

Involvement of an Aberrant Vascular System in Neurodevelopmental, Neuropsychiatric, and Neuro-Degenerative Diseases



{kind=link}
{kind=link}
Abstract
1. Introduction
2. Neural Stem Cells and Vascularization
3. Neurodevelopmental and Neuropsychiatric Disorders Associated with Abnormal Vascularization
3.1. 22q11.2 Deletion Syndrome
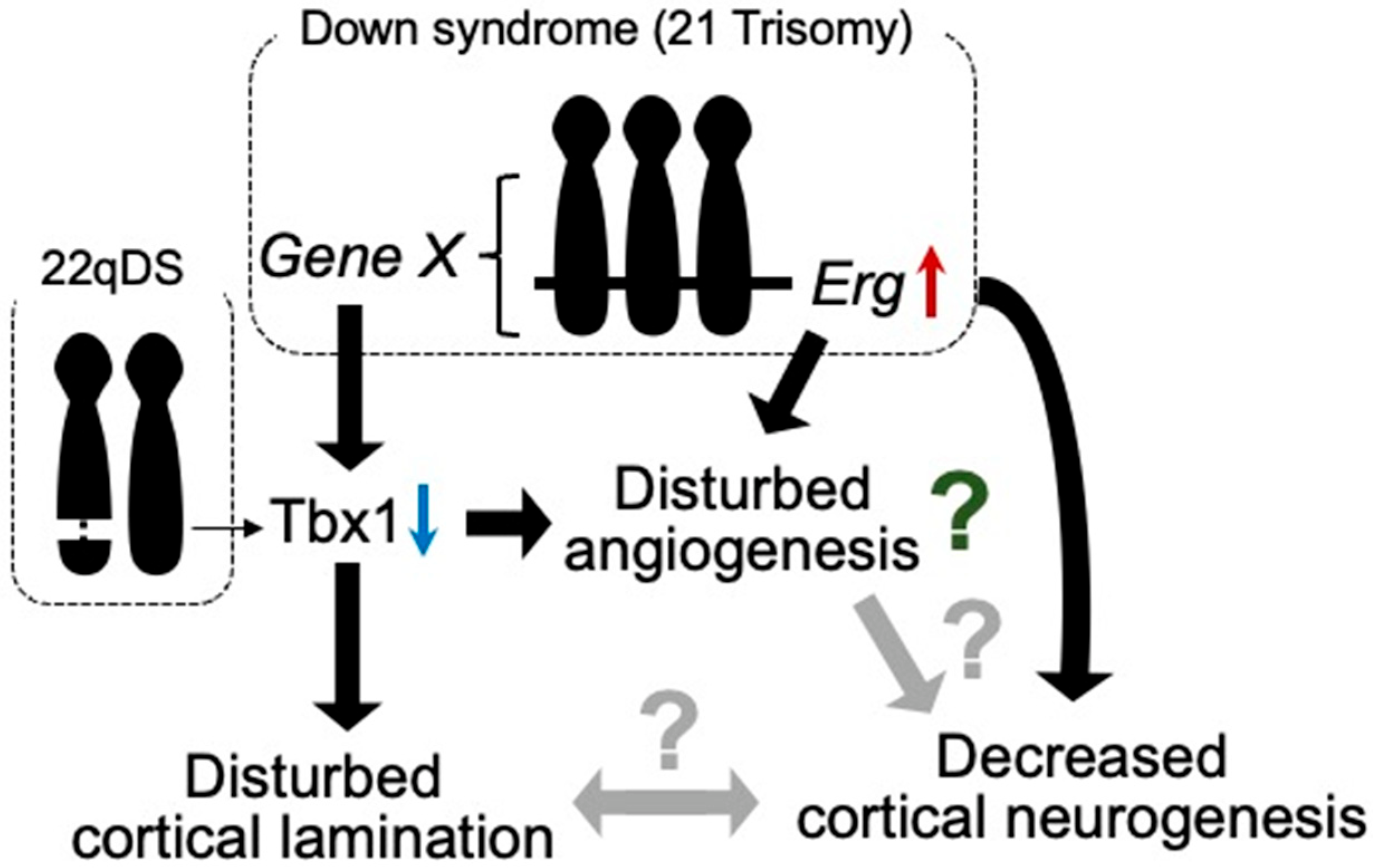
3.2. Down Syndrome (DS)
3.3. Schizophrenia
4. Neurodegenerative Disorders Associated with Abnormal Vascularization
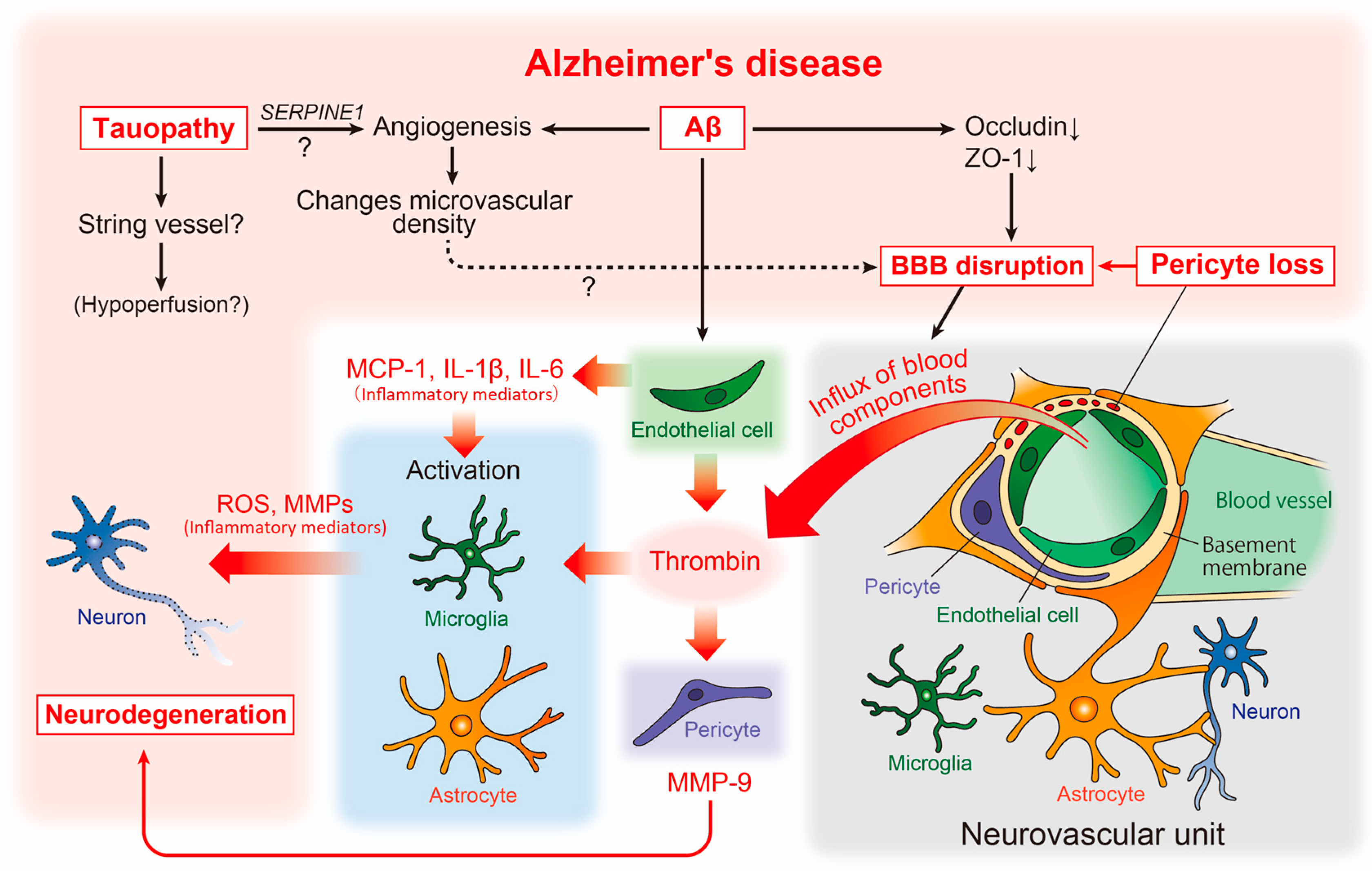
4.1. AD and Cerebrovascular Abnormalities
4.2. Other Neurodegenerative Diseases and Cerebrovascular Abnormalities
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dessaud, E.; McMahon, A.P.; Briscoe, J. Pattern formation in the vertebrate neural tube: A sonic hedgehog morphogen-regulated transcriptional network. Development 2008, 135, 2489–2503. [Google Scholar] [CrossRef]
- Hogan, K.A.; Ambler, C.A.; Chapman, D.L.; Bautch, V.L. The neural tube patterns vessels developmentally using the VEGF signaling pathway. Development 2004, 131, 1503–1513. [Google Scholar] [CrossRef]
- Vasudevan, A.; Long, J.E.; Crandall, J.E.; Rubenstein, J.L.; Bhide, P.G. Compartment-specific transcription factors orchestrate angiogenesis gradients in the embryonic brain. Nat. Neurosci. 2008, 11, 429–439. [Google Scholar] [CrossRef]
- Bjornsson, C.S.; Apostolopoulou, M.; Tian, Y.; Temple, S. It takes a village: Constructing the neurogenic niche. Dev. Cell 2015, 32, 435–446. [Google Scholar] [CrossRef]
- Takashima, S.; Watanabe, C.; Ema, M.; Mizutani, K.I. Interaction of the nervous system and vascular system is required for the proper assembly of the neocortex. Neurochem. Int. 2019, 129, 104481. [Google Scholar] [CrossRef]
- Komabayashi-Suzuki, M.; Yamanishi, E.; Watanabe, C.; Okamura, M.; Tabata, H.; Iwai, R.; Ajioka, I.; Matsushita, J.; Kidoya, H.; Takakura, N.; et al. Spatiotemporally Dependent Vascularization Is Differently Utilized among Neural Progenitor Subtypes during Neocortical Development. Cell Rep. 2019, 29, 1113–1129. [Google Scholar] [CrossRef]
- Toledo, J.B.; Arnold, S.E.; Raible, K.; Brettschneider, J.; Xie, S.X.; Grossman, M.; Monsell, S.E.; Kukull, W.A.; Trojanowski, J.Q. Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer’s Coordinating Centre. Brain 2013, 136, 2697–2706. [Google Scholar] [CrossRef]
- De la Torre, J.C.; Mussivand, T. Can disturbed brain microcirculation cause Alzheimer’s disease? Neurol. Res. 1993, 15, 146–153. [Google Scholar] [CrossRef]
- Viswanathan, A.; Greenberg, S.M. Cerebral amyloid angiopathy in the elderly. Ann. Neurol. 2011, 70, 871–880. [Google Scholar] [CrossRef]
- Di Marco, B.; Crouch, E.E.; Shah, B.; Duman, C.; Paredes, M.F.; Ruiz de Almodovar, C.; Huang, E.J.; Alfonso, J. Reciprocal Interaction between Vascular Filopodia and Neural Stem Cells Shapes Neurogenesis in the Ventral Telencephalon. Cell Rep. 2020, 33, 108256. [Google Scholar] [CrossRef]
- Ihrie, R.A.; Alvarez-Buylla, A. Lake-front property: A unique germinal niche by the lateral ventricles of the adult brain. Neuron 2011, 70, 674–686. [Google Scholar] [CrossRef]
- Tavazoie, M.; Van der Veken, L.; Silva-Vargas, V.; Louissaint, M.; Colonna, L.; Zaidi, B.; Garcia-Verdugo, J.M.; Doetsch, F. A specialized vascular niche for adult neural stem cells. Cell Stem Cell 2008, 3, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Goderie, S.K.; Jin, L.; Karanth, N.; Sun, Y.; Abramova, N.; Vincent, P.; Pumiglia, K.; Temple, S. Endothelial cells stimulate self-renewal and expand neurogenesis of neural stem cells. Science 2004, 304, 1338–1340. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Wang, Y.; Kokovay, E.; Lin, G.; Chuang, S.M.; Goderie, S.K.; Roysam, B.; Temple, S. Adult SVZ stem cells lie in a vascular niche: A quantitative analysis of niche cell-cell interactions. Cell Stem Cell 2008, 3, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Kokovay, E.; Goderie, S.; Wang, Y.; Lotz, S.; Lin, G.; Sun, Y.; Roysam, B.; Shen, Q.; Temple, S. Adult SVZ lineage cells home to and leave the vascular niche via differential responses to SDF1/CXCR4 signaling. Cell Stem Cell 2010, 7, 163–173. [Google Scholar] [CrossRef]
- Johansson, P.A. The choroid plexuses and their impact on developmental neurogenesis. Front. Neurosci. 2014, 8, 340. [Google Scholar] [CrossRef]
- Silva-Vargas, V.; Maldonado-Soto, A.R.; Mizrak, D.; Codega, P.; Doetsch, F. Age-Dependent Niche Signals from the Choroid Plexus Regulate Adult Neural Stem Cells. Cell Stem Cell 2016, 19, 643–652. [Google Scholar] [CrossRef]
