
First Steps towards the Development of Epigenetic Biomarkers in Female Cheetahs (Acinonyx jubatus)

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Animals
2.2. Sample Collection and PBMC Isolation
2.3. DNA Extraction and Enrichment of Methylated DNA
2.4. Library Preparation and High Throughput Sequencing
2.5. Bioinformatics
2.6. Methylation Analysis
3. Results
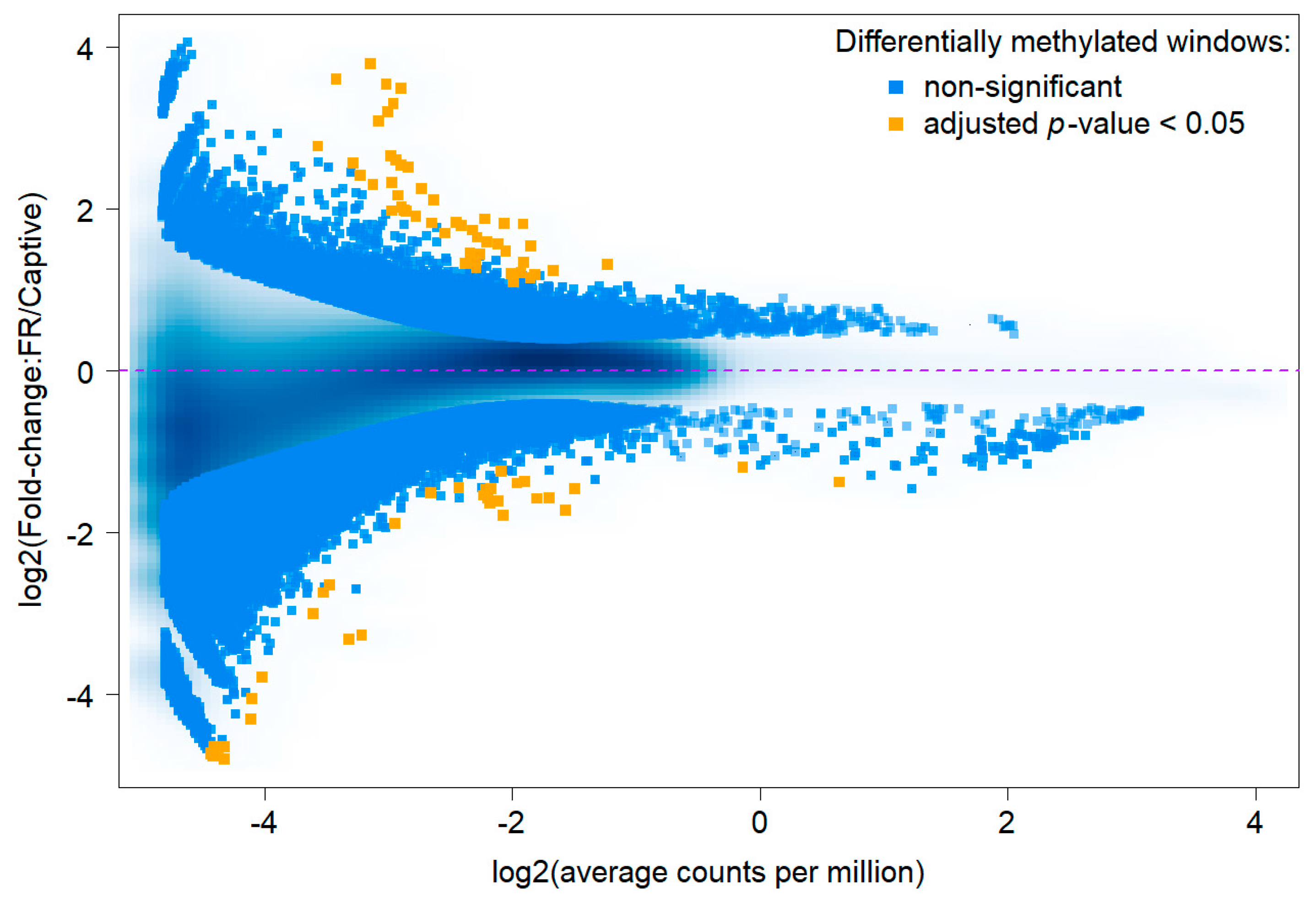
3.1. Sequencing Results and Differentially Methylated Regions
3.2. Comparative Methylation Analysis
