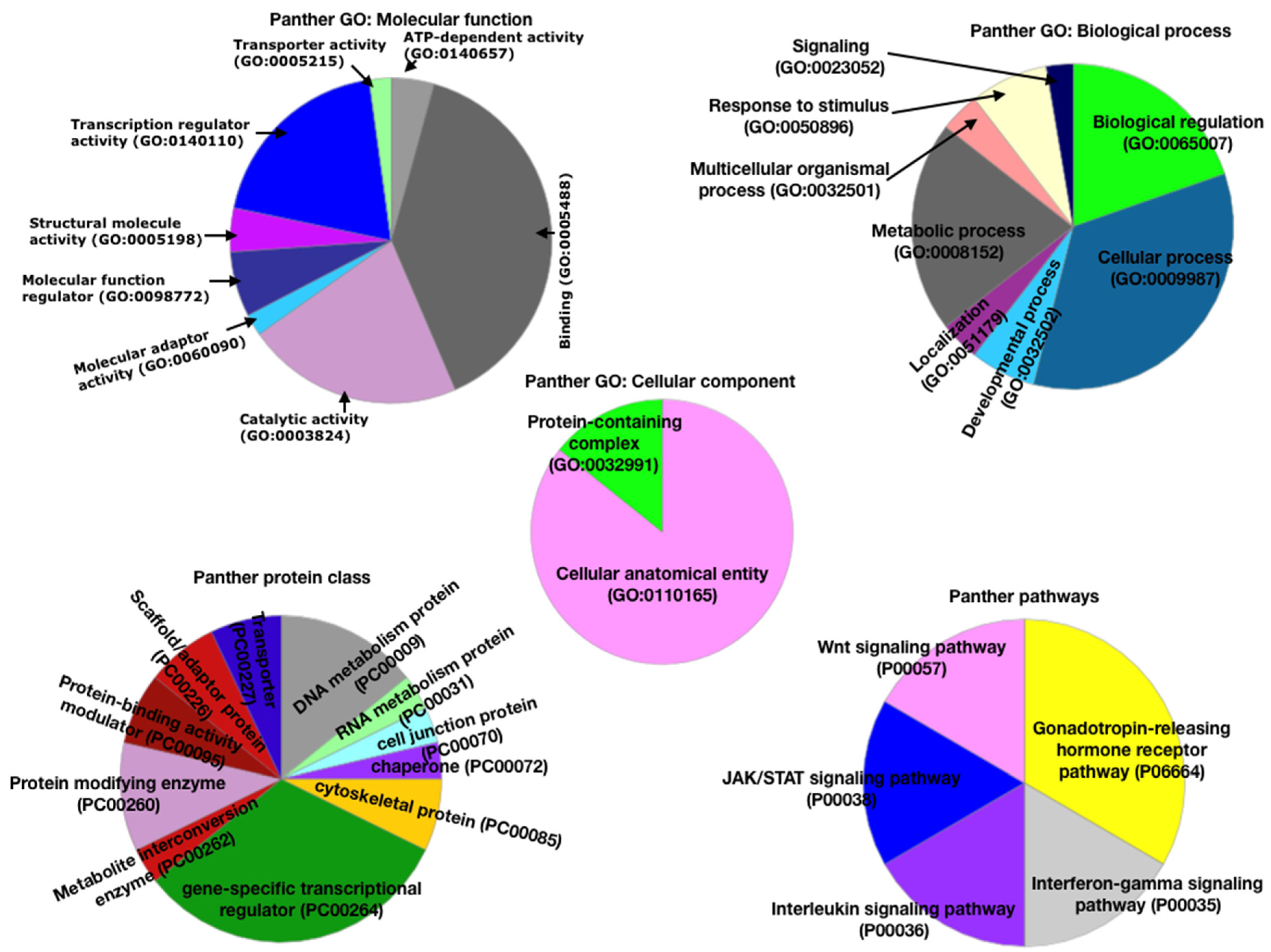
3.2. Functional Analysis Using Panther Database
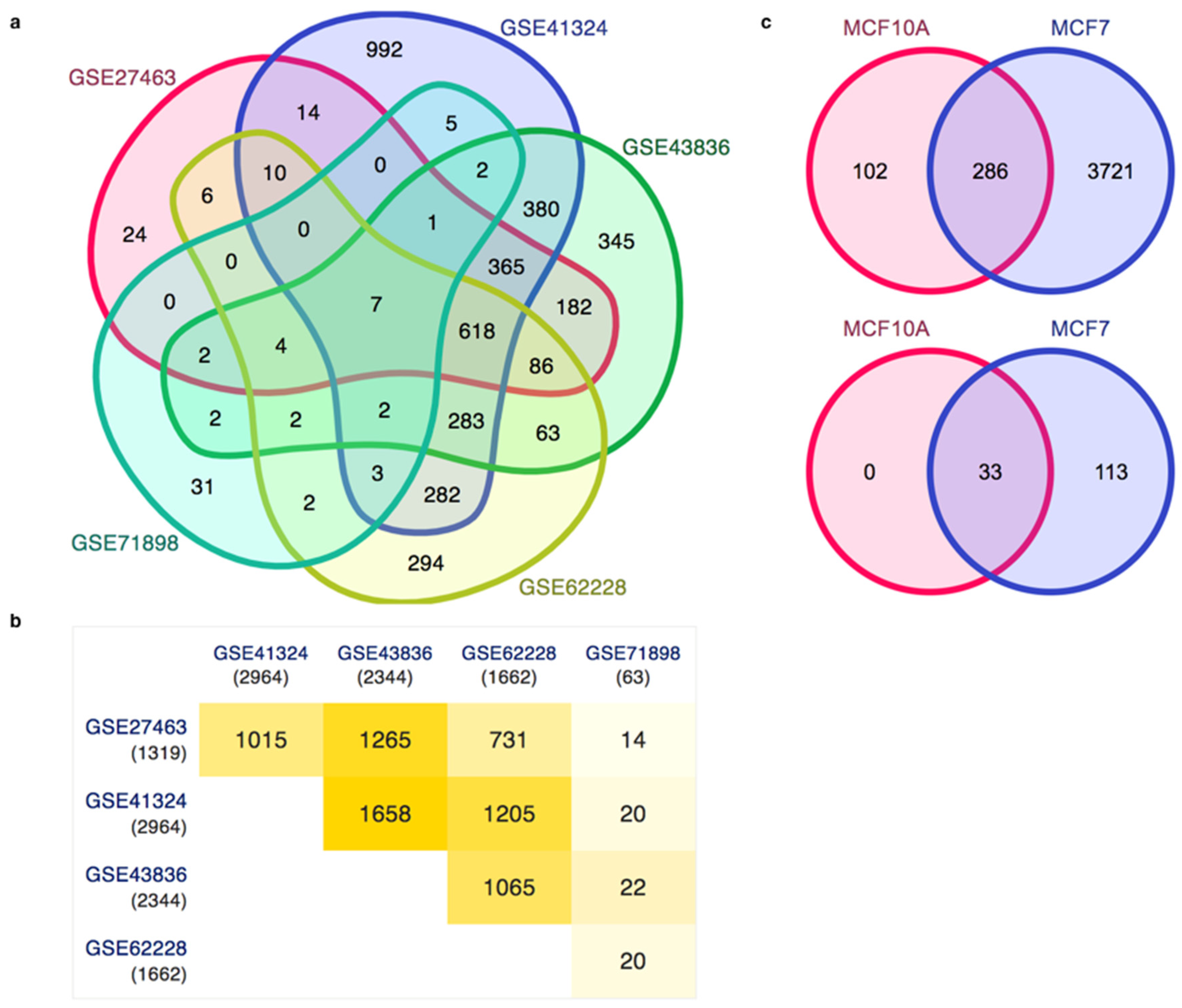
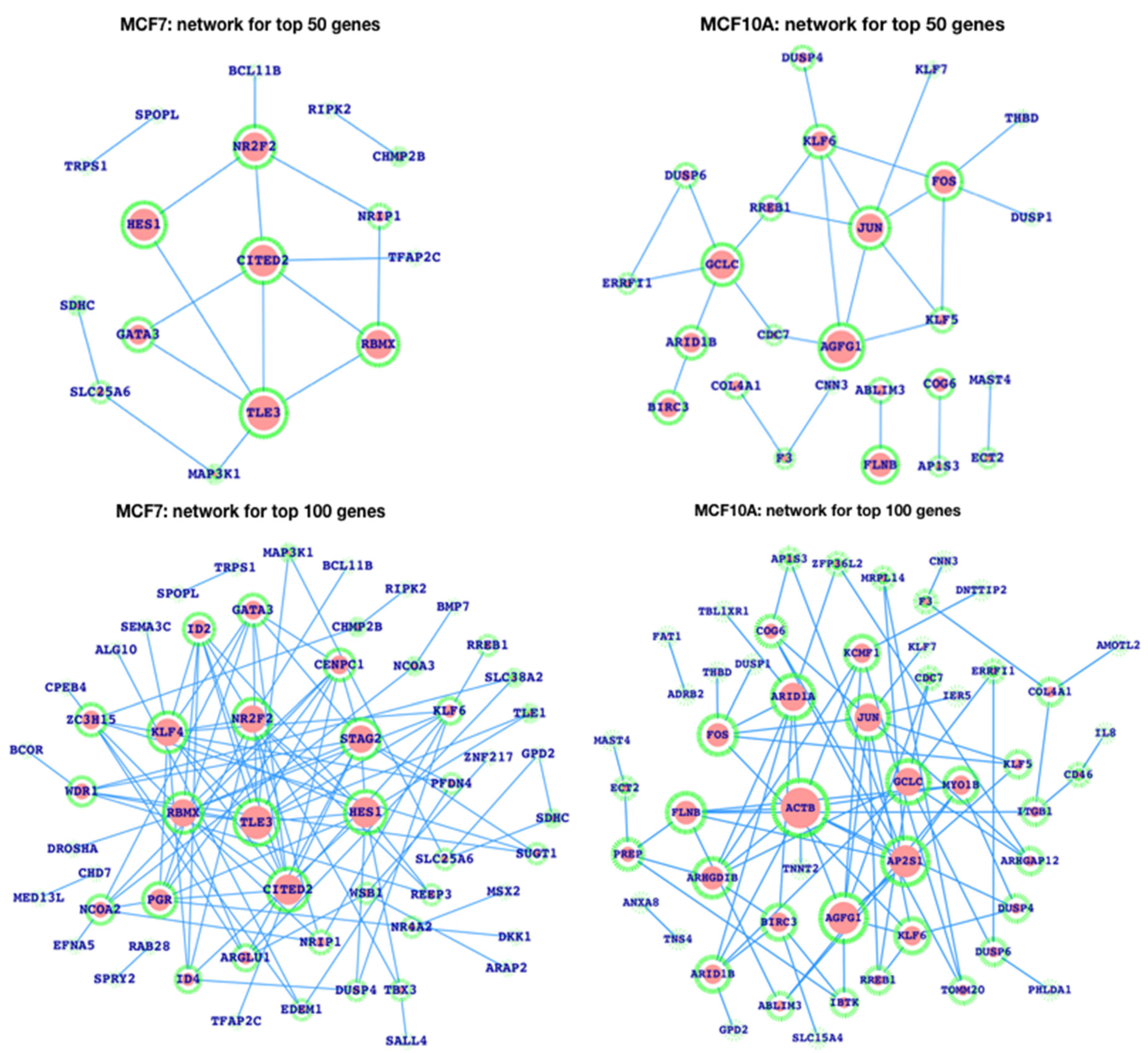
3.3. Gene Expression Patterns in Human Breast Cancer and the Analysis of Enhancers in the Breast Cancer Cell Line
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kamangar, F.; Dores, G.M.; Anderson, W.F. Patterns of Cancer Incidence, Mortality, and Prevalence Across Five Continents: Defining Priorities to Reduce Cancer Disparities in Different Geographic Regions of the World. J. Clin. Oncol. 2006, 24, 2137–2150. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2014, 136, E359–E386. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Laversanne, M.; Brewster, D.H.; Gombe Mbalawa, C.; Kohler, B.; Piñeros, M.; Steliarova-Foucher, E.; Swaminathan, R.; Antoni, S.; et al. Cancer Incidence in Five Continents: Inclusion criteria, highlights from Volume X and the global status of cancer registration. Int. J. Cancer 2015, 137, 2060–2071. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.D.; Holzinger, E.R.; Li, R.; Pendergrass, S.A.; Kim, D. Methods of integrating data to uncover genotype–phenotype interactions. Nat. Rev. Genet. 2015, 16, 85–97. [Google Scholar] [CrossRef]
- Gonda, T.J.; Ramsay, R.G. Directly targeting transcriptional dysregulation in cancer. Nat. Rev. Cancer 2015, 15, 686–694. [Google Scholar] [CrossRef] [PubMed]
- Maley, C.C. Multistage carcinogenesis in Barrett′s esophagus. Cancer Lett. 2007, 245, 22–32. [Google Scholar] [CrossRef]
