
Splicing Modulation as a Promising Therapeutic Strategy for Lysosomal Storage Disorders: The Mucopolysaccharidoses Example

 ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Splicing: How It Works and How It Can Be Modulated
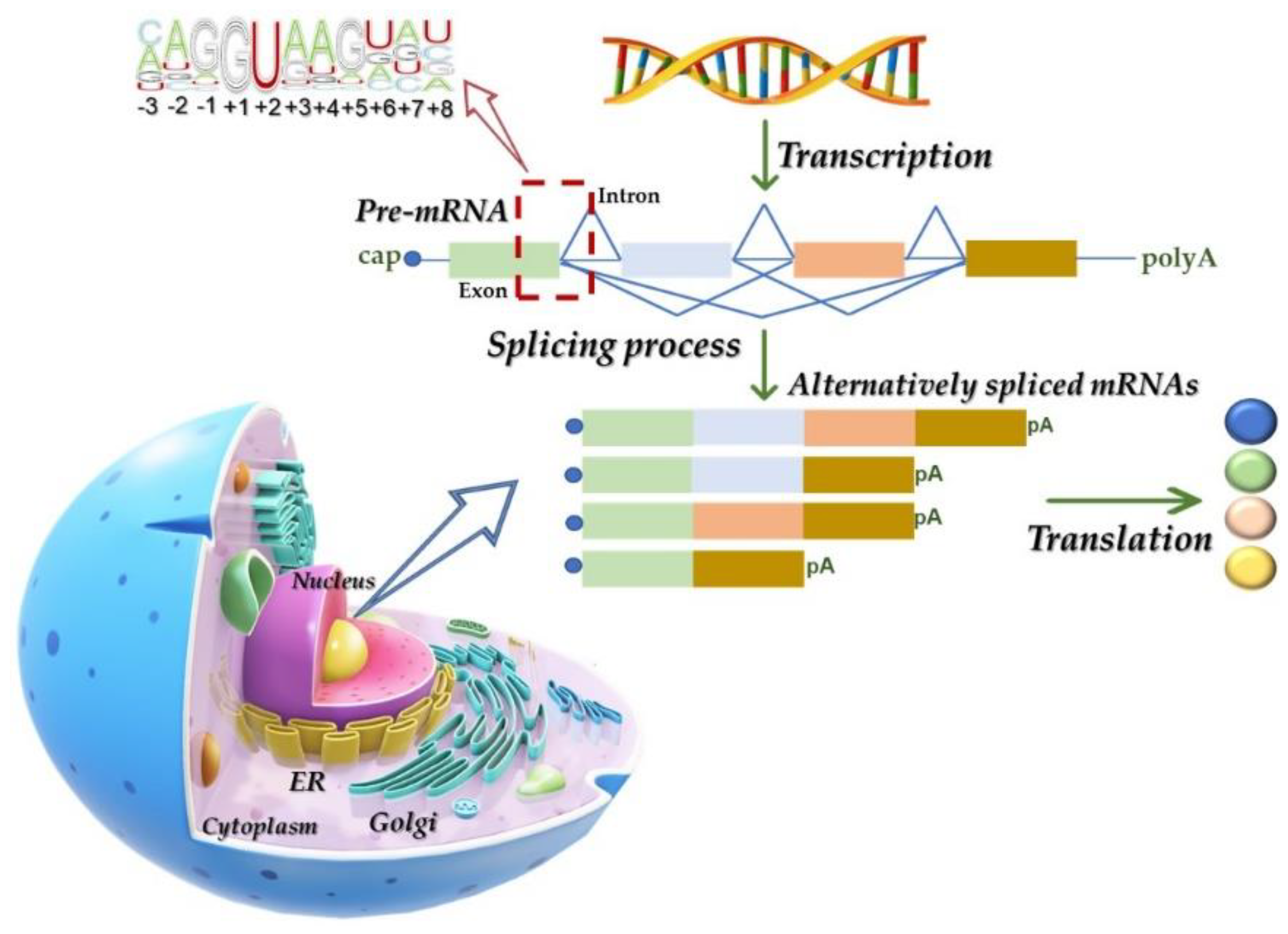
2.1. The Splicing Process: Machinery and Mechanisms
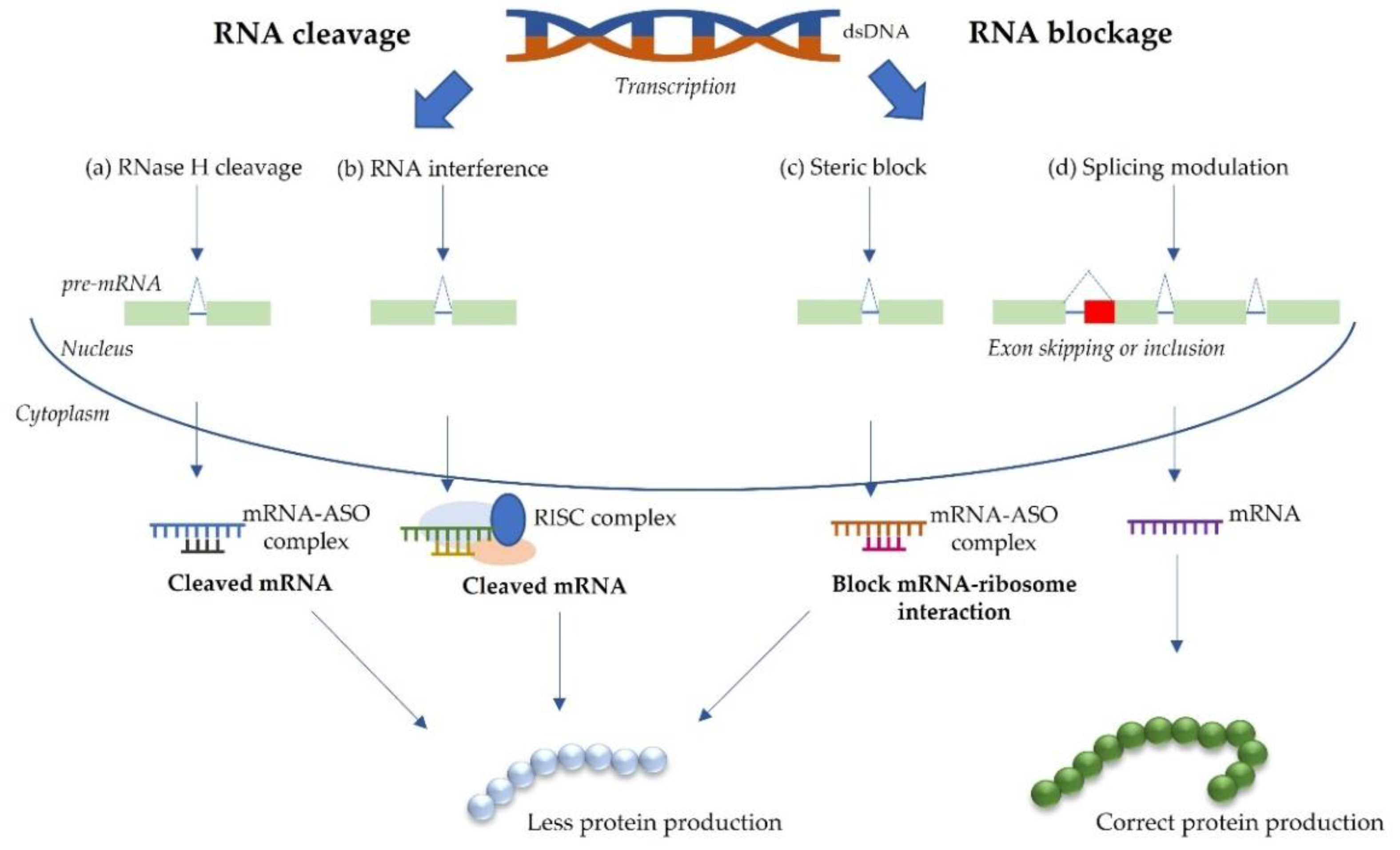
2.2. RNA-Based Approaches for Splice Modulation
2.3. Hurdles
3. Treatment Strategies for LSD Patients: MPSs in the Spotlight
3.1. Lysosomal Storage Diseases
3.2. Mucopolysaccharidoses
4. RNA-Based Therapeutic Approaches for MPS Mutations
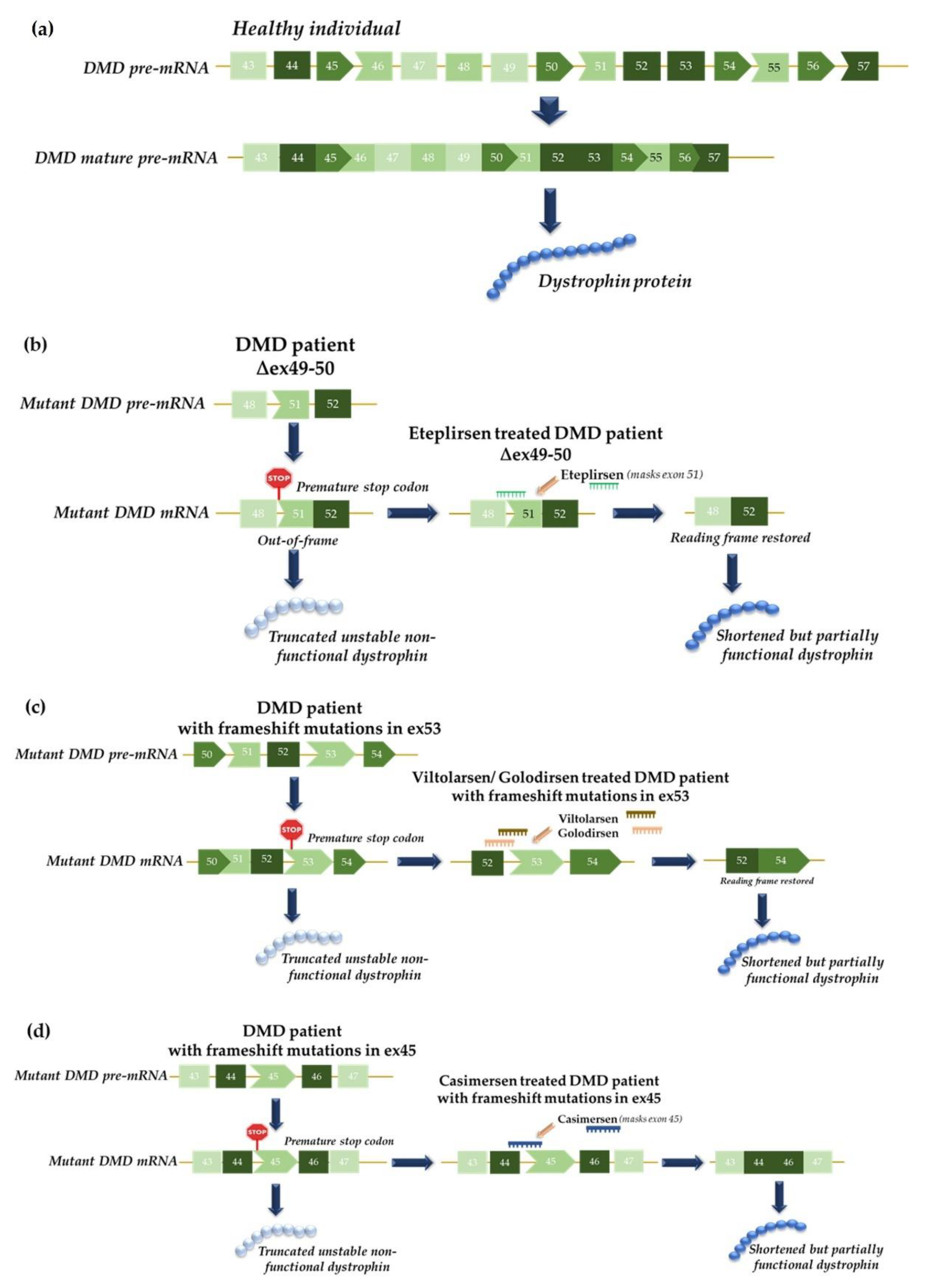
4.1. Functional Studies of Splicing Mutations and Development of Therapeutic Approaches Using Antisense Oligonucleotides: The MPS II Example
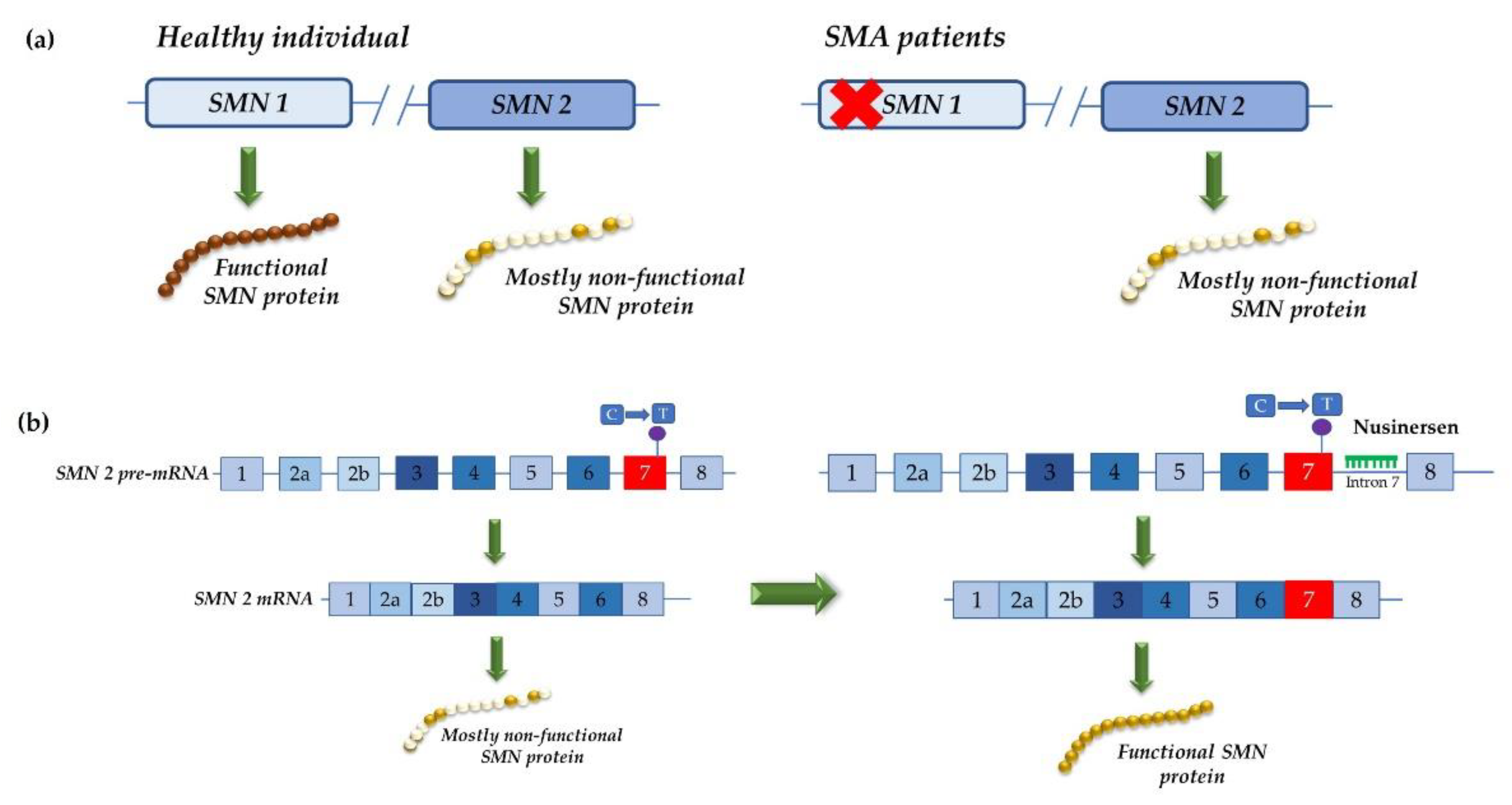
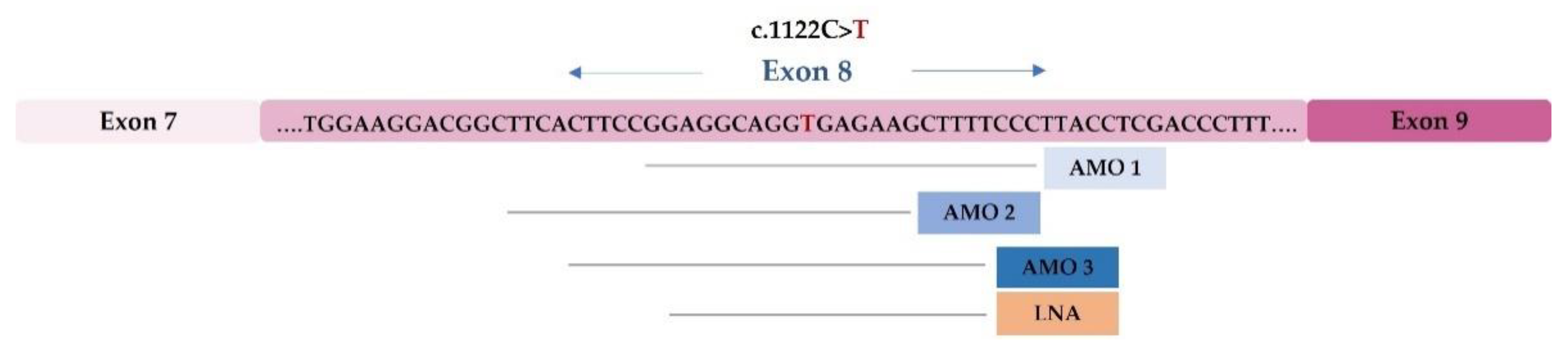
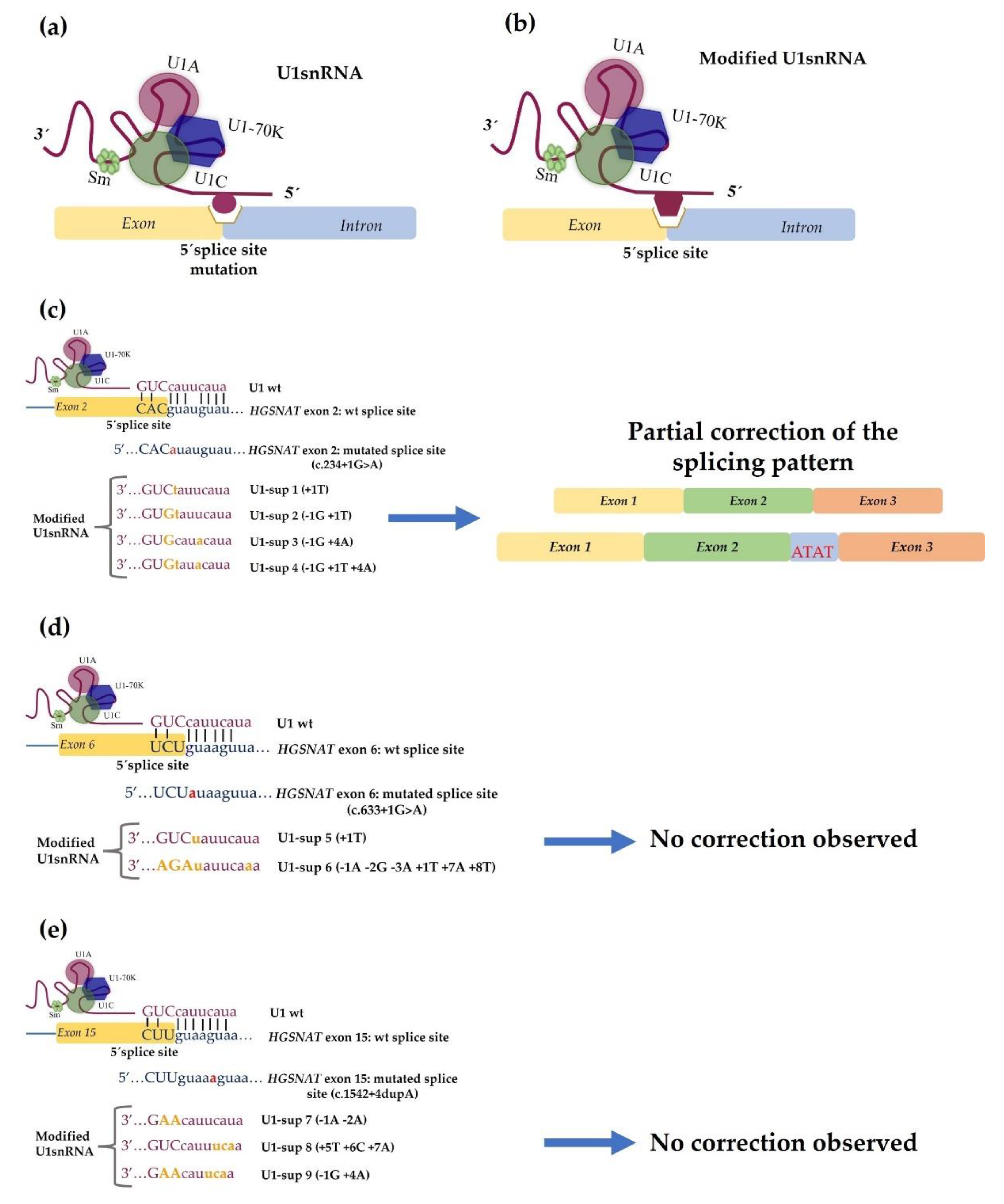
4.2. Development of Therapeutic Approaches Using Modified U1 snRNA Vectors: The MPS IIIC Example
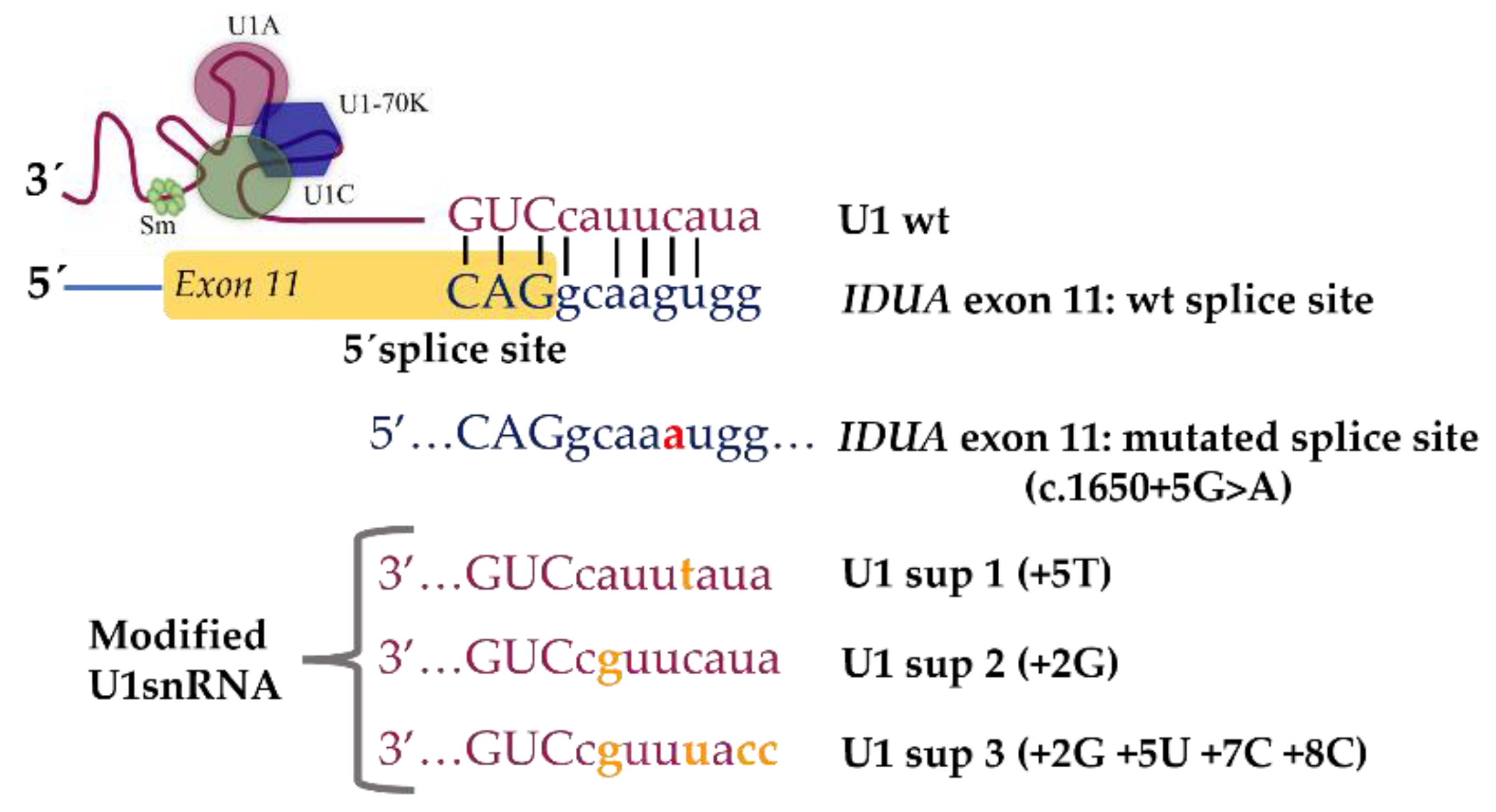
4.3. Identification and Characterization of Novel Splicing Defects and Assessment of Their Amenability for Splicing Correction Therapeutic Approaches: The MPS I Example
5. Challenges for the Development of Splice Modulation Approaches for MPSs
5.1. Existence of Disease-Relevant Models
5.2. Design and Development of Effective Delivery Strategies
5.3. Accurate Characterization of Disease-Causing Variants at mRNA Level
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Lieberman, J. Tapping the RNA world for therapeutics. Nat. Struct. Mol. Biol. 2018, 25, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Dhuri, K.; Bechtold, C.; Quijano, E.; Pham, H.; Gupta, A.; Vikram, A.; Bahal, R. Antisense Oligonucleotides: An Emerging Area in Drug Discovery and Development. J. Clin. Med. 2020, 9, 2004. [Google Scholar] [CrossRef]
- Gagliardi, M.; Ashizawa, A.T. The Challenges and Strategies of Antisense Oligonucleotide Drug Delivery. Biomedicines 2021, 9, 433. [Google Scholar] [CrossRef] [PubMed]
- Anna, A.; Monika, G. Splicing mutations in human genetic disorders: Examples, detection, and confirmation. J. Appl. Genet. 2018, 59, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Pereira, C.; San Millán-Tejado, B.; Gallardo-Gómez, M.; Pérez-Márquez, T.; Alves-Villar, M.; Melcón-Crespo, C.; Fernández-Martín, J.; Ortolano, S. Therapeutic Approaches in Lysosomal Storage Diseases. Biomolecules 2021, 11, 1775. [Google Scholar] [CrossRef] [PubMed]
- van Gool, R.; Tucker-Bartley, A.; Yang, E.; Todd, N.; Guenther, F.; Goodlett, B.; Al-Hertani, W.; Bodamer, O.A.; Upadhyay, J. Targeting neurological abnormalities in lysosomal storage diseases. Trends Pharmacol. Sci. 2021, in press. [Google Scholar] [CrossRef]
