

Suppression of OCT-1 in Metastatic Breast Cancer Cells Reduces Tumor Metastatic Potential, Hypoxia Resistance, and Drug Resistance

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. RNA Isolation and qRT-PCR Analysis
2.2. Western Blot Analysis
2.3. Cell Migration
2.4. Growth Rate Calculation
2.5. Cell Stress
2.6. Constructs Transfection and Dual Luciferase Assay
2.7. Statistics
3. Results
3.1. A Decrease in OCT-1 Expression Levels in the MDA-MB231 Cells
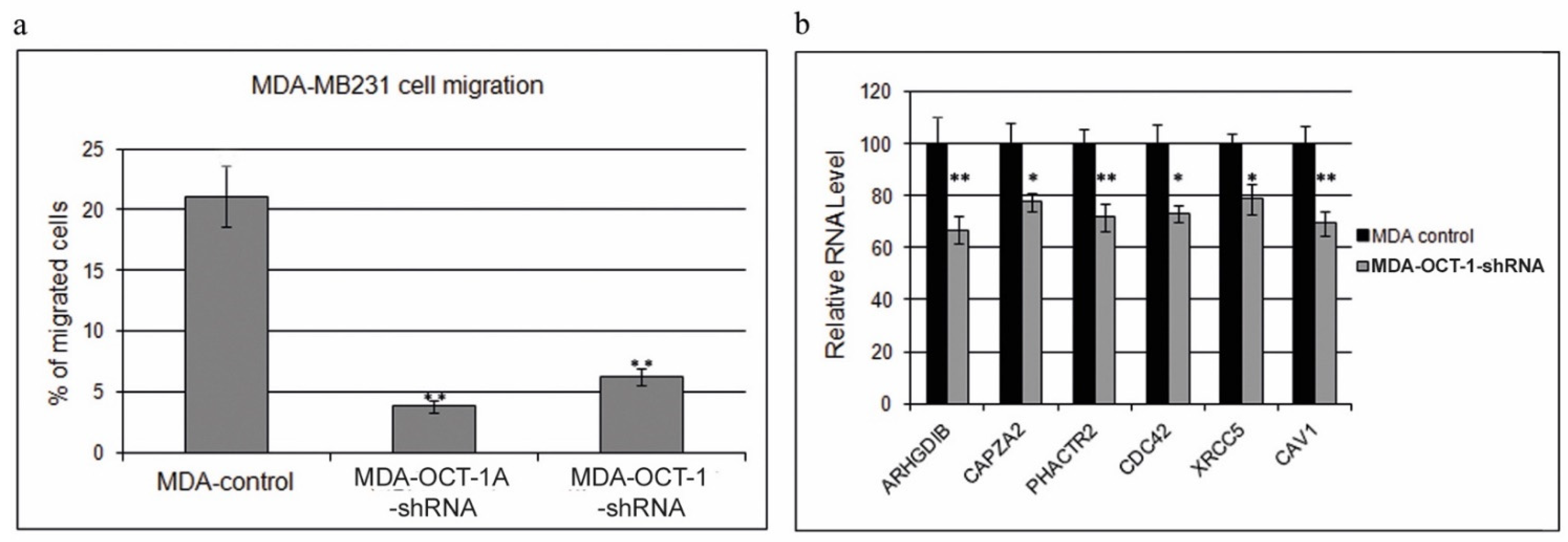
3.2. Knockdown of the Total OCT-1 or Its OCT-1A Isoform Inhibits Breast Cancer Cell Migration and Decreases the Expression of Human Invasion and Metastasis Genes but Does Not Affect Cell Growth Rates
3.3. A Decrease in the OCT-1 and Its OCT-1A Isoform Expression Increases Hypoxia-Associated MDA-MB231 Cell Death
3.4. A Decrease in OCT-1 and OCT-1A Isoform Expression Inhibits Endoplasmic Reticulum Stress Response
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vázquez-Arreguín, K.; Tantin, D. The Oct1 transcription factor and epithelial malignancies: Old protein learns new tricks. Biochim. Et Biophys. Acta 2016, 1859, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Pankratova, E.V.; Stepchenko, A.G.; Portseva, T.; Mogila, V.A.; Georgieva, S.G. Different N-terminal isoforms of Oct-1 control expression of distinct sets of genes and their high levels in Namalwa Burkitt’s lymphoma cells affect a wide range of cellular processes. Nucleic Acids Res. 2016, 44, 9218–9230. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pankratova, E.V.; Stepchenko, A.G.; Krylova, I.D.; Portseva, T.N.; Georgieva, S.G. The regulatory interplay between Oct-1 isoforms contributes to hematopoiesis and the isoforms imbalance correlates with a malignant transformation of B cells. Oncotarget 2018, 9, 29892–29905. [Google Scholar] [CrossRef]
- Stepchenko, A.G.; Georgieva, S.G.; Pankratova, E.V. Multiple Interactions of the Oct-1 (POU2F1) Transcription Factor with PORE and MORE Sites. Mol. Biol. 2019, 53, 430–435. [Google Scholar] [CrossRef]
- Kang, H.-B.; Fan, J.; Lin, R.; Elf, S.; Ji, Q.; Zhao, L.; Jin, L.; Seo, J.H.; Shan, C.; Arbiser, J.L.; et al. Metabolic Rewiring by Oncogenic BRAF V600E Links Ketogenesis Pathway to BRAF-MEK1 Signaling. Mol. Cell. 2015, 59, 345–358. [Google Scholar] [CrossRef]
- Imai, S.I.; Nishibayashi, S.; Takao, K.; Tomifuji, M.; Fujino, T.; Hasegawa, M.; Takano, T. Dissociation of Oct-1 from the nuclear peripheral structure induces the cellular aging-associated collagenase gene expression. Mol. Biol. Cell 1997, 8, 2407–2419. [Google Scholar] [CrossRef] [PubMed]
- Malhas, A.N.; Lee, C.F.; Vaux, D.J. Lamin B1 controls oxidative stress responses via Oct-1. J. Cell Biol. 2009, 184, 45–55. [Google Scholar] [CrossRef]