- Wong, N.; Chan, K.Y.-Y.; Macgregor, P.F.; Lai, P.B.-S.; Squire, J.A.; Beheshti, B.; Albert, M.; Leung, T.W.-T. Transcriptional Profiling Identifies Gene Expression Changes Associated with IFN-α Tolerance in Hepatitis C–Related Hepatocellular Carcinoma Cells. Clin. Cancer Res. 2005, 11, 1319. [Google Scholar] [CrossRef]
- Zlotorynski, E. Transcript elongation: Pause at your peril. Nat. Rev. Cancer 2014, 14, 450–451. [Google Scholar] [CrossRef]
- Magnani, L.; Stoeck, A.; Zhang, X.; Lánczky, A.; Mirabella, A.C.; Wang, T.-L.; Györffy, B.; Lupien, M. Genome-wide reprogramming of the chromatin landscape underlies endocrine therapy resistance in breast cancer. Proc. Natl. Acad. Sci. USA 2013, 110, E1490–E1499. [Google Scholar] [CrossRef]
- McLendon, R.; Friedman, A.; Bigner, D.; Van Meir, E.G.; Brat, D.J.; M Mastrogianakis, G.; Olson, J.J.; Mikkelsen, T.; Lehman, N.; Aldape, K.; et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e11. [Google Scholar] [CrossRef] [PubMed]
- Paek, A.R.; Mun, J.Y.; Hong, K.-M.; Lee, J.; Hong, D.W.; You, H.J. Zinc finger protein 143 expression is closely related to tumor malignancy via regulating cell motility in breast cancer. BMB Rep. 2017, 50, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Izumi, H.; Wakasugi, T.; Shimajiri, S.; Tanimoto, A.; Sasaguri, Y.; Kashiwagi, E.; Yasuniwa, Y.; Akiyama, M.; Han, B.; Wu, Y.; et al. Role of ZNF143 in tumor growth through transcriptional regulation of DNA replication and cell-cycle-associated genes. Cancer Sci. 2010, 101, 2538–2545. [Google Scholar] [CrossRef]