- Wraith, J.E.; Beck, M. Clinical Aspects and Clinical Diagnosis. In Lysosomal Storage Disorders—A Practial Guide; Metha, A., Winchester, B.E., Eds.; John Wiley and Sons: Hoboken, NJ, USA, 2012; pp. 13–19. [Google Scholar]
- Giugliani, R. The Mucopolysaccharidoses. In Lysosomal Storage Disorders—A Practical Guide; Mehta, A.B., Ed.; Wiley-Blackwell: Oxford, UK, 2013; p. 208. [Google Scholar]
- Dardis, A.; Buratti, E. Impact, Characterization, and Rescue of Pre-mRNA Splicing Mutations in Lysosomal Storage Disorders. Genes 2018, 9, 73. [Google Scholar] [CrossRef]
- McBride, K.L.; Flanigan, K.M. Update in the Mucopolysaccharidoses. Semin. Pediatr. Neurol. 2021, 37, 100874. [Google Scholar] [CrossRef]
- Berget, S.M.; Moore, C.; Sharp, P.A. Spliced segments at the 5’ terminus of adenovirus 2 late mRNA. Proc. Natl. Acad. Sci. USA 1977, 74, 3171–3175. [Google Scholar] [CrossRef]
- Chow, L.T.; Gelinas, R.E.; Broker, T.R.; Roberts, R.J. An amazing sequence arrangement at the 5’ ends of adenovirus 2 messenger RNA. Cell 1977, 12, 1–8. [Google Scholar] [CrossRef]
- Chen, M.; Manley, J.L. Mechanisms of alternative splicing regulation: Insights from molecular and genomics approaches. Nat. Rev. Mol. Cell Biol. 2009, 10, 741–754. [Google Scholar] [CrossRef] [PubMed]
- De Conti, L.; Baralle, M.; Buratti, E. Exon and intron definition in pre-mRNA splicing. Wiley Interdiscip Rev. RNA 2013, 4, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Matera, A.G.; Wang, Z. A day in the life of the spliceosome. Nat. Rev. Mol. Cell Biol. 2014, 15, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Will, C.L.; Luhrmann, R. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 2011, 3, a003707. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. Mechanistic insights into precursor messenger RNA splicing by the spliceosome. Nat. Rev. Mol. Cell Biol. 2017, 18, 655–670. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Wan, R.; Shi, Y. Molecular Mechanisms of pre-mRNA Splicing through Structural Biology of the Spliceosome. Cold Spring Harb. Perspect. Biol. 2019, 11, a032409. [Google Scholar] [CrossRef] [PubMed]
- Tazi, J.; Bakkour, N.; Stamm, S. Alternative splicing and disease. Biochim. Biophys. Acta 2009, 1792, 14–26. [Google Scholar] [CrossRef]
- Sune-Pou, M.; Limeres, M.J.; Moreno-Castro, C.; Hernandez-Munain, C.; Sune-Negre, J.M.; Cuestas, M.L.; Sune, C. Innovative Therapeutic and Delivery Approaches Using Nanotechnology to Correct Splicing Defects Underlying Disease. Front. Genet. 2020, 11, 731. [Google Scholar] [CrossRef]
- Kim, H.K.; Pham, M.H.C.; Ko, K.S.; Rhee, B.D.; Han, J. Alternative splicing isoforms in health and disease. Pflugers Arch. 2018, 470, 995–1016. [Google Scholar] [CrossRef]