- Shakya, A.; Cooksey, R.; Cox, J.E.; Wang, V.; McClain, D.A.; Tantin, D. Oct1 loss of function induces a coordinate metabolic shift that opposes tumorigenicity. Nat. Cell Biol. 2009, 11, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Arreguín, K.; Maddox, J.; Kang, J.; Park, D.; Cano, R.R.; Factor, R.E.; Ludwig, T.; Tantin, D. BRCA1 through Its E3 Ligase Activity Regulates the Transcription Factor Oct1 and Carbohydrate Metabolism. Mol. Cancer Res. 2018, 16, 439–452. [Google Scholar] [CrossRef]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef]
- DeSantis, C.E.; Lin, C.C.; Mariotto, A.B.; Siegel, R.L.; Stein, K.D.; Kramer, J.L.; Alteri, R.; Robbins, A.S.; Jemal, A. Cancer treatment and survivorship statistics, 2014. CA Cancer J. Clin. 2014, 64, 252–271. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Kong, X.; Deng, N.; Tan, P.; Chen, H.; Wang, J.; Li, Z.; Hu, Y.; Zou, W.; Xu, J.; et al. OCT1 is a determinant of synbindin-related ERK signalling with independent prognostic significance in gastric cancer. Gut 2015, 64, 37–48. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Finak, G.; Bertos, N.; Pepin, F.; Sadekova, S.; Souleimanova, M.; Zhao, H.; Chen, H.; Omeroglu, G.; Meterissian, S.; Omeroglu, A.; et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat. Med. 2008, 14, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Langerød, A.; Ji, Y.; Nowels, K.W.; Nesland, J.M.; Tibshirani, R.; Bukholm, I.K.; Kåresen, R.; Botstein, D.; Børresen-Dale, A.-L.; et al. Different gene expression patterns in invasive lobular and ductal carcinomas of the breast. Mol. Biol. Cell 2004, 15, 2523–2536. [Google Scholar] [CrossRef]
- Maddox, J.; Shakya, A.; South, S.; Shelton, D.; Andersen, J.N.; Chidester, S.; Kang, J.; Gligorich, K.M.; Jones, D.A.; Spangrude, G.J.; et al. Transcription factor Oct1 is a somatic and cancer stem cell determinant. PLoS Genet. 2012, 8, e1003048. [Google Scholar] [CrossRef]
- Vázquez-Arreguín, K.; Bensard, C.; Schell, J.C.; Swanson, E.; Chen, X.; Rutter, J.; Tantin, D. Oct1/Pou2f1 is selectively required for colon regeneration and regulates colon malignancy. PLoS Genet. 2019, 15, e1007687. [Google Scholar] [CrossRef]
- Hwang-Verslues, W.W.; Chang, P.-H.; Jeng, Y.-M.; Kuo, W.-H.; Chiang, P.-H.; Chang, Y.-C.; Hsieh, T.-H.; Su, F.-Y.; Lin, L.-C.; Abbondante, S.; et al. Loss of corepressor PER2 under hypoxia up-regulates OCT1-mediated EMT gene expression and enhances tumor malignancy. Proc. Natl. Acad. Sci. USA 2013, 110, 12331–12336. [Google Scholar] [CrossRef] [PubMed]
- Almeida, R.; Almeida, J.; Shoshkes, M.; Mendes, N.; Mesquita, P.; Silva, E.; Van Seuningen, I.; Reis, A.C.; Santos-Silva, F.; David, L. OCT-1 is over-expressed in intestinal metaplasia and intestinal gastric carcinomas and binds to, but does not transactivate, CDX2 in gastric cells. J. Pathol. 2005, 207, 396–401. [Google Scholar] [CrossRef][Green Version]