- Hong, M.-G.; Alexeyenko, A.; Lambert, J.-C.; Amouyel, P.; Prince, J.A. Genome-wide pathway analysis implicates intracellular transmembrane protein transport in Alzheimer disease. J. Hum. Genet. 2010, 55, 707–709. [Google Scholar] [CrossRef][Green Version]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2011, 40, D109–D114. [Google Scholar] [CrossRef]
- Mi, H.; Poudel, S.; Muruganujan, A.; Casagrande, J.T.; Thomas, P.D. PANTHER version 10: Expanded protein families and functions, and analysis tools. Nucleic Acids Res. 2016, 44, D336–D342. [Google Scholar] [CrossRef]
- Warsi, M.K.; Kamal, M.A.; Baeshen, M.N.; Izhari, M.A.; Mobashir, A.F.A.M. Comparative Study of Gene Expression Profiling Unravels Functions associated with Pathogenesis of Dengue Infection. Curr. Pharm. Des. 2020, 26, 5293–5299. [Google Scholar] [CrossRef]
- Kamal, M.A.; Warsi, M.K.; Alnajeebi, A.; Ali, H.A.; Helmi, N.; Izhari, M.A.; Mustafa, S.; Mobashir, M. Gene expression profiling and clinical relevance unravel the role hypoxia and immune signaling genes and pathways in breast cancer: Role of hypoxia and immune signaling genes in breast cancer. J. Intern. Med. Sci. 2020, 1, 2–10. [Google Scholar] [CrossRef]
- Bajrai, L.; Sohrab, S.S.; Alandijany, T.A.; Mobashir, M.; Parveen, S.; Kamal, M.A.; Azhar, E.I. Gene expression profiling of early acute febrile stage of dengue infection and its comparative analysis with Streptococcus pneumoniae infection. Front. Cell. Infect. Microbiol. 2021, 1–30. [Google Scholar] [CrossRef]