- Gur, R.E.; Bassett, A.S.; McDonald-McGinn, D.M.; Bearden, C.E.; Chow, E.; Emanuel, B.S.; Owen, M.; Swillen, A.; Van den Bree, M.; Vermeesch, J.; et al. A neurogenetic model for the study of schizophrenia spectrum disorders: The International 22q11.2 Deletion Syndrome Brain Behavior Consortium. Mol. Psychiatry 2017, 22, 1664–1672. [Google Scholar] [CrossRef]
- Zinkstok, J.R.; Boot, E.; Bassett, A.S.; Hiroi, N.; Butcher, N.J.; Vingerhoets, C.; Vorstman, J.A.S.; van Amelsvoort, T.A.M.J. Neurobiological perspective of 22q11.2 deletion syndrome. Lancet Psychiatry 2019, 6, 951–960. [Google Scholar] [CrossRef]
- Bearden, C.E.; van Erp, T.G.; Dutton, R.A.; Tran, H.; Zimmermann, L.; Sun, D.; Geaga, J.A.; Simon, T.J.; Glahn, D.C.; Cannon, T.D.; et al. Mapping cortical thickness in children with 22q11.2 deletions. Cereb. Cortex 2007, 17, 1889–1898. [Google Scholar] [CrossRef]
- Sun, D.; Ching, C.R.K.; Lin, A.; Forsyth, J.K.; Kushan, L.; Vajdi, A.; Jalbrzikowski, M.; Hansen, L.; Villalon-Reina, J.E.; Qu, X.; et al. Large-scale mapping of cortical alterations in 22q11.2 deletion syndrome: Convergence with idiopathic psychosis and effects of deletion size. Mol. Psychiatry 2020, 25, 1822–1834. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Teot, L.; Murdoch, G.; Monaghan-Nichols, A.P.; McFadden, K. Neuropathology of 22q11 deletion syndrome in an infant. Pediatr. Dev. Pathol. 2014, 17, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, S.; Martucciello, S.; Fulcoli, F.G.; Bilio, M.; Ferrentino, R.; Nusco, E.; Illingworth, E. Tbx1 regulates brain vascularization. Hum. Mol. Genet. 2014, 23, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, S.; Flore, G.; Martucciello, S.; Bilio, M.; Turturo, M.G.; Illingworth, E. VEGFR3 modulates brain microvessel branching in a mouse model of 22q11.2 deletion syndrome. Life Sci. Alliance 2022, 5, e202101308. [Google Scholar] [CrossRef] [PubMed]
- Flore, G.; Cioffi, S.; Bilio, M.; Illingworth, E. Cortical Development Requires Mesodermal Expression of Tbx1, a Gene Haploinsufficient in 22q11.2 Deletion Syndrome. Cereb. Cortex 2017, 27, 2210–2225. [Google Scholar]
- Hiramoto, T.; Kang, G.; Suzuki, G.; Satoh, Y.; Kucherlapati, R.; Watanabe, Y.; Hiroi, N. Tbx1: Identification of a 22q11.2 gene as a risk factor for autism spectrum disorder in a mouse model. Hum. Mol. Genet. 2011, 20, 4775–4785. [Google Scholar] [CrossRef]
- Takahashi, T.; Okabe, S.; Broin, P.Ó.; Nishi, A.; Ye, K.; Beckert, M.V.; Izumi, T.; Machida, A.; Kang, G.; Abe, S.; et al. Structure and function of neonatal social communication in a genetic mouse model of autism. Mol. Psychiatry 2016, 21, 1208–1214. [Google Scholar] [CrossRef]
- Hiramoto, T.; Sumiyoshi, A.; Yamauchi, T.; Tanigaki, K.; Shi, Q.; Kang, G.; Ryoke, R.; Nonaka, H.; Enomoto, S.; Izumi, T.; et al. Tbx1, a gene encoded in 22q11.2 copy number variant, is a link between alterations in fimbria myelination and cognitive speed in mice. Mol. Psychiatry 2022, 27, 929–938. [Google Scholar] [CrossRef]
- Chen, L.; Mupo, A.; Huynh, T.; Cioffi, S.; Woods, M.; Jin, C.; McKeehan, W.; Thompson-Snipes, L.; Baldini, A.; Illingworth, E. Tbx1 regulates Vegfr3 and is required for lymphatic vessel development. J. Cell Biol. 2010, 189, 417–424. [Google Scholar] [CrossRef]
- Hoeffding, L.K.; Trabjerg, B.B.; Olsen, L.; Mazin, W.; Sparsø, T.; Vangkilde, A.; Mortensen, P.B.; Pedersen, C.B.; Werge, T. Risk of Psychiatric Disorders Among Individuals With the 22q11.2 Deletion or Duplication: A Danish Nationwide, Register-Based Study. JAMA Psychiatry 2017, 74, 282–290. [Google Scholar] [CrossRef]
- Greene, C.; Kealy, J.; Humphries, M.M.; Gong, Y.; Hou, J.; Hudson, N.; Cassidy, L.M.; Martiniano, R.; Shashi, V.; Hooper, S.R.; et al. Dose-dependent expression of claudin-5 is a modifying factor in schizophrenia. Mol. Psychiatry 2018, 23, 2156–2166. [Google Scholar] [CrossRef] [PubMed]
- Nitta, T.; Hata, M.; Gotoh, S.; Seo, Y.; Sasaki, H.; Hashimoto, N.; Furuse, M.; Tsukita, S. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J. Cell Biol. 2003, 161, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Korenberg, J.R.; Chen, X.N.; Schipper, R.; Sun, Z.; Gonsky, R.; Gerwehr, S.; Carpenter, N.; Daumer, C.; Dignan, P.; Disteche, C. Down syndrome phenotypes: The consequences of chromosomal imbalance. Proc. Natl. Acad. Sci. USA 1994, 91, 4997–5001. [Google Scholar] [CrossRef] [PubMed]
- Ross, M.H.; Galaburda, A.M.; Kemper, T.L. Down’s syndrome: Is there a decreased population of neurons? Neurology 1984, 34, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Sidor, B.; Wisniewski, K.E.; Shepard, T.H.; Sersen, E.A. Brain growth in Down syndrome subjects 15 to 22 weeks of gestational age and birth to 60 months. Clin. Neuropathol. 1990, 4, 181–190. [Google Scholar]