- Jeong, S.-H.; Lee, Y.-J.; Cho, B.-I.; Ha, W.-S.; Choi, S.-K.; Jung, E.-J.; Ju, Y.-T.; Jeong, C.-Y.; Ko, G.H.; Yoo, J.; et al. OCT-1 overexpression is associated with poor prognosis in patients with well-differentiated gastric cancer. Tumour Biol. 2014, 35, 5501–5509. [Google Scholar] [CrossRef]
- Hernández, P.; Solé, X.; Valls, J.; Moreno, V.; Capellá, G.; Urruticoechea, A.; Pujana, M.A. Integrative analysis of a cancer somatic mutome. Mol. Cancer 2007, 6, 13. [Google Scholar] [CrossRef]
- Obinata, D.; Takayama, K.-I.; Urano, T.; Murata, T.; Kumagai, J.; Fujimura, T.; Ikeda, K.; Horie-Inoue, K.; Homma, Y.; Ouchi, Y.; et al. Oct1 regulates cell growth of LNCaP cells and is a prognostic factor for prostate cancer. Int. J. Cancer. 2012, 130, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Kalamohan, K.; Periasamy, J.; Bhaskar Rao, D.; Barnabas, G.D.; Ponnaiyan, S.; Ganesan, K. Transcriptional coexpression network reveals the involvement of varying stem cell features with different dysregulations in different gastric cancer subtypes. Mol. Oncol. 2014, 8, 1306–1325. [Google Scholar] [CrossRef] [PubMed]
- Reymann, S.; Borlak, J. Transcription profiling of lung adenocarcinomas of c-myc-transgenic mice: Identification of the c-myc regulatory gene network. BMC Syst. Biol. 2008, 2, 46. [Google Scholar] [CrossRef]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008, 40, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, E.; Merchant, R.E.; Wiestler, O.D.; Fontana, A. Primary brain tumors differ in their expression of octamer deoxyribonucleic acid-binding transcription factors from long-term cultured glioma cell lines. Neurosurgery 1994, 34, 129–135. [Google Scholar]
- Xu, S.-H.; Huang, J.-Z.; Xu, M.-L.; Yu, G.; Yin, X.-F.; Chen, D.; Yan, G.-R. ACK1 promotes gastric cancer epithelial-mesenchymal transition and metastasis through AKT-POU2F1-ECD signalling. J. Pathol. 2015, 236, 175–185. [Google Scholar] [CrossRef]
- Patsialou, A.; Wang, Y.; Lin, J.; Whitney, K.; Goswami, S.; Kenny, P.A.; Condeelis, J.S. Selective gene-expression profiling of migratory tumor cells in vivo predicts clinical outcome in breast cancer patients. Breast Cancer Res. 2012, 14, R139. [Google Scholar] [CrossRef]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Ma, Y.; Sekiya, M.; Kainoh, K.; Matsuda, T.; Iwasaki, H.; Osaki, Y.; Sugano, Y.; Suzuki, H.; Takeuchi, Y.; Miyamoto, T.; et al. Transcriptional co-repressor CtBP2 orchestrates epithelial-mesenchymal transition through a novel transcriptional holocomplex with OCT1. Biochem. Biophys. Res. Commun. 2020, 523, 354–360. [Google Scholar] [CrossRef]
- Chen, X.; Leung, S.Y.; Yuen, S.T.; Chu, K.-M.; Ji, J.; Li, R.; Chan, A.S.Y.; Law, S.; Troyanskaya, O.G.; Wong, J.; et al. Variation in gene expression patterns in human gastric cancers. Mol. Biol. Cell 2003, 14, 3208–3215. [Google Scholar] [CrossRef]