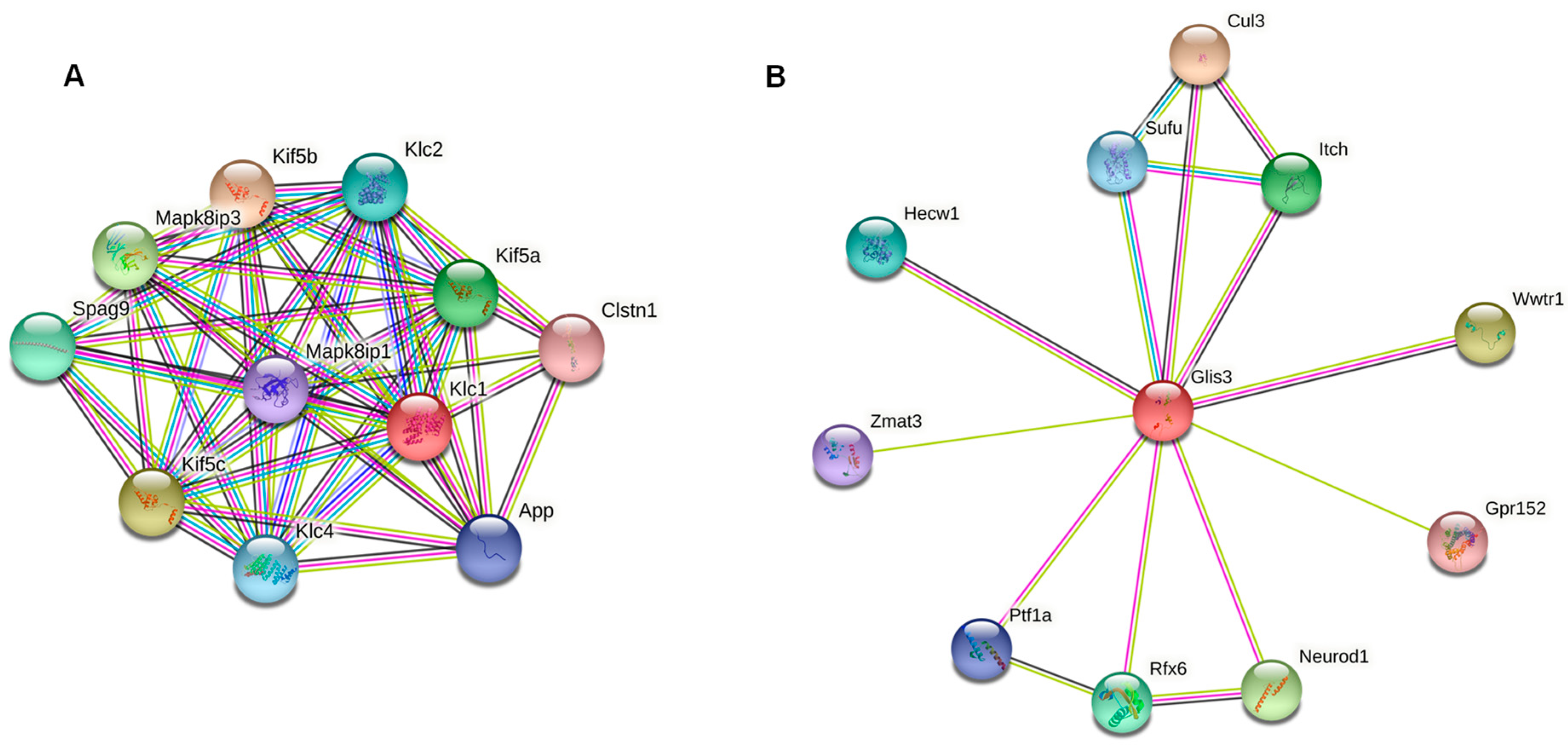
3.3. Genes Overlapped by DMRs and Potential Biomarkers
3.4. DMRs as Potential Biomarkers
4. Discussion
4.1. Differential Methylated Genes in Cheetahs under Human Care
4.2. Biomarker Validation-Long-Term Aim
5. Conclusions
6. Outlook
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Durant, S.; Mitchell, N.; Ipavec, A.; Groom, R. Acinonyx jubatus. In The IUCN Red List of Threatened Species; 2015 (e. T219A50649567). Available online: https://www.iucnredlist.org/details/219/0 (accessed on 1 August 2021).