- Guidi, S.; Bonasoni, P.; Ceccarelli, C.; Santini, D.; Gualtieri, F.; Ciani, E.; Bartesaghi, R. Neurogenesis impairment and increased cell death reduce total neuron number in the hippocampal region of fetuses with Down syndrome. Brain Pathol. 2008, 18, 180–197. [Google Scholar] [CrossRef]
- Chakrabarti, L.; Galdzicki, Z.; Haydar, H.F. Defects in embryonic neurogenesis and initial synapse formation in the forebrain of the Ts65Dn mouse model of Down syndrome. J. Neurosci. 2007, 27, 11483–11495. [Google Scholar] [CrossRef]
- Ishihara, K.; Amano, K.; Takaki, E.; Shimohata, A.; Sago, H.; Epstein, C.J.; Yamakawa, K. Enlarged brain ventricles and impaired neurogenesis in the Ts1Cje and Ts2Cje mouse models of Down syndrome. Cereb. Cortex 2010, 20, 1131–1143. [Google Scholar] [CrossRef]
- Ishihara, K. Genes Associated with Disturbed Cerebral Neurogenesis in the Embryonic Brain of Mouse Models of Down Syndrome. Genes 2021, 12, 1598. [Google Scholar] [CrossRef]
- Baek, K.H.; Zaslavsky, A.; Lynch, R.C.; Britt, C.; Okada, Y.; Siarey, R.J.; Lensch, M.W.; Park, I.H.; Yoon, S.S.; Minami, T.; et al. Down’s syndrome suppression of tumour growth and the role of the calcineurin inhibitor DSCR1. Nature 2009, 459, 1126–1130. [Google Scholar] [CrossRef]
- Reynolds, L.E.; Watson, A.R.; Baker, M.; Jones, T.A.; D’Amico, G.; Robinson, S.D.; Joffre, C.; Garrido-Urbani, S.; Rodriguez-Manzaneque, J.C.; Martino-Echarri, E.; et al. Tumour angiogenesis is reduced in the Tc1 mouse model of Down’s syndrome. Nature 2010, 465, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, K.; Shimizu, R.; Takata, K.; Kawashita, E.; Amano, K.; Shimohata, A.; Low, D.; Nabe, T.; Sago, H.; Alexander, W.S.; et al. Perturbation of the immune cells and prenatal neurogenesis by the triplication of the Erg gene in mouse models of Down syndrome. Brain Pathol. 2020, 30, 75–91. [Google Scholar] [CrossRef] [PubMed]
- Vlaeminck-Guillem, V.; Carrere, S.; Dewitte, F.; Stehelin, D.; Desbiens, X.; Duterque-Coquillaud, M. The Ets family member Erg gene is expressed in mesodermal tissues and neural crests at fundamental steps during mouse embryogenesis. Mech. Dev. 2000, 91, 331–335. [Google Scholar] [CrossRef]
- Shah, A.V.; Birdsey, G.M.; Randi, A.M. Regulation of endothelial homeostasis, vascular development and angiogenesis by the transcription factor ERG. Vascul. Pharmacol. 2016, 86, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, R.; Ishihara, K.; Kawashita, E.; Sago, H.; Yamakawa, K.; Mizutani, K.I.; Akiba, S. Decrease in the T-box1 gene expression in embryonic brain and adult hippocampus of down syndrome mouse models. Biochem. Biophys. Res. Commun. 2021, 535, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.E.; Lawrence, J.B. Chromosome silencing in vitro reveals trisomy 21 causes cell-autonomous deficits in angiogenesis and early dysregulation in Notch signaling. Cell Rep. 2022, 40, 111174. [Google Scholar] [CrossRef]
- Arion, D.; Horváth, S.; Lewis, D.A.; Mirnics, K. Infragranular gene expression disturbances in the prefrontal cortex in schizophrenia: Signature of altered neural development? Neurobiol. Dis. 2010, 37, 738–746. [Google Scholar] [CrossRef]
- Lewis, D.A.; Pierri, J.N.; Volk, D.W.; Melchitzky, D.S.; Woo, T.U. Altered GABA neurotransmission and prefrontal cortical dysfunction in schizophrenia. Biol. Psychiatry 1999, 46, 616–626. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, L.; Gage, F.H. Microglia, complement and schizophrenia. Nat. Neurosci. 2019, 22, 333–334. [Google Scholar] [CrossRef]
- Hakak, Y.; Walker, J.R.; Li, C.; Wong, W.H.; Davis, K.L.; Buxbaum, J.D.; Haroutunian, V.; Fienberg, A.A. Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc. Natl. Acad. Sci. USA 2001, 98, 4746–4751. [Google Scholar] [CrossRef]
- Meyer-Lindenberg, A. From maps to mechanisms through neuroimaging of schizophrenia. Nature 2010, 468, 194–202. [Google Scholar] [CrossRef]
- Kaar, S.J.; Angelescu, I.; Marques, T.R.; Howes, O.D. Pre-frontal parvalbumin interneurons in schizophrenia: A meta-analysis of post-mortem studies. J. Neural. Transm. 2019, 126, 1637–1651. [Google Scholar] [CrossRef] [PubMed]
- Berdenis van Berlekom, A.; Muflihah, C.H.; Snijders, G.J.L.J.; MacGillavry, H.D.; Middeldorp, J.; Hol, E.M.; Kahn, R.S.; de Witte, L.D. Synapse Pathology in Schizophrenia: A Meta-analysis of Postsynaptic Elements in Postmortem Brain Studies. Schizophr. Bull. 2020, 46, 374–386. [Google Scholar] [CrossRef] [PubMed]