- Wang, J.; Yang, Y.-H.; Wang, A.-Q.; Yao, B.; Xie, G.; Feng, G.; Zhang, Y.; Cheng, Z.-S.; Hui, L.; Dai, T.-Z.; et al. Immunohistochemical detection of the Raf kinase inhibitor protein in nonneoplastic gastric tissue and gastric cancer tissue. Med Oncol. 2010, 27, 219–223. [Google Scholar] [CrossRef] [PubMed]
- D’Errico, M.; de Rinaldis, E.; Blasi, M.F.; Viti, V.; Falchetti, M.; Calcagnile, A.; Sera, F.; Saieva, C.; Ottini, L.; Palli, D.; et al. Genome-wide expression profile of sporadic gastric cancers with microsatellite instability. Eur. J. Cancer 2009, 45, 461–469. [Google Scholar] [CrossRef]
- Wang, Y.-P.; Song, G.-H.; Chen, J.; Xiao, C.; Li, C.; Zhong, L.; Sun, X.; Wang, Z.-W.; Deng, G.-L.; Yu, F.-D.; et al. Elevated OCT1 participates in colon tumorigenesis and independently predicts poor prognoses of colorectal cancer patients. Tumour Biol. 2016, 37, 3247–3255. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Leem, S.H.; Lee, S.Y.; Kim, S.C.; Park, E.S.; Kim, S.B.; Kim, S.K.; Kim, Y.J.; Kim, W.J.; Chu, I.S. Expression signature of E2F1 and its associated genes predict superficial to invasive progression of bladder tumors. J. Clin. Oncol. 2010, 28, 2660–2667. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Carbayo, M.; Socci, N.D.; Lozano, J.; Saint, F.; Cordon-Cardo, C. Defining molecular profiles of poor outcome in patients with invasive bladder cancer using oligonucleotide microarrays. J. Clin. Oncol. 2006, 24, 778–789. [Google Scholar] [CrossRef]
- Dyrskjøt, L.; Kruhøffer, M.; Thykjaer, T.; Marcussen, N.; Jensen, J.L.; Møller, K.; Ørntoft, T.F. Gene expression in the urinary bladder: A common carcinoma in situ gene expression signature exists disregarding histopathological classification. Cancer Res. 2004, 64, 4040–4048. [Google Scholar] [CrossRef]
- Xiao, S.; Liao, S.; Zhou, Y.; Jiang, B.; Li, Y.; Xue, M. High expression of octamer transcription factor 1 in cervical cancer. Oncol. Lett. 2014, 7, 1889–1894. [Google Scholar] [CrossRef]
- Sharpe, D.J.; Orr, K.S.; Moran, M.; White, S.J.; McQuaid, S.; Lappin, T.R.; Thompson, A.; James, J.A. POU2F1 activity regulates HOXD10 and HOXD11 promoting a proliferative and invasive phenotype in head and neck cancer. Oncotarget 2014, 5, 8803–8815. [Google Scholar] [CrossRef]
- Roussos, E.T.; Condeelis, J.S.; Patsialou, A. Chemotaxis in cancer. Nat. Rev. Cancer 2011, 11, 573–587. [Google Scholar] [CrossRef]
- Pinner, S.; Sahai, E. Imaging amoeboid cancer cell motility in vivo. J. Microsc. 2008, 231, 441–445. [Google Scholar] [CrossRef]