- Wang, Z.; Burge, C.B. Splicing regulation: From a parts list of regulatory elements to an integrated splicing code. RNA 2008, 14, 802–813. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Huang, B.O.; Xu, Y.M.; Li, J.; Huang, L.F.; Lin, J.; Zhang, J.; Min, Q.H.; Yang, W.M.; et al. Mechanism of alternative splicing and its regulation. Biomed. Rep. 2015, 3, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Dvinge, H. Regulation of alternative mRNA splicing: Old players and new perspectives. FEBS Lett. 2018, 592, 2987–3006. [Google Scholar] [CrossRef] [PubMed]
- Arechavala-Gomeza, V.; Khoo, B.; Aartsma-Rus, A. Splicing modulation therapy in the treatment of genetic diseases. Appl. Clin. Genet. 2014, 7, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, M.L.; Zamecnik, P.C. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef] [PubMed]
- Syed, Y.Y. Eteplirsen: First Global Approval. Drugs 2016, 76, 1699–1704. [Google Scholar] [CrossRef] [PubMed]
- Young, C.S.; Pyle, A.D. Exon Skipping Therapy. Cell 2016, 167, 1144. [Google Scholar] [CrossRef] [PubMed]
- Heo, Y.A. Golodirsen: First Approval. Drugs 2020, 80, 329–333. [Google Scholar] [CrossRef]
- Dhillon, S. Viltolarsen: First Approval. Drugs 2020, 80, 1027–1031. [Google Scholar] [CrossRef]
- Shirley, M. Casimersen: First Approval. Drugs 2021, 81, 875–879. [Google Scholar] [CrossRef]
- Finkel, R.S.; Chiriboga, C.A.; Vajsar, J.; Day, J.W.; Montes, J.; De Vivo, D.C.; Yamashita, M.; Rigo, F.; Hung, G.; Schneider, E.; et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: A phase 2, open-label, dose-escalation study. Lancet 2016, 388, 3017–3026. [Google Scholar] [CrossRef]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef] [PubMed]
- Monaco, A.P.; Bertelson, C.J.; Liechti-Gallati, S.; Moser, H.; Kunkel, L.M. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 1988, 2, 90–95. [Google Scholar] [CrossRef]
- Kole, R.; Krieg, A.M. Exon skipping therapy for Duchenne muscular dystrophy. Adv. Drug Deliv. Rev. 2015, 87, 104–107. [Google Scholar] [CrossRef]
- Mendell, J.R.; Rodino-Klapac, L.R.; Sahenk, Z.; Roush, K.; Bird, L.; Lowes, L.P.; Alfano, L.; Gomez, A.M.; Lewis, S.; Kota, J.; et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann. Neurol. 2013, 74, 637–647. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Krieg, A.M. FDA Approves Eteplirsen for Duchenne Muscular Dystrophy: The Next Chapter in the Eteplirsen Saga. Nucleic Acid Ther. 2017, 27, 1–3. [Google Scholar] [CrossRef]
- FDA. Grants Accelerated Approval to First Targeted Treatment for Rare Duchenne Muscular Dystrophy Mutation. Available online: https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-first-targeted-treatment-rare-duchenne-muscular-dystrophy-mutation (accessed on 15 March 2022).