- Eldakhakhny, B.M.; Al Sadoun, H.; Choudhry, H.; Mobashir, M. In-Silico Study of Immune System Associated Genes in Case of Type-2 Diabetes with Insulin Action and Resistance, and/or Obesity. Front. Endocrinol. 2021, 12, 641888. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.P.; Kamal, M.A.; Warsi, M.K.; Alnajeebi, A.; Ali, H.A.; Helmi, N.; Izhari, M.A.; Mustafa, S.; Firoz, A.; Mobashir, M. In-silico study reveals immunological signaling pathways, their genes, and potential herbal drug targets in ovarian cancer. Inform. Med. Unlocked 2020, 20, 100422. [Google Scholar]
- Helmi, N.; Alammari, D.; Mobashir, M. Role of Potential COVID-19 Immune System Associated Genes and the Potential Pathwayslinkage with Type-2 Diabetes. Comb. Chem. High Throughput Screen. 2021, 24, 2452–2462. [Google Scholar]
- Bajrai, L.H.; Sohrab, S.S.; Mobashir, M.; Kamal, M.A.; Rizvi, M.A.; Azhar, E.I. Understanding the role of potential pathways and its components including hypoxia and immune system in case of oral cancer. Sci. Rep. 2021, 11, 19576. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Mobashir, M.; Al-Keridis, L.A.; Alshammari, N.; Adnan, M.; Abid, M.; Hassan, M.I. A Network-Guided Approach to Discover Phytochemical-Based Anticancer Therapy: Targeting MARK4 for Hepatocellular Carcinoma. Front. Oncol. 2022, 12, 914032. [Google Scholar] [CrossRef]
- Danko, C.G.; Hah, N.; Luo, X.; Martins, A.L.; Core, L.; Lis, J.T.; Siepel, A.; Kraus, W.L. Signaling Pathways Differentially Affect RNA Polymerase II Initiation, Pausing, and Elongation Rate in Cells. Mol. Cell 2013, 50, 212–222. [Google Scholar] [CrossRef]