- Wagstyl, K.; Ronan, L.; Whitaker, K.J.; Goodyer, I.M.; Roberts, N.; Crow, T.J.; Fletcher, P.C. Multiple markers of cortical morphology reveal evidence of supragranular thinning in schizophrenia. Transl. Psychiatry 2016, 6, e780. [Google Scholar] [CrossRef] [PubMed]
- Kempton, M.J.; Stahl, D.; Williams, S.C.; DeLisi, L.E. Progressive lateral ventricular enlargement in schizophrenia: A meta-analysis of longitudinal MRI studies. Schizophr. Res. 2010, 120, 54–62. [Google Scholar] [CrossRef]
- Srikanth, P.; Han, K.; Callahan, D.G.; Makovkina, E.; Muratore, C.R.; Lalli, M.A.; Zhou, H.; Boyd, J.D.; Kosik, K.S.; Selkoe, D.J.; et al. Genomic DISC1 Disruption in hiPSCs Alters Wnt Signaling and Neural Cell Fate. Cell Rep. 2015, 12, 1414–1429. [Google Scholar] [CrossRef]
- Brennand, K.; Simone, A.; Jou, J.; Gelboin-Burkhart, C.; Tran, N.; Sangar, S.; Li, Y.; Mu, Y.; Chen, G.; Yu, D.; et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature 2011, 473, 221–225. [Google Scholar] [CrossRef]
- Srikanth, P.; Lagomarsino, V.N.; Muratore, C.R.; Ryu, S.C.; He, A.; Taylor, W.M.; Zhou, C.; Arellano, M.; Young-Pearse, T.L. Shared effects of DISC1 disruption and elevated WNT signaling in human cerebral organoids. Transl. Psychiatry 2018, 8, 77. [Google Scholar] [CrossRef]
- Topol, A.; Zhu, S.; Tran, N.; Simone, A.; Fang, G.; Brennand, K.J. Altered WNT Signaling in Human Induced Pluripotent Stem Cell Neural Progenitor Cells Derived from Four Schizophrenia Patients. Biol. Psychiatry 2015, 78, e29–e34. [Google Scholar] [CrossRef]
- Hirabayashi, Y.; Itoh, Y.; Tabata, H.; Nakajima, K.; Akiyama, T.; Masuyama, N.; Gotoh, Y. The Wnt/beta-catenin pathway directs neuronal differentiation of cortical neural precursor cells. Development 2004, 131, 2791–2801. [Google Scholar] [CrossRef]
- Mutch, C.A.; Funatsu, N.; Monuki, E.S.; Chenn, A. Beta-catenin signaling levels in progenitors influence the laminar cell fates of projection neurons. J. Neurosci. 2009, 29, 13710–13719. [Google Scholar] [CrossRef] [PubMed]
- Sawada, T.; Chater, T.E.; Sasagawa, Y.; Yoshimura, M.; Fujimori-Tonou, N.; Tanaka, K.; Benjamin, K.J.M.; Paquola, A.C.M.; Erwin, J.A.; Goda, Y.; et al. Developmental excitation-inhibition imbalance underlying psychoses revealed by single-cell analyses of discordant twins-derived cerebral organoids. Mol. Psychiatry 2020, 25, 2695–2711. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.W.; Wayland, M.; Lan, M.; Ryan, M.; Giger, T.; Lockstone, H.; Wuethrich, I.; Mimmack, M.; Wang, L.; Kotter, M.; et al. The cerebral microvasculature in schizophrenia: A laser capture microdissection study. PLoS ONE 2008, 3, e3964. [Google Scholar] [CrossRef]
- Shalev, H.; Serlin, Y.; Friedman, A. Breaching the blood–brain barrier as a gate to psychiatric disorder. Cardiovasc. Psychiatry Neurol. 2009, 2009, 278531. [Google Scholar]
- Bechter, K.; Reiber, H.; Herzog, S.; Fuchs, D.; Tumani, H.; Maxeiner, H.G. Cerebrospinal fluid analysis in affective and schizophrenic spectrum disorders: Identification of subgroups with immune responses and blood-CSF barrier dysfunction. J. Psychiatr. Res. 2010, 44, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Wang, T.; Zhang, T.; Yi, S.; Zhao, S.; Li, N.; Yang, Y.; Zhang, F.; Xu, L.; Shan, B.; et al. Increased blood-brain barrier permeability of the thalamus and the correlation with symptom severity and brain volume alterations in schizophrenia patients. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2022, 7, 1025–1034. [Google Scholar]
- Puvogel, S.; Alsema, A.; Kracht, L.; Webster, M.J.; Weickert, C.S.; Sommer, I.E.C.; Eggen, B.J.L. Single-nucleus RNA sequencing of midbrain blood-brain barrier cells in schizophrenia reveals subtle transcriptional changes with overall preservation of cellular proportions and phenotypes. Mol. Psychiatry 2022, 27, 4731–4740. [Google Scholar] [CrossRef] [PubMed]
- Casas, B.S.; Vitória, G.; Prieto, C.P.; Casas, M.; Chacón, C.; Uhrig, M.; Ezquer, F.; Ezquer, M.; Rehen, S.K.; Palma, V. Schizophrenia-derived hiPSC brain microvascular endothelial-like cells show impairments in angiogenesis and blood-brain barrier function. Mol. Psychiatry 2022, 27, 4731–4740. [Google Scholar] [CrossRef]
- Lichtermann, D.; Ekelund, J.; Pukkala, E.; Tanskanen, A.; Lönnqvist, J. Incidence of cancer among persons with schizophrenia and their relatives. Arch. Gen. Psychiatry 2001, 58, 573–578. [Google Scholar] [CrossRef]
- Barak, Y.; Achiron, A.; Mandel, M.; Mirecki, I.; Aizenberg, D. Reduced cancer incidence among patients with schizophrenia. Cancer 2005, 104, 2817–2821. [Google Scholar] [CrossRef]