- Durant, S.M.; Mitchell, N.; Groom, R.; Pettorelli, N.; Ipavec, A.; Jacobson, A.B.; Woodroffe, R.; Bohm, M.; Hunter, L.T.B.; Becker, M.S.; et al. The global decline of cheetah Acinonyx jubatus and what it means for conservation. Proc. Natl. Acad. Sci. USA 2017, 114, 528–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weise, F.J.; Vijay, V.; Jacobson, A.P.; Schoonover, R.F.; Groom, R.J.; Horgan, J.; Keeping, D.; Klein, R.; Marnewick, K.; Maude, G.; et al. The distribution and numbers of cheetahs (Acinonyx jubatus) in southern Africa. PeerJ 2017, 5, e4096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, S.E.; Hoffman, E.A. Minimizing genetic adaptation in captive breeding programs: A review. Biol. Conserv. 2009, 142, 2388–2400. [Google Scholar] [CrossRef]
- Terrell, K.; Crosier, E.A.; Wildt, E.D.; O’Brien, S.J.; Anthony, N.; Marker, L.; Johnson, W. Continued decline in genetic diversity among wild cheetahs (Acinonyx jubatus) without further loss of semen quality. Biol. Conserv. 2016, 200, 192–199. [Google Scholar] [CrossRef] [Green Version]
- Marker-Kraus, L.; Grisham, J. Captive breeding of cheetahs in North American zoos: 1987–1991. Zoo Biol. 1993, 12, 5–18. [Google Scholar] [CrossRef]
- Wildt, D.E.; Brown, J.L.; Bush, M.; Barone, M.A.; Cooper, K.A.; Grisham, J.; Howard, J.G. Reproductive status of cheetahs (Acinonyx jubatus) in North American zoos: The benefits of physiological surveys for strategic planning. Zoo Biol. 1993, 12, 45–80. [Google Scholar] [CrossRef]
- Bertschinger, H.J.; Meltzer, D.G.A.; Dyk, A. Captive breeding of cheetahs in South Africa- 30 years of data from the de Wildt Cheetah and Wildlife Centre. Reprod. Domest. Anim. 2008, 43, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.L.; Wildt, D.E.; Wielebnowski, N.; Goodrowe, K.L.; Graham, L.H.; Wells, S.; Howard, J.G. Reproductive activity in captive female cheetahs (Acinonyx jubatus) assessed by faecal steroids. Reproduction 1996, 106, 337–346. [Google Scholar] [CrossRef] [Green Version]
- Versteege, L. European Studbook Southern Cheetah (Acinonyx jubatus); Beekse Bergen: Hilvarenbeek, The Netherlands, 2013. [Google Scholar]
- Wachter, B.; Thalwitzer, S.; Hofer, H.; Lonzer, J.; Hildebrandt, T.B.; Hermes, R. Reproductive history and absence of predators are important determinants of reproductive fitness: The cheetah controversy revisited. Conserv. Lett. 2011, 4, 47–54. [Google Scholar] [CrossRef]
- Evermann, J.F.; Heeney, J.L.; Roelke, M.E.; McKeirnan, A.J.; O’Brien, S.J. Biological and pathological consequences of feline infectious peritonitis virus infection in the cheetah. Arch. Virol. 1988, 102, 155–171. [Google Scholar] [CrossRef]
- Munson, L.; Terio, A.K.; Worley, M.; Jago, M.; Bagot-Smith, A.; Marker, L. Extrinsic factors significantly affect patterns of disease in free-ranging and captive cheetah (Acinonyx jubatus) populations. J. Wildl. Dis. 2005, 41, 542–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munson, L. Disease of captive cheetahs (Acinonyx jubatus): Results of the cheetah research council pathology survey, 1989–1992. Zoo Biol. 1993, 12, 105–124. [Google Scholar] [CrossRef]
- Brower, A.I.; Munson, L.; Radcliffe, R.W.; Citino, S.B.; Lackey, L.B.; van Winkle, T.J.; Stalis, I.; Terio, K.A.; Summers, B.A.; de Lahunta, A. Leukoencephalomyelopathy of Mature Captive Cheetahs and Other Large Felids: A Novel Neurodegenerative Disease That Came and Went? Vet. Pathol. 2014, 51, 1013–1021. [Google Scholar] [CrossRef] [Green Version]
- Franklin, A.D.; Schmidt-Küntzel, A.; Terio, K.A.; Marker, L.L.; Crosier, A.E. Serum Amyloid A protein concentration in blood is influenced by genetic differences in the cheetah (Acinonyx jubatus). J. Hered. 2015, 107, 115–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caro, T.M. Cheetahs of the Serengeti Plains: Group Living in an Asocial Species; University of Chicago Press: Chicago, IL, USA, 1994; p. 478. [Google Scholar]