- Wang, J.; Dai, X.; Berry, L.D.; Cogan, J.D.; Liu, Q.; Shyr, Y. HACER: An atlas of human active enhancers to interpret regulatory variants. Nucleic Acids Res. 2018, 47, D106–D112. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Quackenbush, J. Microarray data normalization and transformation. Nat. Genet. 2002, 32, 496–501. [Google Scholar] [CrossRef]
- Baechler, E.C.; Batliwalla, F.M.; Karypis, G.; Gaffney, P.M.; Ortmann, W.A.; Espe, K.J.; Shark, K.B.; Grande, W.J.; Hughes, K.M.; Kapur, V.; et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. USA 2003, 100, 2610–2615. [Google Scholar] [CrossRef]
- Simon, R. Microarray-based expression profiling and informatics. Curr. Opin. Biotechnol. 2008, 19, 26–29. [Google Scholar] [CrossRef] [PubMed]
- Hornberg, J.J.; Bruggeman, F.J.; Westerhoff, H.V.; Lankelma, J. Cancer: A Systems Biology disease. Biosystems 2006, 83, 81–90. [Google Scholar] [CrossRef]
- Ideker, T.; Thorsson, V.; Siegel, A.F.; Hood, L.E. Testing for differentially-expressed genes by maximum-likelihood analysis of microarray data. J. Comput. Biol. 2000, 7, 805–817. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Ernberg, I.; Kauffman, S. Cancer attractors: A systems view of tumors from a gene network dynamics and developmental perspective. Semin. Cell Dev. Biol. 2009, 20, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Zou, J.; Zaman, N.; Beitel, L.K.; Trifiro, M.; Paliouras, M. Seminars in Cancer Biology. Semin. Cancer Biol. 2013, 23, 279–285. [Google Scholar] [CrossRef]
- Girke, T. Microarray Analysis. 2011, pp. 1–42. Available online: https://docplayer.net/14940736-Microarray-analysis-the-basics-thomas-girke-december-9-2011-microarray-analysis-slide-1-42.html (accessed on 13 November 2022).