- Hippisley-Cox, J.; Vinogradova, Y.; Coupland, C.; Parker, C. Risk of malignancy in patients with schizophrenia or bipolar disorder: Nested case-control study. Arch. Gen. Psychiatry 2007, 64, 1368–1376. [Google Scholar] [CrossRef] [PubMed]
- Chou, F.H.; Tsai, K.Y.; Su, C.Y.; Lee, C.C. The incidence and relative risk factors for developing cancer among patients with schizophrenia: A nine-year follow-up study. Schizophr. Res. 2011, 129, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Sundquist, K.; Ning, Y.; Kendler, K.S.; Sundquist, J.; Chen, X. Incidence of cancer in patients with schizophrenia and their first-degree relatives: A population-based study in Sweden. Schizophr. Bull. 2013, 39, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.M.; Chen, Y.J.; Kuo, D.J.; Jaiteh, L.E.; Wu, Y.C.; Lo, T.S.; Li, Y.H. Cancer incidence in patients with schizophrenia or bipolar disorder: A nationwide population-based study in Taiwan, 1997-2009. Schizophr. Bull. 2013, 39, 407–416. [Google Scholar] [CrossRef]
- Corrada, M.M.; Brookmeyer, R.; Paganini-Hill, A.; Berlau, D.; Kawas, C.H. Dementia incidence continues to increase with age in the oldest old: The 90+ study. Ann. Neurol. 2010, 67, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Kugler, E.C.; Greenwood, J.; MacDonald, R.B. The “Neuro-Glial-Vascular” Unit: The Role of Glia in Neurovascular Unit Formation and Dysfunction. Front. Cell Dev. Biol. 2021, 27, 732820. [Google Scholar] [CrossRef]
- Buée, L.; Hof, P.R.; Bouras, C.; Delacourte, A.; Perl, D.P.; Morrison, J.H.; Fillit, H.M. Pathological alterations of the cerebral microvasculature in Alzheimer’s disease and related dementing disorders. Acta Neuropathol. 1994, 87, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Biron, K.E.; Dickstein, D.L.; Gopaul, R.; Jefferies, W.A. Amyloid triggers extensive cerebral angiogenesis causing blood brain barrier permeability and hypervascularity in Alzheimer’s disease. PLoS ONE 2011, 6, e23789. [Google Scholar] [CrossRef]
- Bennett, R.E.; Robbins, A.B.; Hu, M.; Cao, X.; Betensky, R.A.; Clark, T.; Das, S.; Hyman, B.T. Tau induces blood vessel abnormalities and angiogenesis-related gene expression in P301L transgenic mice and human Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, E1289–E1298. [Google Scholar] [CrossRef]
- Brown, W.R. A review of string vessels or collapsed, empty basement membrane tubes. J. Alzheimers Dis. 2010, 21, 725–739. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.P.; Bae, D.G.; Kang, H.J.; Gwag, B.J.; Gho, Y.S.; Chae, C.B. Co-accumulation of vascular endothelial growth factor with beta-amyloid in the brain of patients with Alzheimer’s disease. Neurobiol. Aging 2004, 25, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Religa, P.; Cao, R.; Religa, D.; Xue, Y.; Bogdanovic, N.; Westaway, D.; Marti, H.H.; Winblad, B.; Cao, Y. VEGF significantly restores impaired memory behavior in Alzheimer’s mice by improvement of vascular survival. Sci. Rep. 2013, 3, 2053. [Google Scholar] [CrossRef]
- Alitalo, K.; Tammela, T.; Petrova, T.V. Lymphangiogenesis in development and human disease. Nature 2005, 438, 946–953. [Google Scholar] [CrossRef]
- Aspelund, A.; Antila, S.; Proulx, S.T.; Karlsen, T.V.; Karaman, S.; Detmar, M.; Wiig, H.; Alitalo, K. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J. Exp. Med. 2015, 212, 991–999. [Google Scholar] [CrossRef]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef]
- Sengillo, J.D.; Winkler, E.A.; Walker, C.T.; Sullivan, J.S.; Johnson, M.; Zlokovic, B.V. Deficiency in mural vascular cells coincides with blood-brain barrier disruption in Alzheimer’s disease. Brain Pathol. 2013, 23, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Miners, J.S.; Schulz, I.; Love, S. Differing associations between Aβ accumulation, hypoperfusion, blood-brain barrier dysfunction and loss of PDGFRB pericyte marker in the precuneus and parietal white matter in Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2018, 38, 103–115. [Google Scholar] [CrossRef]
- Van de Haar, H.J.; Burgmans, S.; Jansen, J.F.; van Osch, M.J.; van Buchem, M.A.; Muller, M.; Hofman, P.A.; Verhey, F.R.; Backes, W.H. Blood-Brain Barrier Leakage in Patients with Early Alzheimer Disease. Radiology 2016, 281, 527–535. [Google Scholar] [CrossRef]
- Choi, S.H.; Lee, D.Y.; Kim, S.U.; Jin, B.K. Thrombin-induced oxidative stress contributes to the death of hippocampal neurons in vivo: Role of microglial NADPH oxidase. J. Neurosci. 2005, 25, 4082–4090. [Google Scholar] [CrossRef]