- Caro, T.M.; Laurenson, M.K. Ecological and genetic factors in conservation: A cautionary tale. Science 1994, 263, 485–486. [Google Scholar] [CrossRef]
- Thalwitzer, S.; Wachter, B.; Robert, N.; Wibbelt, G.; Müller, T.; Lonzer, J.; Meli, M.L.; Bay, G.; Hofer, H.; Lutz, H. Seroprevalences to viral pathogens in free-ranging and captive cheetahs (Acinonyx jubatus) on Namibian farmland. Clin. Vaccine Immunol. 2010, 17, 232–238. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, S.J.; Roelke, M.E.; Marker, L.; Newman, A.; Winkler, C.A.; Meltzer, D.; Colly, L.; Evermann, J.F.; Bush, M.; Wildt, D.E. Genetic basis for species vulnerability in the cheetah. Science 1985, 227, 1428–1434. [Google Scholar] [CrossRef] [Green Version]
- Menotti-Raymond, M.A.; O’Brien, S.J. Evolutionary conservation of ten microsatellite loci in four species of Felidae. J. Hered. 1995, 86, 319–322. [Google Scholar] [CrossRef]
- Castro-Prieto, A.; Wachter, B.; Sommer, S. Cheetah paradigm revisited: MHC diversity in the world’s largest free-ranging population. Mol. Biol. Evol. 2011, 28, 1455–1468. [Google Scholar] [CrossRef]
- Dobrynin, P.; Liu, S.; Tamazian, G.; Xiong, Z.; Yurchenko, A.A.; Krasheninnikova, K.; Kliver, S.; Schmidt-Küntzel, A.; Koepfli, K.P.; Johnson, W.; et al. Genomic legacy of the African cheetah, Acinonyx jubatus. Genome Biol. 2015, 16, 277. [Google Scholar] [CrossRef] [Green Version]
- Sommer, S. The importance of immune gene variability (MHC) in evolutionary ecology and conservation. Front. Zool. 2005, 2, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charruau, P.; Fernandes, C.; Orozcoter Wengel, P.; Peters, J.; Hunter, L.; Ziaie, H.; Jourabchian, A.; Jowkar, H.; Schaller, G.; Ostrowski, S.; et al. Phylogeography, genetic structure and population divergence time of cheetahs in Africa and Asia: Evidence for long-term geographic isolates. Mol. Ecol. 2011, 20, 706–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wielebnowski, N. Reassessing the relationship between juvenile mortality and genetic monomorphism in captive cheetahs. Zoo Biol. 1996, 15, 353–369. [Google Scholar] [CrossRef]
- Czamara, D.; Tissink, E.; Tuhkanen, J.; Martins, J.; Awaloff, Y.; Drake, A.J.; Khulan, B.; Palotie, A.; Winter, S.M.; Nemeroff, C.B.; et al. Combined effects of genotype and childhood adversity shape variability of DNA methylation across age. Transl. Psychiatry 2021, 11, 88. [Google Scholar] [CrossRef]
- Weyrich, A.; Jeschek, M.; Schrapers, K.T.; Lenz, D.; Chung, T.H.; Rübensam, K.; Yasar, S.; Schneemann, M.; Ortmann, S.; Jewgenow, K.; et al. Diet changes alter paternally inherited epigenetic patterns in male wild guinea pigs. Environ. Epigenetics 2018, 4, dvy011. [Google Scholar] [CrossRef]
- Weyrich, A.; Lenz, D.; Jeschek, M.; Chung, T.H.; Rübensam, K.; Göritz, F.; Jewgenow, K.; Fickel, J. Paternal intergenerational epigenetic response to heat exposure in male wild guinea pigs. Mol. Ecol. 2016, 25, 1729–1740. [Google Scholar] [CrossRef]
- Weyrich, A.; Yasar, S.; Lenz, D.; Fickel, J. Tissue-specific epigenetic inheritance after paternal heat exposure in male wild guinea pigs. Mammal. Genome 2020, 31, 157–169. [Google Scholar] [CrossRef] [Green Version]
- Tung, J.; Barreiro, L.B.; Johnson, Z.P.; Hansen, K.D.; Michopoulos, V.; Toufexis, D.; Michelini, K.; Wilson, M.E.; Gilad, Y. Social environment is associated with gene regulatory variation in the rhesus macaque immune system. Proc. Natl. Acad. Sci. USA 2012, 109, 6490–6495. [Google Scholar] [CrossRef] [Green Version]
- Lea, A.J.; Altmann, J.; Alberts, S.C.; Tung, J. Resource base influences genome-wide DNA methylation levels in wild baboons (Papio cynocephalus). Mol. Ecol. 2016, 25, 1681–1696. [Google Scholar] [CrossRef] [Green Version]