- FDA. Approves Targeted Treatment for Rare Duchenne Muscular Dystrophy Mutation. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-targeted-treatment-rare-duchenne-muscular-dystrophy-mutation (accessed on 15 March 2022).
- Dzierlega, K.; Yokota, T. Optimization of antisense-mediated exon skipping for Duchenne muscular dystrophy. Gene Ther. 2020, 27, 407–416. [Google Scholar] [CrossRef]
- Wilton, S.D.; Fletcher, S. Splice modification to restore functional dystrophin synthesis in Duchenne muscular dystrophy. Curr. Pharm. Des. 2010, 16, 988–1001. [Google Scholar] [CrossRef]
- Hua, Y.; Sahashi, K.; Hung, G.; Rigo, F.; Passini, M.A.; Bennett, C.F.; Krainer, A.R. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010, 24, 1634–1644. [Google Scholar] [CrossRef]
- Faravelli, I.; Nizzardo, M.; Comi, G.P.; Corti, S. Spinal muscular atrophy--recent therapeutic advances for an old challenge. Nat. Rev. Neurol. 2015, 11, 351–359. [Google Scholar] [CrossRef]
- Verma, A. Recent Advances in Antisense Oligonucleotide Therapy in Genetic Neuromuscular Diseases. Ann. Indian Acad. Neurol. 2018, 21, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Cartegni, L.; Krainer, A.R. Correction of disease-associated exon skipping by synthetic exon-specific activators. Nat. Struct. Biol. 2003, 10, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Sahashi, K.; Rigo, F.; Hung, G.; Horev, G.; Bennett, C.F.; Krainer, A.R. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 2011, 478, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A. FDA Approval of Nusinersen for Spinal Muscular Atrophy Makes 2016 the Year of Splice Modulating Oligonucleotides. Nucleic Acid Ther. 2017, 27, 67–69. [Google Scholar] [CrossRef]
- Chaytow, H.; Faller, K.M.E.; Huang, Y.T.; Gillingwater, T.H. Spinal muscular atrophy: From approved therapies to future therapeutic targets for personalized medicine. Cell Rep. Med. 2021, 2, 100346. [Google Scholar] [CrossRef]
- Balestra, D.; Scalet, D.; Ferrarese, M.; Lombardi, S.; Ziliotto, N.; Croes, C.C.; Petersen, N.; Bosma, P.; Riccardi, F.; Pagani, F.; et al. A Compensatory U1snRNA Partially Rescues FAH Splicing and Protein Expression in a Splicing-Defective Mouse Model of Tyrosinemia Type I. Int. J. Mol. Sci. 2020, 21, 2136. [Google Scholar] [CrossRef]
- Breuel, S.; Vorm, M.; Brauer, A.U.; Owczarek-Lipska, M.; Neidhardt, J. Combining Engineered U1 snRNA and Antisense Oligonucleotides to Improve the Treatment of a BBS1 Splice Site Mutation. Mol. Ther. Nucleic Acids 2019, 18, 123–130. [Google Scholar] [CrossRef]
- Daguenet, E.; Dujardin, G.; Valcarcel, J. The pathogenicity of splicing defects: Mechanistic insights into pre-mRNA processing inform novel therapeutic approaches. EMBO Rep. 2015, 16, 1640–1655. [Google Scholar] [CrossRef]