- Sidani, M.; Wyckoff, J.; Xue, C.; Segall, J.E.; Condeelis, J. Probing the microenvironment of mammary tumors using multiphoton microscopy. J. Mammary Gland. Biol. Neoplasia. 2006, 11, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bi, X.; Huang, X.; Wang, B.; Guo, Q.; Wu, Z. Systematic investigation of biomarker-like role of ARHGDIB in breast cancer. Cancer Biomark. 2020, 28, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, J.; Lai, X.N.; Jiao, X.Q.; Xiong, J.P.; Xiong, L.X. Focus on Cdc42 in Breast Cancer: New Insights, Target Therapy Development and Non-Coding RNAs. Cells 2019, 8, 146. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.N.; Yang, J.T.; Huang, C.; Huang, H.C.; Wu, Y.P.; Chen, J.C. Elevated XRCC5 expression level can promote temozolomide resistance and predict poor prognosis in glioblastoma. Oncol. Lett. 2021, 21, 443. [Google Scholar] [CrossRef]
- Campos, A.; Salomon, C.; Bustos, R.; Díaz, J.; Martínez, S.; Silva, V.; Reyes, C.; Díaz-Valdivia, N.; Varas-Godoy, M.; Lobos-González, L.; et al. Caveolin-1-containing extracellular vesicles transport adhesion proteins and promote malignancy in breast cancer cell lines. Nanomedicine 2018, 13, 2597–2609. [Google Scholar] [CrossRef]
- Ogura, T.; Azuma, K.; Sato, J.; Kinowaki, K.; Takayama, K.-I.; Takeiwa, T.; Kawabata, H.; Inoue, S. OCT1 Is a Poor Prognostic Factor for Breast Cancer Patients and Promotes Cell Proliferation via Inducing NCAPH. Int. J. Mol. Sci. 2021, 22, 11505. [Google Scholar] [CrossRef]
- Wang, P.; Jin, T. Oct-1 functions as a sensor for metabolic and stress signals. Islets 2010, 2, 46–48. [Google Scholar] [CrossRef]
- Minassian, L.M.; Cotechini, T.; Huitema, E.; Graham, C.H. Hypoxia-Induced Resistance to Chemotherapy in Cancer. Adv. Exp. Med. Biol. 2019, 1136, 123–139. [Google Scholar]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stepchenko, A.G.; Bulavkina, E.V.; Portseva, T.N.; Georgieva, S.G.; Pankratova, E.V. Suppression of OCT-1 in Metastatic Breast Cancer Cells Reduces Tumor Metastatic Potential, Hypoxia Resistance, and Drug Resistance. Life 2022, 12, 1435. https://doi.org/10.3390/life12091435
Stepchenko AG, Bulavkina EV, Portseva TN, Georgieva SG, Pankratova EV. Suppression of OCT-1 in Metastatic Breast Cancer Cells Reduces Tumor Metastatic Potential, Hypoxia Resistance, and Drug Resistance. Life. 2022; 12(9):1435. https://doi.org/10.3390/life12091435
Chicago/Turabian StyleStepchenko, Alexander G., Elizaveta V. Bulavkina, Tatiana N. Portseva, Sofia G. Georgieva, and Elizaveta V. Pankratova. 2022. "Suppression of OCT-1 in Metastatic Breast Cancer Cells Reduces Tumor Metastatic Potential, Hypoxia Resistance, and Drug Resistance" Life 12, no. 9: 1435. https://doi.org/10.3390/life12091435
APA StyleStepchenko, A. G., Bulavkina, E. V., Portseva, T. N., Georgieva, S. G., & Pankratova, E. V. (2022). Suppression of OCT-1 in Metastatic Breast Cancer Cells Reduces Tumor Metastatic Potential, Hypoxia Resistance, and Drug Resistance. Life, 12(9), 1435. https://doi.org/10.3390/life12091435