- Turner, B.M. Epigenetic responses to environmental change and their evolutionary implications. Philos. Trans. R. Soc. B Biol. Sci. 2009, 364, 3403–3418. [Google Scholar] [CrossRef] [Green Version]
- Bateson, P.; Gluckman, P. Plasticity and robustness in development and evolution. Int. J. Epidemiol. 2012, 41, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Jablonka, E.; Raz, G. Transgenerational epigenetic inheritance: Prevalence, mechanisms, and implications for the study of heredity and evolution. Q. Rev. Biol. 2009, 84, 131–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szyf, M. Epigenetics, DNA methylation and chromatin modifying drugs. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 243–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skinner, M.K.; Ben Maamar, M.; Sadler-Riggleman, I.; Beck, D.; Nilsson, E.; McBirney, M.; Klukovich, R.; Xie, Y.; Tang, C.; Yan, W. Alterations in sperm DNA methylation, non-coding RNA and histone retention associate with DDT-induced epigenetic transgenerational inheritance of disease. Epigenet Chromatin 2018, 11, 8. [Google Scholar] [CrossRef] [Green Version]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Develop. 2002, 16, 6–21. [Google Scholar] [CrossRef] [Green Version]
- Egger, G.; Liang, G.; Aparicio, A.; Jones, P.A. Epigenetics under human disease and prospects for epigenetic therapy. Nature 2004, 429, 457–463. [Google Scholar] [CrossRef]
- Hernández-Aguilera, A.; Fernández-Arroyo, S.; Cuyàs, E.; Luciano-Mateo, F.; Cabre, N.; Camps, J.; Lopez-Miranda, J.; Menendez, J.A.; Joven, J. Epigenetics and nutrition-related epidemics of metabolic diseases: Current perspectives and challenges. Food Chem. Toxicol. 2016, 96, 191–204. [Google Scholar] [CrossRef]
- Siddle, H.V.; Kreiss, A.; Tovar, C.; Yuen, C.K.; Cheng, Y.; Belov, K.; Swift, K.; Pearse, A.M.; Hamede, R.; Jones, M.E.; et al. Reversible epigenetic down-regulation of MHC molecules by devil facial tumour disease illustrates immune escape by a contagious cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 5103–5108. [Google Scholar] [CrossRef] [Green Version]
- Pavlou, M.P.; Diamandis, E.P.; Blasutig, I.M. The long journey of cancer biomarkers from the bench to the clinic. Clin. Chem. 2013, 59, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Melzheimer, J.; Streif, S.; Wasiolka, B.; Fischer, M.; Thalwitzer, S.; Heinrich, S.K.; Weigold, A.; Hofer, H.; Wachter, B. Queuing, takeovers, and becoming a fat cat: Long-term data reveal two distinct male spatial tactics at different life-history stages in Namibian cheetahs. Ecosphere 2018, 9, e02308. [Google Scholar] [CrossRef]
- Heinrich, S.K.; Hofer, H.; Courtiol, A.; Melzheimer, J.; Dehnhard, M.; Czirják, G.Á.; Wachter, B. Cheetahs have a stronger constitutive innate immunity than leopards. Sci. Rep. 2017, 7, 44837. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, J.; Ning, C.; Zheng, X.; Fu, J.; Wang, A.; Zhang, Q.; Liu, J.F. Genome-wide DNA methylation and transcriptome analyses reveal genes involved in immune responses of pig peripheral blood mononuclear cells to poly I:C. Sci. Rep. 2017, 7, 9709. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhou, Y.; Lin, N.; Lowdon, R.F.; Hong, C.; Nagarajan, R.P.; Cheng, J.B.; Li, D.; Stevens, M.; Lee, H.J.; et al. Functional DNA methylation differences between tissues, cell types, and across individuals discovered using the M&M algorithm. Genome Res. 2013, 23, 1522–1540. [Google Scholar] [PubMed] [Green Version]