- Damase, T.R.; Sukhovershin, R.; Boada, C.; Taraballi, F.; Pettigrew, R.I.; Cooke, J.P. The Limitless Future of RNA Therapeutics. Front. Bioeng. Biotechnol. 2021, 9, 628137. [Google Scholar] [CrossRef]
- Arechavala-Gomeza, V.; Garanto, A. Antisense RNA Therapeutics: A Brief Overview. Methods Mol. Biol. 2022, 2434, 33–49. [Google Scholar] [CrossRef]
- Vázquez-Domínguez, I.; Garanto, A. Considerations for Generating Humanized Mouse Models to Test Efficacy of Antisense Oligonucleotides. Methods Mol. Biol. 2022, 2434, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Abril, J.F.; Castelo, R.; Guigó, R. Comparison of splice sites in mammals and chicken. Genome Res. 2005, 15, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, C.; Desviat, L.R.; Smedsrod, B.; Pietri-Rouxel, F.; Denti, M.A.; Disterer, P.; Lorain, S.; Nogales-Gadea, G.; Sardone, V.; Anwar, R.; et al. Delivery is key: Lessons learnt from developing splice-switching antisense therapies. EMBO Mol. Med. 2017, 9, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Schoch, K.M.; Miller, T.M. Antisense Oligonucleotides: Translation from Mouse Models to Human Neurodegenerative Diseases. Neuron 2017, 94, 1056–1070. [Google Scholar] [CrossRef]
- Heon-Roberts, R.; Nguyen, A.L.A.; Pshezhetsky, A.V. Molecular Bases of Neurodegeneration and Cognitive Decline, the Major Burden of Sanfilippo Disease. J. Clin. Med. 2020, 9, 344. [Google Scholar] [CrossRef]
- Anthony, K. RNA-based therapeutics for neurological diseases. RNA Biol. 2022, 19, 176–190. [Google Scholar] [CrossRef]
- Hammond, S.M.; Aartsma-Rus, A.; Alves, S.; Borgos, S.E.; Buijsen, R.A.M.; Collin, R.W.J.; Covello, G.; Denti, M.A.; Desviat, L.R.; Echevarría, L.; et al. Delivery of oligonucleotide-based therapeutics: Challenges and opportunities. EMBO Mol. Med. 2021, 13, e13243. [Google Scholar] [CrossRef]
- Fuller, M.; Meikle, P.J.; Hopwood, J.J. Epidemiology of lysosomal storage disorders: An overview. In Fabry Disease: Perspectives from 5 Years of FOS; Mehta, A., Beck, M., Sunder-Plassmann, G., Eds.; Oxford PharmaGenesis: Oxford, UK, 2006. [Google Scholar]
- Fernández-Marmiesse, A.; Morey, M.; Pineda, M.; Eiris, J.; Couce, M.L.; Castro-Gago, M.; Fraga, J.M.; Lacerda, L.; Gouveia, S.; Pérez-Poyato, M.S.; et al. Assessment of a targeted resequencing assay as a support tool in the diagnosis of lysosomal storage disorders. Orphanet J. Rare Dis. 2014, 9, 59. [Google Scholar] [CrossRef]
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Primers 2018, 4, 27. [Google Scholar] [CrossRef]
- Fecarotta, S.; Gasperini, S.; Parenti, G. New treatments for the mucopolysaccharidoses: From pathophysiology to therapy. Ital. J. Pediatr. 2018, 44, 124. [Google Scholar] [CrossRef]
- Bellettato, C.M.; Scarpa, M. Pathophysiology of neuropathic lysosomal storage disorders. J. Inherit. Metab. Dis. 2010, 33, 347–362. [Google Scholar] [CrossRef] [PubMed]