- Choi, M.S.; Kim, Y.E.; Lee, W.J.; Choi, J.W.; Park, G.H.; Kim, S.D.; Jeon, S.J.; Go, H.S.; Shin, S.M.; Kim, W.K.; et al. Activation of protease-activated receptor1 mediates induction of matrix metalloproteinase-9 by thrombin in rat primary astrocytes. Brain Res. Bull. 2008, 76, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Grammas, P.; Samany, P.G.; Thirumangalakudi, L. Thrombin and inflammatory proteins are elevated in Alzheimer’s disease microvessels: Implications for disease pathogenesis. J. Alzheimers Dis. 2006, 9, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Wright, J.; Wall, T.; Grammas, P. Brain endothelial cells synthesize neurotoxic thrombin in Alzheimer’s disease. Am. J. Pathol. 2010, 176, 1600–1606. [Google Scholar] [CrossRef]
- Machida, T.; Takata, F.; Matsumoto, J.; Takenoshita, H.; Kimura, I.; Yamauchi, A.; Dohgu, S.; Kataoka, Y. Brain pericytes are the most thrombin-sensitive matrix metalloproteinase-9-releasing cell type constituting the blood-brain barrier in vitro. Neurosci. Lett. 2015, 599, 109–114. [Google Scholar] [CrossRef]
- Vukic, V.; Callaghan, D.; Walker, D.; Lue, L.F.; Liu, Q.Y.; Couraud, P.O.; Romero, I.A.; Weksler, B.; Stanimirovic, D.B.; Zhang, W. Expression of inflammatory genes induced by beta-amyloid peptides in human brain endothelial cells and in Alzheimer’s brain is mediated by the JNK-AP1 signaling pathway. Neurobiol. Dis. 2009, 34, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.; Maloney, A.J. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 1976, 2, 1403. [Google Scholar] [CrossRef]
- Bowen, D.M.; Smith, C.B.; White, P.; Davison, A.N. Neurotransmitter-related enzymes and indices of hypoxia in senile dementia and other abiotrophies. Brain 1976, 99, 459–496. [Google Scholar] [CrossRef]
- Shimohama, S.; Taniguchi, T.; Fujiwara, M.; Kameyama, M. Changes in nicotinic and muscarinic cholinergic receptors in Alzheimer-type dementia. J. Neurochem. 1986, 46, 288–293. [Google Scholar] [CrossRef]
- Whitehouse, P.J.; Price, D.L.; Struble, R.G.; Clark, A.W.; Coyle, J.T.; Delon, M.R. Alzheimer’s disease and senile dementia: Loss of neurons in the basal forebrain. Science 1982, 215, 1237–1239. [Google Scholar] [CrossRef]
- Giacobini, E.; Cuello, A.C.; Fisher, A. Reimagining cholinergic therapy for Alzheimer’s disease. Brain 2022, 145, 2250–2275. [Google Scholar] [CrossRef]
- Bartus, R.T.; Dean, R.L.; Beer, B.; Lippa, A.S. The cholinergic hypothesis of geriatric memory dysfunction. Science 1982, 217, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Khachaturian, A.S.; Vergallo, A.; Farlow, M.R.; Snyder, P.J.; Giacobini, E.; Khachaturian, Z.S. Revisiting the Cholinergic Hypothesis in Alzheimer’s Disease: Emerging Evidence from Translational and Clinical Research. J. Prev. Alzheimers Dis. 2019, 6, 2–15. [Google Scholar] [PubMed]
- Pozzi, F.E.; Conti, E.; Appollonio, I.; Ferrarese, C.; Tremolizzo, L. Predictors of response to acetylcholinesterase inhibitors in dementia: A systematic review. Front. Neurosci. 2022, 20, 998224. [Google Scholar] [CrossRef] [PubMed]
- Grothe, M.J.; Ewers, M.; Krause, B.; Heinsen, H.; Teipel, S.J. Alzheimer’s Disease Neuroimaging Initiative. Basal forebrain atrophy and cortical amyloid deposition in nondemented elderly subjects. Alzheimers. Dement. 2014, 10, S344–S353. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Rodriguez, J.J.; Pacheco-Herrero, M.; Thyssen, D.; Murillo-Carretero, M.I.; Berrocoso, E.; Spires-Jones, T.L.; Bacskai, B.J.; Garcia-Alloza, M. Rapid β-amyloid deposition and cognitive impairment after cholinergic denervation in APP/PS1 mice. J. Neuropathol. Exp. Neurol. 2013, 72, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R.; Hunter, R.; Wyper, D.J.; Patterson, J.; Kelly, R.C.; Montaldi, D.; McCullouch, J. Longitudinal changes in cognitive function and regional cerebral function in Alzheimer’s disease: A SPECT blood flow study. J. Psychiatr. Res. 1996, 30, 109–126. [Google Scholar] [CrossRef] [PubMed]
- Austin, S.A.; Katusic, Z.S. Partial loss of endothelial nitric oxide leads to increased cerebrovascular beta amyloid. J. Cereb. Blood Flow Metab. 2020, 40, 392–403. [Google Scholar] [CrossRef]
- Zecchin, H.G.; Priviero, F.B.; Souza, C.T.; Zecchin, K.G.; Prada, P.O.; Carvalheira, J.B.; Velloso, L.A.; Antunes, E.; Saad, M.J. Defective insulin and acetylcholine induction of endothelial cell-nitric oxide synthase through insulin receptor substrate/Akt signaling pathway in aorta of obese rats. Diabetes 2007, 56, 1014–1024. [Google Scholar] [CrossRef]