- Bergman, Y.; Cedar, H. DNA methylation dynamics in health and disease. Nat. Struct. Mol. Biol. 2013, 20, 274–281. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; A McCarthy, S.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Lienhard, M.; Grimm, C.; Morkel, M.; Herwig, R.; Chavez, L. MEDIPS: Genome-wide differential coverage analysis of sequencing data derived from DNA enrichment experiments. Bioinformatics 2014, 30, 284–286. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR; a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Lienhard, M.; Chavez, L. Quantitative Comparison of Large-Scale DNA Enrichment Sequencing Data. In Statistical Genomics; Humana Press: New York, NY, USA, 2016; pp. 191–208. [Google Scholar]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Snel, B.; Lehmann, G.; Bork, P.; Huynen, M.A. STRING: A web-server to retrieve and display the repeatedly occurring neighbourhood of a gene. Nucleic Acids Res. 2000, 28, 3442–3444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef] [PubMed]
- Christie, M.R.; Marine, M.L.; French, R.A.; Blouin, M.S. Genetic adaptation to captivity can occur in a single generation. Proc. Natl. Acad. Sci. USA 2011, 109, 238–242. [Google Scholar] [CrossRef] [Green Version]
- Courtney Jones, S.K.; Byrne, P.G. What role does heritability play in transgenerational phenotypic responses to captivity? Implications for managing captive populations. Zoo Biol. 2017, 36, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Courtney Jones, S.K.; Munn, A.J.; Byrne, P.G. Effect of captivity on morphology: Negligible changes in external morphology mask significant changes in internal morphology. Open Sci. R. Soc. 2018, 5, 172470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bird, A.P.; Wolffe, A.P. Methylation-induced repression—Belts, braces, and chromatin. Cell 1999, 99, 451–454. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, S.F.; Epel, D. Ecological Developmental Biology: Integrating Epigenetics, Medicine, and Evolution; Sinauer Associates: Sunderland, UK, 2008. [Google Scholar]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharm 2013, 38, 23–38. [Google Scholar] [CrossRef] [Green Version]
- Carpentier, D.C.; Gao, W.N.; Ewles, H.; Morgan, G.W.; Smith, G.L. Vaccinia virus protein complex F12/E2 interacts with kinesin light chain isoform 2 to engage the kinesin-1 motor complex. PLoS Pathog. 2015, 11, e1004723. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.; Carpentier, D.; Ewles, H.A.; Lee, S.A.; Smith, G.L. Vaccinia virus proteins A36 and F12/E2 show strong preferences for different kinesin light chain isoforms. Traffic 2017, 18, 505–518. [Google Scholar] [CrossRef]
- Fang, Z.; Méresse, S. Endomembrane remodelling and dynamics in Salmonella infection. Microb. Cell 2021, 9, 24–41. [Google Scholar] [CrossRef]
- Anastasiadi, D.; Esteve-Codina, A.; Piferrer, F. Consistent inverse correlation between DNA methylation of the first intron and gene expression across tissues and species. Epigenetics Chromatin 2018, 11, 37. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.; Cook, B.; Zhou, T.; Ghazizadeh, Z.; Lis, R.; Zhang, T.; Khalaj, M.; Crespo, M.; Khalaj, M.; Crespo, M.; et al. Discovery of a drug candidate for GLIS3-associated diabetes. Nat. Commun. 2018, 9, 2681. [Google Scholar] [CrossRef] [PubMed]
- Papendick, R.E.; Munson, L.; O’Brien, T.D.; Johnson, K.H. Systemic AA amyloidosis in captive cheetahs (Acinonyx jubatus). Vet. Pathol. 1997, 34, 549–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terrell, S.P.; Fontenot, D.K.; Miller, M.A.; Weber, M.A. Chylous ascites in a cheetah (Acinonyx jubatus) with venoocclusive liver disease. J. Zoo Wildl. Med. 2003, 34, 380–384. [Google Scholar] [CrossRef]
- Benelli, M.; Franceschini, G.M.; Magi, A.; Romagnoli, D.; Biagioni, C.; Migliaccio, I.; Malorn, L.; Demichelis, F. Charting differentially methylated regions in cancer with Rocker-meth. Commun. Biol. 2021, 4, 1249. [Google Scholar] [CrossRef]
- Blanco, F.C.; Soria, M.A.; Klepp, L.I.; Bigi, F. ERAP1 and PDE8A are downregulated in cattle protected against bovine tuberculosis. Microbiol. Physiol. 2017, 27, 237–245. [Google Scholar]