- Mokhtariye, A.; Hagh-Nazari, L.; Varasteh, A.R.; Keyfi, F. Diagnostic methods for Lysosomal Storage Disease. Rep. Biochem. Mol. Biol. 2019, 7, 119–128. [Google Scholar] [PubMed]
- Zanetti, A.; D’Avanzo, F.; Bertoldi, L.; Zampieri, G.; Feltrin, E.; De Pascale, F.; Rampazzo, A.; Forzan, M.; Valle, G.; Tomanin, R. Setup and Validation of a Targeted Next-Generation Sequencing Approach for the Diagnosis of Lysosomal Storage Disorders. J. Mol. Diagn. 2020, 22, 488–502. [Google Scholar] [CrossRef] [PubMed]
- Viana, G.M.; Priestman, D.A.; Platt, F.M.; Khan, S.; Tomatsu, S.; Pshezhetsky, A.V. Brain Pathology in Mucopolysaccharidoses (MPS) Patients with Neurological Forms. J. Clin. Med. 2020, 9, 396. [Google Scholar] [CrossRef]
- Coutinho, M.F.; Lacerda, L.; Alves, S. Glycosaminoglycan storage disorders: A review. Biochem. Res. Int. 2012, 2012, 471325. [Google Scholar] [CrossRef]
- Rapoport, D.M.; Mitchell, J.J. Pathophysiology, evaluation, and management of sleep disorders in the mucopolysaccharidoses. Mol. Genet. Metab. 2017, 122S, 49–54. [Google Scholar] [CrossRef]
- Kubaski, F.; Osago, H.; Mason, R.W.; Yamaguchi, S.; Kobayashi, H.; Tsuchiya, M.; Orii, T.; Tomatsu, S. Glycosaminoglycans detection methods: Applications of mass spectrometry. Mol. Genet. Metab. 2017, 120, 67–77. [Google Scholar] [CrossRef]
- Peters, H.; Ellaway, C.; Nicholls, K.; Reardon, K.; Szer, J. Treatable lysosomal storage diseases in the advent of disease-specific therapy. Intern. Med. J. 2020, 50 (Suppl. 4), 5–27. [Google Scholar] [CrossRef]
- Filocamo, M.; Tomanin, R.; Bertola, F.; Morrone, A. Biochemical and molecular analysis in mucopolysaccharidoses: What a paediatrician must know. Ital. J. Pediatr. 2018, 44, 129. [Google Scholar] [CrossRef]
- HGMD. Available online: https://my.qiagendigitalinsights.com/ (accessed on 14 March 2022).
- Giugliani, R.; Muschol, N.; Keenan, H.A.; Dant, M.; Muenzer, J. Improvement in time to treatment, but not time to diagnosis, in patients with mucopolysaccharidosis type I. Arch. Dis. Child 2020, 106, 674–679. [Google Scholar] [CrossRef]
- Parini, R.; Deodato, F.; Di Rocco, M.; Lanino, E.; Locatelli, F.; Messina, C.; Rovelli, A.; Scarpa, M. Open issues in Mucopolysaccharidosis type I-Hurler. Orphanet J. Rare Dis. 2017, 12, 112. [Google Scholar] [CrossRef] [PubMed]
- Kubaski, F.; de Oliveira Poswar, F.; Michelin-Tirelli, K.; Matte, U.D.S.; Horovitz, D.D.; Barth, A.L.; Baldo, G.; Vairo, F.; Giugliani, R. Mucopolysaccharidosis Type I. Diagnostics 2020, 10, 161. [Google Scholar] [CrossRef] [PubMed]
- Hashmi, M.S.G.V. Mucopolysaccharidosis Type II. Available online: https://www.ncbi.nlm.nih.gov/books/NBK560829/ (accessed on 15 March 2022).