- Lapointe, J.; Li, C.; Higgins, J.P.; van de Rijn, M.; Bair, E.; Montgomery, K.; Ferrari, M.; Egevad, L.; Rayford, W.; Bergerheim, U.; et al. Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc. Natl. Acad. Sci. USA 2004, 101, 811–816. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545. [Google Scholar] [CrossRef] [PubMed]
- Okawa, S.; Angarica, V.E.; Lemischka, I.; Moore, K.; del Sol, A. A differential network analysis approach for lineagespeci. Nat. Publ. Group 2015, 1, 15012. [Google Scholar]
- Alexeyenko, A.; Sonnhammer, E.L.L. Global networks of functional coupling in eukaryotes from comprehensive data integration. Genome Res. 2009, 19, 1107–1116. [Google Scholar] [CrossRef]
- El-Kafrawy, S.A.; El-Daly, M.M.; Bajrai, L.H.; Alandijany, T.A.; Faizo, A.A.; Mobashir, M.; Ahmed, S.S.; Ahmed, S.; Alam, S.; Jeet, R.; et al. Genomic profiling and network-level understanding uncover the potential genes and the pathways in hepatocellular carcinoma. Front. Genet. 2022, 13, 880440. [Google Scholar] [CrossRef] [PubMed]
- Khouja, H.I.; Ashankyty, I.M.; Bajrai, L.H.; Kumar, P.K.P.; Kamal, M.A.; Firoz, A.; Mobashir, M. Multi-staged gene expressionprofiling reveals potential genesand the critical pathways in kidneycancer. Sci. Rep. 2022, 12, 7240. [Google Scholar] [CrossRef]
- Anwer, S.T.; Mobashir, M.; Fantoukh, O.I.; Khan, B.; Imtiyaz, K.; Naqvi, I.H.; Rizvi, M.M.A. Synthesis of Silver Nano Particles Using Myricetin and the In-Vitro Assessment of Anti-Colorectal Cancer Activity: In-Silico Integration. IJMS 2022, 23, 11024. [Google Scholar] [CrossRef]
- Mobashir, M.; Turunen, S.P.; Izhari, M.A.; Ashankyty, I.M.; Helleday, T.; Lehti, K. An Approach for Systems-Level Understanding of Prostate Cancer from High-Throughput Data Integration to Pathway Modeling and Simulation. Cells 2022, 11, 4121. [Google Scholar] [CrossRef]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e10. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Aliabadi, H.M.; Uludağ, H.; Han, J. Identification of Potential Drug Targets in Cancer Signaling Pathways using Stochastic Logical Models. Sci. Rep. 2016, 6, 23078. [Google Scholar] [CrossRef]
- Guttridge, K.H.F.D.G.D.; Glass, D.J.; Guttridge, D.C. Cancer Cachexia: Mediators, Signaling, and Metabolic Pathways. Cell Metab. 2012, 16, 153–166. [Google Scholar]
- Brechbiel, J.; Miller-Moslin, K.; Adjei, A.A. Crosstalk Between Hedgehog and Other Signaling Pathways as a Basis for Combination Therapies in Cancer. Cancer Treat. Rev. 2014, 40, 750–759. [Google Scholar] [CrossRef]
- Eroles, P.; Bosch, A.; Pérez-Fidalgo, J.A.; Lluch, A. Cancer Treatment Reviews. Cancer Treat. Rev. 2012, 38, 698–707. [Google Scholar] [CrossRef]
- Almowallad, S.; Alqahtani, L.S.; Mobashir, A.M. NF-kB in Signaling Patterns and Its Temporal Dynamics Encode/Decode Human Diseases. Life 2022, 12, 2012. [Google Scholar] [CrossRef]
- Wang, E. Cancer Letters. Cancer Lett. 2013, 340, 261–269. [Google Scholar] [CrossRef]