- Ye, B.; Liu, B.; Hao, L.; Zhu, X.; Yang, L.; Wang, S.; Xia, P.; Du, Y.; Meng, S.; Huang, G.; et al. Klf4 glutamylation is required for cell reprogramming and early embryonic development in mice. Nat. Commun. 2018, 9, 1261. [Google Scholar] [CrossRef]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef]
- Ludwig, C.; Dehnhard, M.; Pribbenow, S.; Silinski-Mehr, S.; Hofer, H.; Wachter, B. Asymmetric reproductive aging in cheetahs (Acinonyx jubatus) females in European zoos. JZAR 2019, 7, 87–93. [Google Scholar]
- Ahn, J.; Heo, S.; Lee, J.; Bang, D. Introduction to Single-cell DNA methylation profiling methods. Biomolecules 2021, 11, 1013. [Google Scholar] [CrossRef]
- Szyf, M. The early life social environment and DNA methylation: DNA methylation mediating the long-term impact of social environments early in life. Epigenetics 2011, 6, 971–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massart, R.; Suderman, M.J.; Nemoda, Z.; Sutti, S.; Ruggiero, A.M.; Dettmer, A.M.; Suomi, S.J.; Szyf, M. The signature of maternal social rank in placenta deoxyribonucleic acid methylation profiles in rhesus monkeys. Child Dev. 2016, 88, 900–918. [Google Scholar] [CrossRef]
- Guerrero, T.P.; Fickel, J.; Benhaiem, S.; Weyrich, A. Epigenomics and gene regulation in mammalian social systems. Curr. Zool. 2020, 66, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Ecker, S.; Beck, S. Epigenetic variation taking center stage in immunological research. Epigenomics 2017, 9, 375–378. [Google Scholar] [CrossRef] [Green Version]
- Thompson, M.J.; von Holdt, B.; Horvath, S.; Pellegrini, M. An epigenetic aging clock for dogs and wolves. Aging 2017, 9, 1055–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, J.A.; Johnston, R.A.; Lea Amanda, J.; Campos, F.A.; Voyles, T.N.; Akinyi, M.Y.; Alberts, S.C.; Archie, E.A.; Tung, J. High social status males experience accelerated epigenetic aging in wild baboons. eLife 2021, 10, e66128. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Animal_ID | Origin | Age | Clinical Status | Reproductive State | Relatedness | Date of Sampling (dd.mm.yyyy) | Batch |
|---|---|---|---|---|---|---|---|
| F_1 | Namibia | 3.5–7.0 y | clinically healthy | lactating mother with 4 cubs (21 days old) | unknown | 28.04.2016 | 1 |
| F_2 | Namibia | 3.5–7.0 y | clinically healthy | in oestrus | unknown | 11.05.2013 | 1 |
| F_3 | Namibia | 3.5–7.0 y | clinically healthy | mother with 3 cubs (5–6 months old) | unknown | 09.06.2012 | 1 |
| F_4 | Namibia | 3.5–7.0 y | clinically healthy | not breeding | unknown | 14.10.2011 | 2 |
| F_5 | Namibia | 2.0–3.5 y | clinically healthy | mother with 3 cubs (8–9 months old) | unknown | 24.07.2012 | 1 |
| F_6 | Namibia | 2.0–3.5 y | clinically healthy | in oestrus | unknown | 16.10.2014 | 1 |
| F_7 | Namibia | 2.0–3.5 y | clinically healthy | not breeding | unknown | 13.12.2011 | 2 |
| F_8 | Namibia | 13–23 m | clinically healthy | not breeding | unknown | 16.04.2014 | 2 |
| C_1 | EEP, Zoo A | 11 y | clinically healthy | not breeding | aunt of C_4 and C_5, great-aunt of C_7 | 23.04.2019 | 1 |
| C_2 | EEP, Zoo B | 10 y 1 m | pendulous abdomen and emesis | not breeding, but bred successfully previously | mother of C_6 | 01.08.2019 | 2 |
| C_3 | EEP, Zoo C | 9 y 9 m | arthrosis, euthanized | not breeding, but bred successfully previously | unrelated up to 3rd degree to the other females | 24.09.2019 | 2 |
| C_4 | EEP, Zoo C | 7 y 4 m | chronic kidney disease, euthanized | not breeding | niece of C_1 and cousin of C_5 | 24.09.2019 | 2 |
| C_5 | EEP, Zoo D | 3 y 9 m | clinically healthy | in oestrus | niece of C_1, aunt of C_7 and cousin of C_4 | 10.12.2018 | 1 |
| C_6 | EEP, Zoo B | 7 m | degenerated cerebrum, euthanized | immature | daughter of C_2 | 19.12.2018 | 1 |