- Pearse, Y.; Iacovino, M. A Cure for Sanfilippo Syndrome? A Summary of Current Therapeutic Approaches and their Promise. Med. Res. Arch. 2020, 8, 2045. [Google Scholar] [CrossRef] [PubMed]
- Valstar, M.J.; Ruijter, G.J.; van Diggelen, O.P.; Poorthuis, B.J.; Wijburg, F.A. Sanfilippo syndrome: A mini-review. J. Inherit. Metab. Dis. 2008, 31, 240–252. [Google Scholar] [CrossRef]
- Jones, S.A.; Breen, C.; Heap, F.; Rust, S.; de Ruijter, J.; Tump, E.; Marchal, J.P.; Pan, L.; Qiu, Y.; Chung, J.K.; et al. A phase 1/2 study of intrathecal heparan-N-sulfatase in patients with mucopolysaccharidosis IIIA. Mol. Genet. Metab. 2016, 118, 198–205. [Google Scholar] [CrossRef]
- Wijburg, F.A.; Whitley, C.B.; Muenzer, J.; Gasperini, S.; Del Toro, M.; Muschol, N.; Cleary, M.; Sevin, C.; Shapiro, E.; Bhargava, P.; et al. Intrathecal heparan-N-sulfatase in patients with Sanfilippo syndrome type A: A phase IIb randomized trial. Mol. Genet. Metab. 2019, 126, 121–130. [Google Scholar] [CrossRef]
- Whitley, C.B.; Vijay, S.; Yao, B.; Pineda, M.; Parker, G.J.M.; Rojas-Caro, S.; Zhang, X.; Dai, Y.; Cinar, A.; Bubb, G.; et al. Final results of the phase 1/2, open-label clinical study of intravenous recombinant human N-acetyl-α-d-glucosaminidase (SBC-103) in children with mucopolysaccharidosis IIIB. Mol. Genet. Metab. 2019, 126, 131–138. [Google Scholar] [CrossRef]
- Seker Yilmaz, B.; Davison, J.; Jones, S.A.; Baruteau, J. Novel therapies for mucopolysaccharidosis type III. J. Inherit. Metab. Dis. 2021, 44, 129–147. [Google Scholar] [CrossRef]
- Muenzer, J. Overview of the mucopolysaccharidoses. Rheumatology 2011, 50 (Suppl. 5), v4–v12. [Google Scholar] [CrossRef]
- Sawamoto, K.; Alvarez Gonzalez, J.V.; Piechnik, M.; Otero, F.J.; Couce, M.L.; Suzuki, Y.; Tomatsu, S. Mucopolysaccharidosis IVA: Diagnosis, Treatment, and Management. Int. J. Mol. Sci. 2020, 21, 1517. [Google Scholar] [CrossRef]
- Harmatz, P.; Shediac, R. Mucopolysaccharidosis VI: Pathophysiology, diagnosis and treatment. Front. Biosci. (Landmark Ed.) 2017, 22, 385–406. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.; Khan, S.; Stapleton, M.; Wang, J.; Chen, J.; Wynn, R.; Yabe, H.; Chinen, Y.; Boelens, J.J.; Mason, R.W.; et al. Hematopoietic Stem Cell Transplantation for Mucopolysaccharidoses: Past, Present, and Future. Biol. Blood Marrow Transplant. 2019, 25, e226–e246. [Google Scholar] [CrossRef] [PubMed]
- Montano, A.M.; Lock-Hock, N.; Steiner, R.D.; Graham, B.H.; Szlago, M.; Greenstein, R.; Pineda, M.; Gonzalez-Meneses, A.; Coker, M.; Bartholomew, D.; et al. Clinical course of sly syndrome (mucopolysaccharidosis type VII). J. Med. Genet. 2016, 53, 403–418. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Lin, J.; Leung, W.T.; Wang, L. A basic understanding of mucopolysaccharidosis: Incidence, clinical features, diagnosis, and management. Intractable Rare Dis. Res. 2020, 9, 1–9. [Google Scholar] [CrossRef]
- Qi, Y.; McKeever, K.; Taylor, J.; Haller, C.; Song, W.; Jones, S.A.; Shi, J. Pharmacokinetic and Pharmacodynamic Modeling to Optimize the Dose of Vestronidase Alfa, an Enzyme Replacement Therapy for Treatment of Patients with Mucopolysaccharidosis Type VII: Results from Three Trials. Clin. Pharm. 2019, 58, 673–683. [Google Scholar] [CrossRef]
- Sawamoto, K.; Stapleton, M.; Almeciga-Diaz, C.J.; Espejo-Mojica, A.J.; Losada, J.C.; Suarez, D.A.; Tomatsu, S. Therapeutic Options for Mucopolysaccharidoses: Current and Emerging Treatments. Drugs 2019, 79, 1103–1134. [Google Scholar] [CrossRef]
- Triggs-Raine, B.; Salo, T.J.; Zhang, H.; Wicklow, B.A.; Natowicz, M.R. Mutations in HYAL1, a member of a tandemly distributed multigene family encoding disparate hyaluronidase activities, cause a newly described lysosomal disorder, mucopolysaccharidosis IX. Proc. Natl. Acad. Sci. USA 1999, 96, 6296–6300. [Google Scholar] [CrossRef]
- Imundo, L.; Leduc, C.A.; Guha, S.; Brown, M.; Perino, G.; Gushulak, L.; Triggs-Raine, B.; Chung, W.K. A complete deficiency of Hyaluronoglucosaminidase 1 (HYAL1) presenting as familial juvenile idiopathic arthritis. J. Inherit. Metab. Dis. 2011, 34, 1013–1022. [Google Scholar] [CrossRef]
- Sato, Y.; Okuyama, T. Novel Enzyme Replacement Therapies for Neuropathic Mucopolysaccharidoses. Int. J. Mol. Sci. 2020, 21, 400. [Google Scholar] [CrossRef]
- Spahiu, L.; Behluli, E.; Peterlin, B.; Nefic, H.; Hadziselimovic, R.; Liehr, T.; Temaj, G. Mucopolysaccharidosis III: Molecular basis and treatment. Pediatr. Endocrinol. Diabetes Metab. 2021, 27, 201–208. [Google Scholar] [CrossRef]
- Matos, L.; Gonçalves, V.; Pinto, E.; Laranjeira, F.; Prata, M.J.; Jordan, P.; Desviat, L.R.; Pérez, B.; Alves, S. Functional analysis of splicing mutations in the IDS gene and the use of antisense oligonucleotides to exploit an alternative therapy for MPS II. Biochim. Biophys. Acta 2015, 1852, 2712–2721. [Google Scholar] [CrossRef] [PubMed]
- Matos, L.; Goncalves, V.; Pinto, E.; Laranjeira, F.; Prata, M.J.; Jordan, P.; Desviat, L.R.; Perez, B.; Alves, S. Data in support of a functional analysis of splicing mutations in the IDS gene and the use of antisense oligonucleotides to exploit an alternative therapy for MPS II. Data Brief 2015, 5, 810–817. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Brusius-Facchin, A.C.; Schwartz, I.V.; Zimmer, C.; Ribeiro, M.G.; Acosta, A.X.; Horovitz, D.; Monlleo, I.L.; Fontes, M.I.; Fett-Conte, A.; Sobrinho, R.P.; et al. Mucopolysaccharidosis type II: Identification of 30 novel mutations among Latin American patients. Mol. Genet. Metab. 2014, 111, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Soukarieh, O.; Gaildrat, P.; Hamieh, M.; Drouet, A.; Baert-Desurmont, S.; Frebourg, T.; Tosi, M.; Martins, A. Exonic Splicing Mutations Are More Prevalent than Currently Estimated and Can Be Predicted by Using In Silico Tools. PLoS Genet. 2016, 12, e1005756. [Google Scholar] [CrossRef]
- Matos, L.; Canals, I.; Dridi, L.; Choi, Y.; Prata, M.J.; Jordan, P.; Desviat, L.R.; Pérez, B.; Pshezhetsky, A.V.; Grinberg, D.; et al. Therapeutic strategies based on modified U1 snRNAs and chaperones for Sanfilippo C splicing mutations. Orphanet J. Rare Dis. 2014, 9, 180. [Google Scholar] [CrossRef]
- Raponi, M.; Baralle, D. Can donor splice site recognition occur without the involvement of U1 snRNP? Biochem. Soc. Trans. 2008, 36, 548–550. [Google Scholar] [CrossRef]
- Valadkhan, S.; Gunawardane, L.S. Role of small nuclear RNAs in eukaryotic gene expression. Essays Biochem. 2013, 54, 79–90. [Google Scholar] [CrossRef]