- Werner, H.M.J.; Mills, G.B.; Ram, P.T. Cancer Systems Biology: A peek into the future of patient care? Nat. Rev. Clin. Oncol. 2014, 11, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Huwait, E.; Mobashir, M. Potential and Therapeutic Roles of Diosmin in Human Diseases. Biomedicines 2022, 10, 1076. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.; Schwalie, P.C.; Ross-Innes, C.S.; Hurtado, A.; Brown, G.D.; Carroll, J.S.; Flicek, P.; Odom, D.T. A CTCF-independent role for cohesin in tissue-specific transcription. Genome Res. 2010, 20, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.-K.; Iyer, V.R. Genome-wide Studies of CCCTC-binding Factor (CTCF) and Cohesin Provide Insight into Chromatin Structure and Regulation: FIGURE 1. J. Biol. Chem. 2012, 287, 30906–30913. [Google Scholar] [CrossRef] [PubMed]
- Ye, B.Y.; Shen, W.L.; Wang, D.; Li, P.; Zhang, Z.; Shi, M.L.; Zhang, Y.; Zhang, F.X.; Zhao, Z.H. ZNF143 is involved in CTCF-mediated chromatin interactions by cooperation with cohesin and other partners. Mol. Biol. 2016, 50, 431–437. [Google Scholar] [CrossRef]
- Phillips-Cremins, J.E.; Sauria, M.E.G.; Sanyal, A.; Gerasimova, T.I.; Lajoie, B.R.; Bell, J.S.K.; Ong, C.-T.; Hookway, T.A.; Guo, C.; Sun, Y.; et al. Architectural Protein Subclasses Shape 3D Organization of Genomesduring Lineage Commitment. Cell 2013, 153, 1281–1295. [Google Scholar] [CrossRef] [PubMed]
- Zhou, V.W.; Goren, A.; Bernstein, B.E. Charting histone modificationsand the functional organization ofmammalian genomes. Nat. Rev. Genet. 2010, 12, 7–18. [Google Scholar] [CrossRef]
- Ngondo-Mbongo, R.P.; Myslinski, E.; Aster, J.C.; Carbon, P. Modulation of gene expression via overlapping binding sites exerted by ZNF143, Notch1 and THAP11. Nucleic Acids Res. 2013, 41, 4000–4014. [Google Scholar] [CrossRef]
- Gusmao, E.G.; Allhoff, M.; Zenke, M.; Costa, I.G. Analysis of computational footprinting methods for dnase sequencing experiments. Nat. Meth. 2016, 13, 303–309. [Google Scholar] [CrossRef]
- Dixon, J.R.; Gorkin, D.U.; Ren, B. Chromatin Domains: The Unit of Chromosome Organization. Mol. Cell 2016, 62, 668–680. [Google Scholar] [CrossRef]
- Vinckevicius, A.; Parker, J.B.; Chakravarti, D. Genomic Determinants of THAP11/ZNF143/HCFC1 Complex Recruitment to Chromatin. Mol. Cell. Biol. 2015, 35, 4135–4146. [Google Scholar] [CrossRef] [PubMed]
- Florea, A.-M.; Büsselberg, D. Cisplatin as an Anti-Tumor Drug: Cellular Mechanisms of Activity, Drug Resistance and Induced Side Effects. Cancers 2011, 3, 1351–1371. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saddeek, S.; Almassabi, R.; Mobashir, M. Role of ZNF143 and Its Association with Gene Expression Patterns, Noncoding Mutations, and the Immune System in Human Breast Cancer. Life 2023, 13, 27. https://doi.org/10.3390/life13010027
Saddeek S, Almassabi R, Mobashir M. Role of ZNF143 and Its Association with Gene Expression Patterns, Noncoding Mutations, and the Immune System in Human Breast Cancer. Life. 2023; 13(1):27. https://doi.org/10.3390/life13010027
Chicago/Turabian StyleSaddeek, Salma, Rehab Almassabi, and Mohammad Mobashir. 2023. "Role of ZNF143 and Its Association with Gene Expression Patterns, Noncoding Mutations, and the Immune System in Human Breast Cancer" Life 13, no. 1: 27. https://doi.org/10.3390/life13010027
APA StyleSaddeek, S., Almassabi, R., & Mobashir, M. (2023). Role of ZNF143 and Its Association with Gene Expression Patterns, Noncoding Mutations, and the Immune System in Human Breast Cancer. Life, 13(1), 27. https://doi.org/10.3390/life13010027