| C_7 | EEP, Zoo E | 6 m | severe ataxia, euthanized | immature | niece of C_5, great-niece of C_1 | 11.07.2018 | 1 |
| Female Groups | N | Average Raw Single End Reads | Read Length [bp] | Average %GC | Clean Single End Reads | Average Mapping Rate [%] |
|---|---|---|---|---|---|---|
| Free-ranging (F) | 8 | 31,435,731 | 20–76 | 52.5 | 30,972,291 | 96.33 |
| Human care (C) | 7 | 41,310,770 | 20–76 | 52.7 | 40,927,482 | 96.08 |
| Number of Differentially Methylated | DMRs Overlapping with Annotated Genomic Regions | ||||
|---|---|---|---|---|---|
| F vs. C | Windows | Regions (DMRs) | Intergenic | Introns | Promoters |
| Hypermethylated | 50 | 22 | 13 | 8 | 1 |
| Hypomethylated | 35 | 23 | 17 | 6 | 0 |
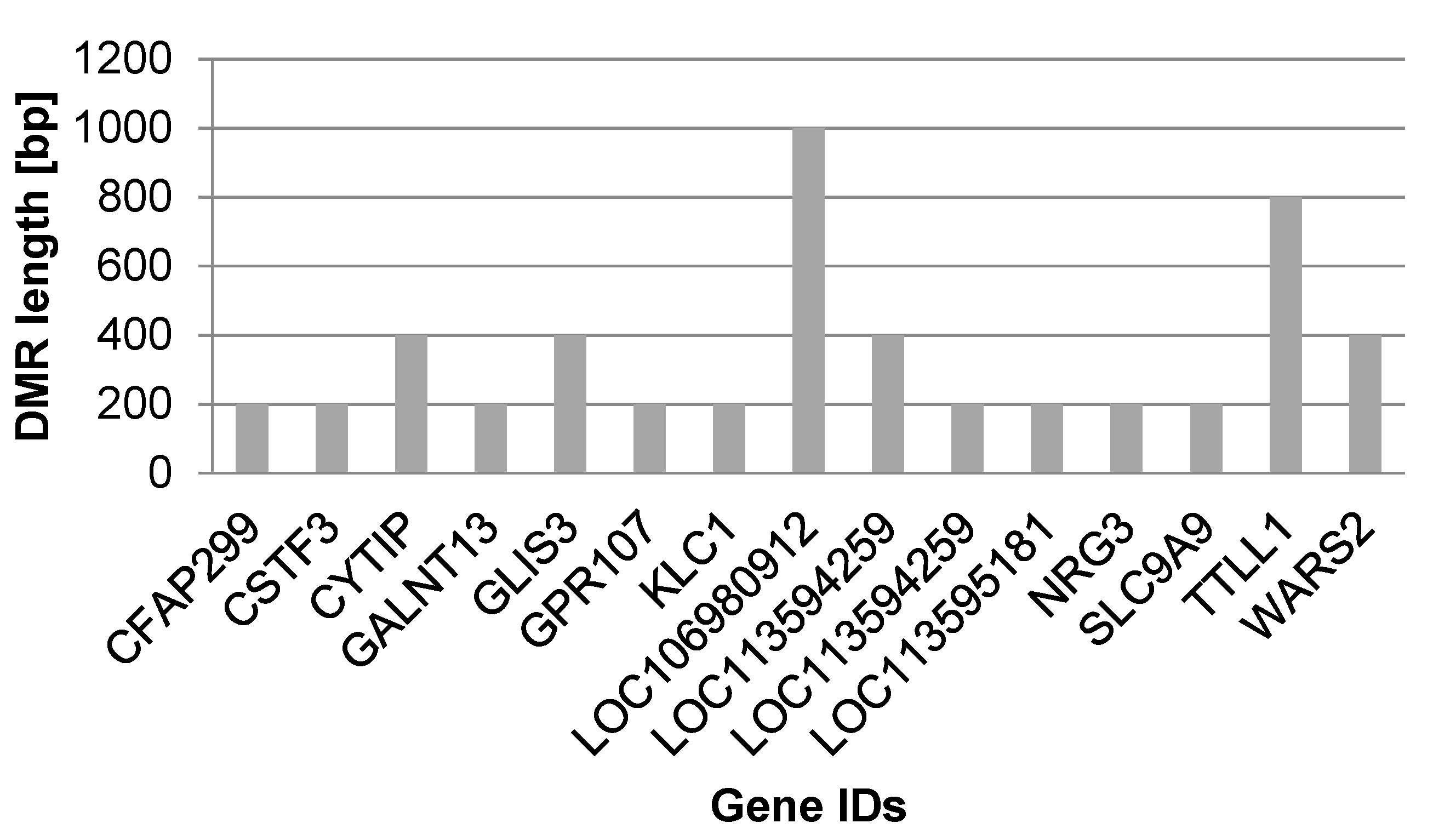
| Gene ID | Gene Name | Direction of Methylation in Captive Animals | Annotation | DMR Length [bp] |
|---|---|---|---|---|
| CFAP299 | Cilia and flagella associated protein 299 | hypo | intron | 200 |
| CSTF3 | Cleavage stimulation factor, 3′ pre-RNA, subunit 3, 77 kDa | hyper | intron | 200 |
| CYTIP | Cytohesin-interacting protein isoform ×1; Cytohesin 1 interacting protein | hyper | intron | 400 |
| GALNT13c | Polypeptide n-acetylgalactosaminyltransferase 13 isoform ×3; Polypeptide N-acetylgalactosaminyltransferase 13 | hyper | intron | 200 |
| GLIS3 a | Zinc finger protein glis3 isoform ×1; Uncharacterized protein; GLIS family zinc finger 3 | hyper | intron | 400 |
| GPR107 | G protein-coupled receptor 107 | hypo | intron | 200 |
| KLC1 a | Kinesin light chain 1 isoform ×1; Uncharacterized protein; Kinesin light chain 1 | hyper | promoter | 200 |
| LOC106980912 | Zinc finger protein 708-like isoform ×1 | hyper | intron | 1000 |
| LOC113594259 | uncharacterized LOC113594259 | hypo | intron | 400 |
| LOC113594259 | uncharacterized LOC113594259 | hypo | intron | 200 |
| LOC113595181 | uncharacterized LOC113595181, ncRNA | hyper | intron | 200 |
| NRG3 | Pro-neuregulin-3, membrane-bound isoform isoform ×5; Uncharacterized protein; Neuregulin 3 | hypo | intron | 200 |
| SLC9A9c | Solute carrier family 9 (sodium/hydrogen exchanger), member 9; Belongs to the monovalent cation:proton antiporter 1 (CPA1) transporter (TC 2.A.36) family | hyper | intron | 200 |
| TTLL1 a,b | Low quality protein: probable tubulin polyglutamylase ttll1; Tubulin tyrosine ligase-like family, member 1 | hyper | intron 1 | 800 |
| WARS2b | Tryptophanyl tRNA synthetase 2, mitochondrial; Belongs to the class-I aminoacyl-tRNA synthetase family | hypo | intron | 400 |
| Intergenic DMR | Number of DMRs | Mean DMR Length | SD DMR Length | Minimum Length | Maximum Length |
|---|---|---|---|---|---|
| Hypermethylated | 13 | 492.31 | 499.94 | 200.00 | 2000.00 |
| Hypomethylated | 17 | 282.35 | 138.20 | 200.00 | 600.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weyrich, A.; Guerrero-Altamirano, T.P.; Yasar, S.; Czirják, G.Á.; Wachter, B.; Fickel, J. First Steps towards the Development of Epigenetic Biomarkers in Female Cheetahs (Acinonyx jubatus). Life 2022, 12, 920. https://doi.org/10.3390/life12060920
Weyrich A, Guerrero-Altamirano TP, Yasar S, Czirják GÁ, Wachter B, Fickel J. First Steps towards the Development of Epigenetic Biomarkers in Female Cheetahs (Acinonyx jubatus). Life. 2022; 12(6):920. https://doi.org/10.3390/life12060920
Chicago/Turabian StyleWeyrich, Alexandra, Tania P. Guerrero-Altamirano, Selma Yasar, Gábor Á. Czirják, Bettina Wachter, and Jörns Fickel. 2022. "First Steps towards the Development of Epigenetic Biomarkers in Female Cheetahs (Acinonyx jubatus)" Life 12, no. 6: 920. https://doi.org/10.3390/life12060920
APA StyleWeyrich, A., Guerrero-Altamirano, T. P., Yasar, S., Czirják, G. Á., Wachter, B., & Fickel, J. (2022). First Steps towards the Development of Epigenetic Biomarkers in Female Cheetahs (Acinonyx jubatus). Life, 12(6), 920. https://doi.org/10.3390/life12060920