- Schmid, F.; Hiller, T.; Korner, G.; Glaus, E.; Berger, W.; Neidhardt, J. A gene therapeutic approach to correct splice defects with modified U1 and U6 snRNPs. Hum. Gene Ther. 2013, 24, 97–104. [Google Scholar] [CrossRef]
- Glaus, E.; Schmid, F.; Da Costa, R.; Berger, W.; Neidhardt, J. Gene therapeutic approach using mutation-adapted U1 snRNA to correct a RPGR splice defect in patient-derived cells. Mol. Ther. 2011, 19, 936–941. [Google Scholar] [CrossRef]
- Hartmann, L.; Neveling, K.; Borkens, S.; Schneider, H.; Freund, M.; Grassman, E.; Theiss, S.; Wawer, A.; Burdach, S.; Auerbach, A.D.; et al. Correct mRNA processing at a mutant TT splice donor in FANCC ameliorates the clinical phenotype in patients and is enhanced by delivery of suppressor U1 snRNAs. Am. J. Hum. Genet. 2010, 87, 480–493. [Google Scholar] [CrossRef]
- Schmid, F.; Glaus, E.; Barthelmes, D.; Fliegauf, M.; Gaspar, H.; Nürnberg, G.; Nürnberg, P.; Omran, H.; Berger, W.; Neidhardt, J. U1 snRNA-mediated gene therapeutic correction of splice defects caused by an exceptionally mild BBS mutation. Hum. Mutat. 2011, 32, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Kandels-Lewis, S.; Seraphin, B. Involvement of U6 snRNA in 5′ splice site selection. Science 1993, 262, 2035–2039. [Google Scholar] [CrossRef] [PubMed]
- Lesser, C.F.; Guthrie, C. Mutations in U6 snRNA that alter splice site specificity: Implications for the active site. Science 1993, 262, 1982–1988. [Google Scholar] [CrossRef] [PubMed]
- Schueler, U.H.; Kolter, T.; Kaneski, C.R.; Zirzow, G.C.; Sandhoff, K.; Brady, R.O. Correlation between enzyme activity and substrate storage in a cell culture model system for Gaucher disease. J. Inherit. Metab. Dis. 2004, 27, 649–658. [Google Scholar] [CrossRef]
- Parenti, G. Treating lysosomal storage diseases with pharmacological chaperones: From concept to clinics. EMBO Mol. Med. 2009, 1, 268–279. [Google Scholar] [CrossRef]
- Vuolo, D.; Do Nascimento, C.C.; D’Almeida, V. Reproduction in Animal Models of Lysosomal Storage Diseases: A Scoping Review. Front. Mol. Biosci. 2021, 8, 773384. [Google Scholar] [CrossRef]
- Rigon, L.; De Filippis, C.; Napoli, B.; Tomanin, R.; Orso, G. Exploiting the potential of drosophila models in lysosomal storage disorders: Pathological mechanisms and drug discovery. Biomedicines 2021, 9, 268. [Google Scholar] [CrossRef]
- Benetó, N.; Vilageliu, L.; Grinberg, D.; Canals, I. Sanfilippo Syndrome: Molecular Basis, Disease Models and Therapeutic Approaches. Int. J. Mol. Sci. 2020, 21, 7819. [Google Scholar] [CrossRef]
- Hartley, T.; Lemire, G.; Kernohan, K.D.; Howley, H.E.; Adams, D.R.; Boycott, K.M. New Diagnostic Approaches for Undiagnosed Rare Genetic Diseases. Annu. Rev. Genomics Hum. Genet. 2020, 21, 351–372. [Google Scholar] [CrossRef]
- Encarnacao, M.; Coutinho, M.F.; Cho, S.M.; Cardoso, M.T.; Ribeiro, I.; Chaves, P.; Santos, J.I.; Quelhas, D.; Lacerda, L.; Teles, E.L.; et al. NPC1 silent variant induces skipping of exon 11 (p.V562V) and unfolded protein response was found in a specific Niemann-Pick type C patient. Mol. Genet. Genom. Med. 2020, 8, e1451. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Brand Name | Drug | Year of Approval | Target Molecule | Treatment Result | Target Disease |
|---|---|---|---|---|---|
| Spinraza®, Biogen | Nusinersen | 2016 | SMN2 mRNA | Induces the inclusion of exon 7 in the SMN2 mRNA | Spinal muscular atrophy |
| Exondys 51™, Sarepta Therapeutics | Eteplirsen | 2016 | Dystrophin mRNA | Induces the exclusion of exon 51 of dystrophin mRNA | Duchenne muscular dystrophy |
| Vyondys 53™, Sarepta Therapeutics | Golodirsen | 2019 | Dystrophin mRNA | Induces the exclusion of exon 53 of dystrophin mRNA | Duchenne muscular dystrophy |
| Viltepso®, NS Pharma | Viltolarsen | 2020 | Dystrophin mRNA | Induces the exclusion of exon 53 of dystrophin mRNA | Duchenne muscular dystrophy |
| Amondys 45™, Sarepta Therapeutics | Casimersen | 2021 | Dystrophin mRNA | Induces the exclusion of exon 45 of dystrophin mRNA | Duchenne muscular dystrophy |
| MPS Type | Common Name(s) | Associated Gene | Enzyme Deficiency | Number of Mutations | % of Splicing Mutations | Treatment Options Available |
|---|---|---|---|---|---|---|
| I | Hurler, Scheie and Hurler–Scheie syndromes | IDUA | Alpha-L-iduronidase | 320 | 15.3 | ERT, HSCT |
| II | Hunter syndrome | IDS | Iduronate-2-sulfatase | 739 | 8.8 | ERT, HSCT |
| IIIA | Sanfilippo syndrome type A | SGSH | Heparan-N-sulfatase | 163 | 2.5 | - |
| IIIB | Sanfilippo syndrome type B | NAGLU | N-acetylglucosaminidase | 256 | 3.1 | - |
| IIIC | Sanfilippo syndrome type C | HGSNAT | Acetyl CoA glucosamine N-acetyltransferase | 91 | 17.6 | - |
| IIID | Sanfilippo syndrome type D | GNS | N-acetyl-glucosamine-6-sulfatase | 28 | 14.3 | - |
| IVA | Morquio syndrome type A | GALNS | N-acetylgalactosamine-6-sulfate sulfatase | 378 | 10.3 | ERT, HSCT |
| IVB | Morquio syndrome type B | GLB1 | β -galactosidase | 265 | 8.3 | - |
| VI | Maroteaux–Lamy syndrome | ARSB | Arylsulfatase B | 229 | 5.7 | ERT |
| VII | Sly syndrome | GUSB | β-glucuronidase | 81 | 7.4 | ERT |
| IX | Hyaluronidasedeficiency | HYAL1 | Hyaluronidase | 7 | 0 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santos, J.I.; Gonçalves, M.; Matos, L.; Moreira, L.; Carvalho, S.; Prata, M.J.; Coutinho, M.F.; Alves, S. Splicing Modulation as a Promising Therapeutic Strategy for Lysosomal Storage Disorders: The Mucopolysaccharidoses Example. Life 2022, 12, 608. https://doi.org/10.3390/life12050608
Santos JI, Gonçalves M, Matos L, Moreira L, Carvalho S, Prata MJ, Coutinho MF, Alves S. Splicing Modulation as a Promising Therapeutic Strategy for Lysosomal Storage Disorders: The Mucopolysaccharidoses Example. Life. 2022; 12(5):608. https://doi.org/10.3390/life12050608
Chicago/Turabian StyleSantos, Juliana Inês, Mariana Gonçalves, Liliana Matos, Luciana Moreira, Sofia Carvalho, Maria João Prata, Maria Francisca Coutinho, and Sandra Alves. 2022. "Splicing Modulation as a Promising Therapeutic Strategy for Lysosomal Storage Disorders: The Mucopolysaccharidoses Example" Life 12, no. 5: 608. https://doi.org/10.3390/life12050608
APA StyleSantos, J. I., Gonçalves, M., Matos, L., Moreira, L., Carvalho, S., Prata, M. J., Coutinho, M. F., & Alves, S. (2022). Splicing Modulation as a Promising Therapeutic Strategy for Lysosomal Storage Disorders: The Mucopolysaccharidoses Example. Life, 12(5), 608. https://doi.org/10.3390/life12050608