- Rikitake, Y.; Kim, H.H.; Huang, Z.; Seto, M.; Yano, K.; Asano, T.; Moskowitz, M.A.; Liao, J.K. Inhibition of Rho kinase (ROCK) leads to increased cerebral blood flow and stroke protection. Stroke 2005, 36, 2251–2257. [Google Scholar] [CrossRef]
- Nizari, S.; Wells, J.A.; Carare, R.O.; Romero, I.A.; Hawkes, C.A. Loss of cholinergic innervation differentially affects eNOS-mediated blood flow, drainage of Aβ and cerebral amyloid angiopathy in the cortex and hippocampus of adult mice. Acta Neuropathol. Commun. 2021, 9, 12. [Google Scholar] [CrossRef]
- Arber, C.; Toombs, J.; Lovejoy, C.; Ryan, N.S.; Paterson, R.W.; Willumsen, N.; Gkanatsiou, E.; Portelius, E.; Blennow, K.; Heslegrave, A.; et al. Familial Alzheimer’s disease patient-derived neurons reveal distinct mutation-specific effects on amyloid beta. Mol. Psychiatry. 2020, 25, 2919–2931. [Google Scholar] [CrossRef] [PubMed]
- Novikova, G.; Kapoor, M.; Tcw, J.; Abud, E.M.; Efthymiou, A.G.; Chen, S.X.; Cheng, H.; Fullard, J.F.; Bendl, J.; Liu, Y.; et al. Integration of Alzheimer’s disease genetics and myeloid genomics identifies disease risk regulatory elements and genes. Nat. Commun. 2021, 12, 1610. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.F.; Smith, A.V.; Aspelund, T.; Betensky, R.A.; Smoller, J.W.; Gudnason, V.; Launer, L.J.; Blacker, D. Genetic overlap between vascular pathologies and Alzheimer’s dementia and potential causal mechanisms. Alzheimers Dement. 2019, 15, 65–75. [Google Scholar] [CrossRef]
- Yang, A.C.; Vest, R.T.; Kern, F.; Lee, D.P.; Agam, M.; Maat, C.A.; Losada, P.M.; Chen, M.B.; Schaum, N.; Khoury, N.; et al. A human brain vascular atlas reveals diverse mediators of Alzheimer’s risk. Nature 2022, 603, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.J.; Raghavan, N.S.; Bhattarai, P.; Siddiqui, T.; Sariya, S.; Reyes-Dumeyer, D.; Flowers, X.E.; Cardoso, S.A.L.; De Jager, P.L.; Bennett, D.A.; et al. FMNL2 regulates gliovascular interactions and is associated with vascular risk factors and cerebrovascular pathology in Alzheimer’s disease. Acta Neuropathol. 2022, 144, 59–79. [Google Scholar] [CrossRef]
- Ngandu, T.; Lehtisalo, J.; Solomon, A.; Levälahti, E.; Ahtiluoto, S.; Antikainen, R.; Bäckman, L.; Hänninen, T.; Jula, A.; Laatikainen, T.; et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): A randomised controlled trial. Lancet 2015, 385, 2255–2263. [Google Scholar] [CrossRef] [PubMed]
- Garbuzova-Davis, S.; Sanberg, P.R. Blood-CNS Barrier Impairment in ALS patients versus an animal model. Front. Cell Neurosci. 2014, 8, 21. [Google Scholar] [CrossRef]
- Winkler, E.A.; Sengillo, J.D.; Sagare, A.P.; Zhao, Z.; Ma, Q.; Zuniga, E.; Wang, Y.; Zhong, Z.; Sullivan, J.S.; Griffin, J.H.; et al. Blood-spinal cord barrier disruption contributes to early motor-neuron degeneration in ALS-model mice. Proc. Natl. Acad. Sci. USA 2014, 111, E1035–E1042. [Google Scholar] [CrossRef]
- Padel, T.; Roth, M.; Gaceb, A.; Li, J.Y.; Bjorkqvist, M.; Paul, G. Brain pericyte activation occurs early in Huntington’s disease. Exp. Neurol. 2018, 305, 139–150. [Google Scholar] [CrossRef]
- Faucheux, B.A.; Agid, Y.; Hirsch, E.C.; Bonnet, A.-M. Blood vessels change in the mesencephalon of patients with Parkinson’s disease. Lancet 1999, 353, 981–982. [Google Scholar] [CrossRef]
- Zhong, Z.; Deane, R.; Ali, Z.; Parisi, M.; Shapovalov, Y.; O’Banion, M.K.; Stojanovic, K.; Sagare, A.; Boillee, S.; Cleveland, D.W.; et al. ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat. Neurosci. 2008, 11, 420–422. [Google Scholar] [CrossRef] [PubMed]
- Drouin-Ouellet, J.; Sawiak, S.J.; Cisbani, G.; Lagace, M.; Kuan, W.L.; Saint-Pierre, M.; Dury, R.J.; Alata, W.; St-Amour, I.; Mason, S.L.; et al. Cerebrovascular and blood-brain barrier impairments in Huntington’s disease: Potential implications for its pathophysiology. Ann. Neurol. 2015, 78, 160–177. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Hsu, Y.H.; Lin, M.H.; Yang, T.H.; Chen, H.M.; Chen, Y.C.; Hsiao, H.Y.; Chen, C.C.; Chern, Y.; Chang, C. Neurovascular abnormalities in humans and mice with Huntington’s disease. Exp. Neurol. 2013, 250, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, H.Y.; Chen, Y.C.; Huang, C.H.; Chen, C.C.; Hsu, Y.H.; Chen, H.M.; Chiu, F.L.; Kuo, H.C.; Chang, C.; Chern, Y. Aberrant astrocytes impair vascular reactivity in Huntington disease. Ann. Neurol. 2015, 78, 178–192. [Google Scholar] [CrossRef]
- Di Pardo, A.; Amico, E.; Scalabri, F.; Pepe, G.; Castaldo, S.; Elifani, F.; Capocci, L.; De Sanctis, C.; Comerci, L.; Pompeo, F.; et al. Impairment of blood-brain barrier is an early event in R6/2 mouse model of Huntington Disease. Sci. Rep. 2017, 7, 41316. [Google Scholar] [CrossRef]
- Lim, R.G.; Quan, C.; Reyes-Ortiz, A.M.; Lutz, S.E.; Kedaigle, A.J.; Gipson, T.A.; Wu, J.; Vatine, G.D.; Stocksdale, J.; Casale, M.S.; et al. Huntington’s disease iPSC-derived brain microvascular endothelial cells reveal WNT-mediated angiogenic and blood-brain barrier deficits. Cell Rep. 2017, 19, 1365–1377. [Google Scholar] [CrossRef] [PubMed]
- Desai Bradaric, B.; Patel, A.; Schneider, J.A.; Carvey, P.M.; Hendey, B. Evidence for angiogenesis in Parkinson’s disease, incidental Lewy body disease, and progressive supranuclear palsy. J. Neural Transm. 2012, 119, 59–71. [Google Scholar] [CrossRef]
- Carvey, P.M.; Zhao, C.H.; Hendey, B.; Lum, H.; Trachtenberg, J.; Desai, B.S.; Snyder, J.; Zhu, Y.G.; Ling, Z.D. 6-Hydroxydopamine-induced alterations in blood-brain barrier permeability. Eur. J. Neurosci. 2005, 22, 1158–1168. [Google Scholar] [CrossRef]
- Elabi, O.F.; Cunha, J.; Gaceb, A.; Fex, M.; Paul, G. High-fat diet-induced diabetes leads to vascular alterations, pericyte reduction, and perivascular depletion of microglia in a 6-OHDA toxin model of Parkinson disease. J. Neuroinflam. 2021, 18, 175. [Google Scholar] [CrossRef]
- Westin, J.E.; Lindgren, H.S.; Gardi, J.; Nyengaard, J.R.; Brundin, P.; Mohapel, P.; Cenci, M.A. Endothelial proliferation and increased blood-brain barrier permeability in the basal ganglia in a rat model of 3,4-dihydroxyphenyl-L-alanine-induced dyskinesia. J. Neurosci. 2006, 26, 9448–9461. [Google Scholar] [CrossRef]
- Zhao, C.H.; Ling, Z.D.; Newman, M.B.; Bhatia, A.; Carvey, P.M. TNF-alpha knockout and minocycline treatment attenuates blood-brain barrier leakage in MPTP-treated mice. Neurobiol. Dis. 2007, 26, 36–46. [Google Scholar] [CrossRef]
- Chen, X.; Lan, X.; Roche, I.; Liu, R.; Geiger, J.D. Caffeine protects against MPTP-induced blood-brain barrier dysfunction in mouse striatum. J. Neurochem. 2008, 107, 1147–1157. [Google Scholar] [CrossRef] [PubMed]
- Kortekaas, R.; Leenders, K.L.; van Oostrom, J.C.; Vaalburg, W.; Bart, J.; Willemsen, A.T.; Hendrikse, N.H. Blood-brain barrier dysfunction in parkinsonian midbrain in vivo. Ann. Neurol. 2005, 57, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.T.; Woulfe, J.M. Striatal blood-brain barrier permeability in Parkinson’s disease. J. Cereb. Blood Flow Metab. 2015, 35, 747–750. [Google Scholar] [CrossRef] [PubMed]
- Al-Bachari, S.; Naish, J.H.; Parker, G.J.M.; Emsley, H.C.A.; Parkes, L.M. Blood-Brain Barrier Leakage Is Increased in Parkinson’s Disease. Front. Physiol. 2020, 11, 593026. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Peng, S.; Ma, Y.; Tang, C.C.; Hellman, M.; Feigin, A.; Eidelberg, D.; Dhawan, V. Blood-brain barrier permeability in Parkinson’s disease patients with and without dyskinesia. J. Neurol. 2021, 268, 2246–2255. [Google Scholar] [CrossRef]
- Ping, L.Y.; Chuang, Y.A.; Hsu, S.H.; Tsai, H.Y.; Cheng, M.C. Screening for Mutations in the TBX1 Gene on Chromosome 22q11.2 in Schizophrenia. Genes 2016, 7, 102. [Google Scholar] [CrossRef]
- Hartley, D.; Blumenthal, T.; Carrillo, M.; DiPaolo, G.; Esralew, L.; Gardiner, K.; Granholm, A.C.; Iqbal, K.; Krams, M.; Lemere, C.; et al. Down syndrome and Alzheimer’s disease: Common pathways, common goals. Alzheimers. Dement. 2015, 11, 700–709. [Google Scholar] [CrossRef]
- Lai, F.; Williams, R.S. A prospective study of Alzheimer disease in Down syndrome. Arch. Neurol. 1989, 46, 849–853. [Google Scholar] [CrossRef]
- Abi-Ghanem, C.; Robison, L.S.; Zuloaga, K.L. Androgens’ effects on cerebrovascular function in health and disease. Biol. Sex Differ. 2020, 11, 35. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ishihara, K.; Takata, K.; Mizutani, K.-i. Involvement of an Aberrant Vascular System in Neurodevelopmental, Neuropsychiatric, and Neuro-Degenerative Diseases. Life 2023, 13, 221. https://doi.org/10.3390/life13010221
Ishihara K, Takata K, Mizutani K-i. Involvement of an Aberrant Vascular System in Neurodevelopmental, Neuropsychiatric, and Neuro-Degenerative Diseases. Life. 2023; 13(1):221. https://doi.org/10.3390/life13010221
Chicago/Turabian StyleIshihara, Keiichi, Kazuyuki Takata, and Ken-ichi Mizutani. 2023. "Involvement of an Aberrant Vascular System in Neurodevelopmental, Neuropsychiatric, and Neuro-Degenerative Diseases" Life 13, no. 1: 221. https://doi.org/10.3390/life13010221
APA StyleIshihara, K., Takata, K., & Mizutani, K.-i. (2023). Involvement of an Aberrant Vascular System in Neurodevelopmental, Neuropsychiatric, and Neuro-Degenerative Diseases. Life, 13(1), 221. https://doi.org/10.3390/life13010221